

The HSF1-CPT1a Pathway Is Differentially Regulated in NAFLD Progression


 and
and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. Patient Samples
2.3. Immunohistochemistry
2.4. Oil-Red-O Staining
2.5. Quantification of Intracellular Fat Accumulation
2.6. RNA Preparation and Real-Time PCR
2.7. siRNA Transfection
2.8. Western Blotting
2.9. Gel Shift Assays
2.10. Statistics
3. Results
3.1. HSF1 Is Abundant in Nuclei of Hepatocytes before and after Bariatric Surgery
3.2. Cultivation with Palmitic and Oleic Acid Induces Fat Accumulation in Hepatic Cell Lines
3.3. HSF1 Is Activated during Fat Accumulation in the NAFLD Model
3.4. Expression Analysis Revealed Strong Upregulation of CPT1a during Fat Accumulation
3.5. HSF1 Binds to a Putative Binding Site in the CPT1a Promotor
3.6. Knockdown of HSF1 Increases Fat Accumulation in HepG2 and Decreases CPT1a Expression
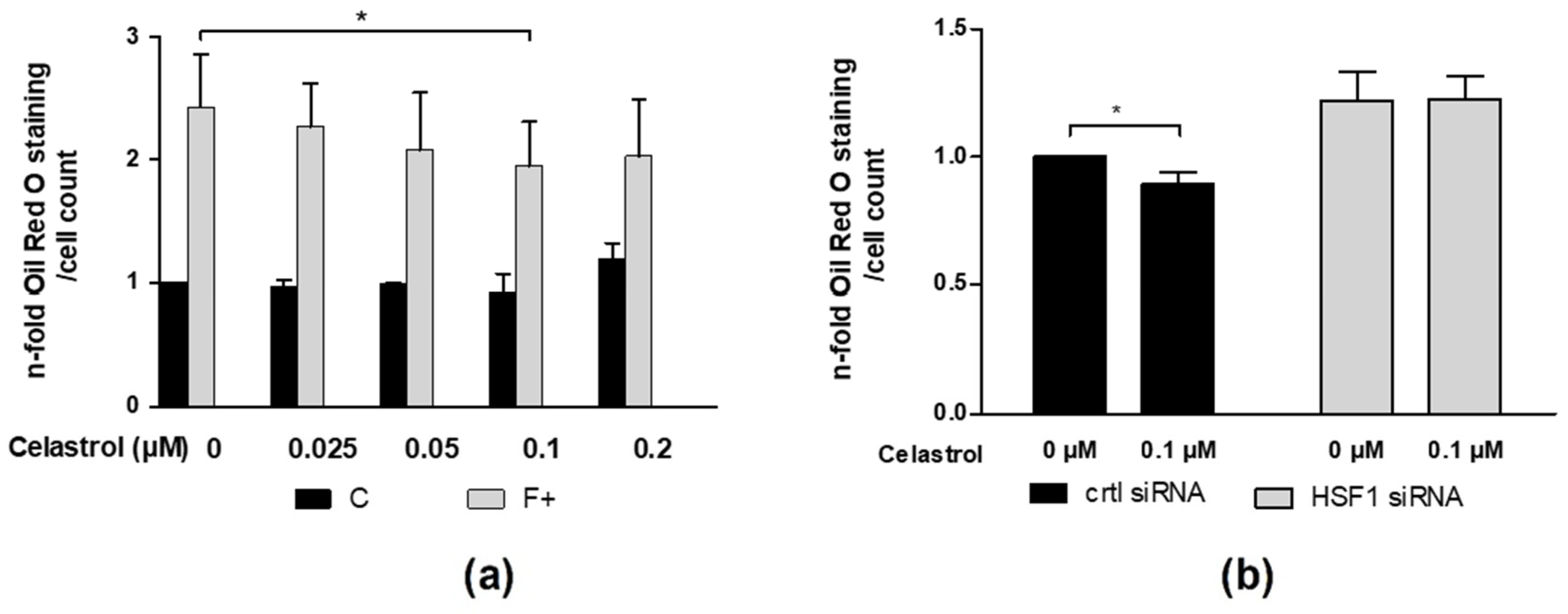
3.7. Activation of HSF1 by Celastrol Diminishes Fat Accumulation in HepG2 Cells
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Zhang, C.; Yang, M. Current Options and Future Directions for NAFLD and NASH Treatment. Int. J. Mol. Sci. 2021, 22, 7571. [Google Scholar] [CrossRef] [PubMed]
- Perumpail, B.J.; Khan, M.A.; Yoo, E.R.; Cholankeril, G.; Kim, D.; Ahmed, A. Clinical epidemiology and disease burden of nonalcoholic fatty liver disease. World J. Gastroenterol. 2017, 23, 8263–8276. [Google Scholar] [CrossRef] [PubMed]
- Buzzetti, E.; Pinzani, M.; Tsochatzis, E.A. The multiple-hit pathogenesis of non-alcoholic fatty liver disease (NAFLD). Metabolism 2016, 65, 1038–1048. [Google Scholar] [CrossRef]
- Aller, R.; Fernandez-Rodriguez, C.; Lo Iacono, O.; Banares, R.; Abad, J.; Carrion, J.A.; Garcia-Monzon, C.; Caballeria, J.; Be-renguer, M.; Rodriguez-Peralvarez, M.; et al. Consensus document. Management of non-alcoholic fatty liver disease (NAFLD). Clinical practice guideline. Gastroenterol. Hepatol. 2018, 41, 328–349. [Google Scholar] [CrossRef]
- Ampuero, J.; Sánchez-Torrijos, Y.; Aguilera, V.; Bellido, F.; Romero-Gómez, M. New therapeutic perspectives in non-alcoholic steatohepatitis. Gastroenterol. Hepatol. 2018, 41, 128–142. [Google Scholar] [CrossRef]
- Angulo, P. NAFLD, obesity, and bariatric surgery. Gastroenterology 2006, 130, 1848–1852. [Google Scholar] [CrossRef] [PubMed]
- Bedossa, P.; Tordjman, J.; Aron-Wisnewsky, J.; Poitou, C.; Oppert, J.-M.; Torcivia, A.; Bouillot, J.-L.; Paradis, V.; Ratziu, V.; Clément, K. Systematic review of bariatric surgery liver biopsies clarifies the natural history of liver disease in patients with severe obesity. Gut 2016, 66, 1688–1696. [Google Scholar] [CrossRef]
- Marchesini, G.; Petta, S.; Grave, R.D. Diet, weight loss, and liver health in nonalcoholic fatty liver disease: Pathophysiology, evidence, and practice. Hepatology 2016, 63, 2032–2043. [Google Scholar] [CrossRef]
- Sheka, A.C.; Adeyi, O.; Thompson, J.; Hameed, B.; Crawford, P.A.; Ikramuddin, S. Nonalcoholic Steatohepatitis: A Review. JAMA 2020, 323, 1175–1183. [Google Scholar] [CrossRef]
- Ahrens, M.; Ammerpohl, O.; von Schönfels, W.; Kolarova, J.; Bens, S.; Itzel, T.; Teufel, A.; Herrmann, A.; Brosch, M.; Hinrichsen, H.; et al. DNA Methylation Analysis in Nonalcoholic Fatty Liver Disease Suggests Distinct Disease-Specific and Remodeling Signatures after Bariatric Surgery. Cell Metab. 2013, 18, 296–302. [Google Scholar] [CrossRef]
- Gomez-Pastor, R.; Burchfiel, E.T.; Thiele, D.J. Regulation of heat shock transcription factors and their roles in physiology and disease. Nat. Rev. Mol. Cell Biol. 2017, 19, 4–19. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Yang, W.; Wang, J.; Meng, Y.; Guan, Y.; Yang, J. FAM3 gene family: A promising therapeutical target for NAFLD and type 2 diabetes. Metabolism 2017, 81, 71–82. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zheng, X.; Xu, F.; Liang, H.; Cao, H.; Cai, M.; Xu, W.; Weng, J. SIRT1/HSF1/HSP pathway is essential for exenatide-alleviated, lipid-induced hepatic endoplasmic reticulum stress. Hepatology 2017, 66, 809–824. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, X.; Xu, L.; Alberobello, A.T.; Gavrilova, O.; Bagattin, A.; Skarulis, M.; Liu, J.; Finkel, T.; Mueller, E. Celastrol Protects against Obesity and Metabolic Dysfunction through Activation of a HSF1-PGC1α Transcriptional Axis. Cell Metab. 2015, 22, 695–708. [Google Scholar] [CrossRef] [Green Version]
- Pappas, A.; Anthonavage, M.; Gordon, J.S. Metabolic Fate and Selective Utilization of Major Fatty Acids in Human Sebaceous Gland. J. Investig. Dermatol. 2002, 118, 164–171. [Google Scholar] [CrossRef] [Green Version]
- Kleiner, D.E.; Brunt, E.M.; Van Natta, M.; Behling, C.; Contos, M.J.; Cummings, O.W.; Ferrell, L.D.; Liu, Y.-C.; Torbenson, M.S.; Unalp-Arida, A.; et al. Design and validation of a histological scoring system for nonalcoholic fatty liver disease. Hepatology 2005, 41, 1313–1321. [Google Scholar] [CrossRef]
- Mehlem, A.; Hagberg, C.; Muhl, L.; Eriksson, U.; Falkevall, A. Imaging of neutral lipids by oil red O for analyzing the metabolic status in health and disease. Nat. Protoc. 2013, 8, 1149–1154. [Google Scholar] [CrossRef] [Green Version]
- Chazotte, B. Labeling nuclear DNA using DAPI. Cold Spring Harb. Protoc. 2011, 2011, pdb.prot5556. [Google Scholar] [CrossRef] [Green Version]
- Geismann, C.; Erhart, W.; Grohmann, F.; Schreiber, S.; Schneider, G.; Schäfer, H.; Arlt, A. TRAIL/NF-kappaB/CX3CL1 Mediated Onco-Immuno Crosstalk Leading to TRAIL Resistance of Pancreatic Cancer Cell Lines. Int. J. Mol. Sci. 2018, 19, 1661. [Google Scholar] [CrossRef] [Green Version]
- Geismann, C.; Grohmann, F.; Dreher, A.; Häsler, R.; Rosenstiel, P.; Legler, K.; Hauser, C.; Egberts, J.; Sipos, B.; Schreiber, S. Role of CCL20 mediated immune cell recruitment in NF-kappaB mediated TRAIL resistance of pancreatic cancer. Biochim. Biophys. Acta Mol. Cell Res. 2017, 1864, 782–796. [Google Scholar] [CrossRef]
- Tsai, T.-H.; Chen, E.; Li, L.; Saha, P.; Lee, H.-J.; Huang, L.-S.; Shelness, G.S.; Chan, L.; Chang, B.H.-J. The constitutive lipid droplet protein PLIN2 regulates autophagy in liver. Autophagy 2017, 13, 1130–1144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Z.; Ding, L.; Yang, W.; Wang, J.; Chen, L.; Chang, Y.; Geng, B.; Cui, Q.; Guan, Y.; Yang, J. Hepatic Activation of the FAM3C-HSF1-CaM Pathway Attenuates Hyperglycemia of Obese Diabetic Mice. Diabetes 2017, 66, 1185–1197. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jin, X.; Moskophidis, D.; Mivechi, N.F. Heat Shock Transcription Factor 1 Is a Key Determinant of HCC Development by Regulating Hepatic Steatosis and Metabolic Syndrome. Cell Metab. 2011, 14, 91–103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rao, Y.; Li, C.; Hu, Y.; Xu, Y.; Song, B.; Guo, S.; Jiang, Z.; Zhao, D.; Chen, S.; Tan, J.; et al. A novel HSF1 activator ameliorates non-alcoholic steatohepatitis by stimulating mitochondrial adaptive oxidation. J. Cereb. Blood Flow Metab. 2022, 179, 1411–1432. [Google Scholar] [CrossRef]
- Westerheide, S.D.; Bosman, J.D.; Mbadugha, B.N.; Kawahara, T.L.; Matsumoto, G.; Kim, S.; Gu, W.; Devlin, J.P.; Silverman, R.B.; Morimoto, R.I. Celastrols as Inducers of the Heat Shock Response and Cytoprotection. J. Biol. Chem. 2004, 279, 56053–56060. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Ahn, Y.H.; Benjamin, I.J.; Honda, T.; Hicks, R.J.; Calabrese, V.; Cole, P.A.; Dinkova-Kostova, A.T. HSF1-dependent upregulation of Hsp70 by sulfhydryl-reactive inducers of the KEAP1/NRF2/ARE pathway. Chem Biol. 2011, 18, 1355–1361. [Google Scholar] [CrossRef] [Green Version]
- Di Naso, F.C.; Porto, R.R.; Fillmann, H.S.; Maggioni, L.; Padoin, A.V.; Ramos, R.J.; Mottin, C.C.; Bittencourt, A.; Marroni, N.A.; de Bittencourt, P.I., Jr. Obesity depresses the anti-inflammatory HSP70 pathway, contributing to NAFLD progression. Obesity 2015, 23, 120–129. [Google Scholar] [CrossRef]
- Stefanovic-Racic, M.; Perdomo, G.; Mantell, B.S.; Sipula, I.J.; Brown, N.F.; O’Doherty, R.M. A moderate increase in carnitine palmitoyltransferase 1a activity is sufficient to substantially reduce hepatic triglyceride levels. Am. J. Physiol. Metab. 2008, 294, E969–E977. [Google Scholar] [CrossRef] [Green Version]
- Orellana-Gavaldà, J.M.; Herrero, L.; Malandrino, M.I.; Pañeda, A.; Rodríguez-Peña, M.S.; Petry, H.; Asins, G.; Van Deventer, S.; Hegardt, F.G.; Serra, D. Molecular therapy for obesity and diabetes based on a long-term increase in hepatic fatty-acid oxidation. Hepatology 2010, 53, 821–832. [Google Scholar] [CrossRef]
- Weber, M.; Mera, P.; Casas, J.; Salvador, J.; Rodríguez, A.; Alonso, S.; Sebastián, D.; Soler-Vázquez, M.C.; Montironi, C.; Recalde, S.; et al. Liver CPT1A gene therapy reduces diet-induced hepatic steatosis in mice and highlights potential lipid biomarkers for human NAFLD. FASEB J. 2020, 34, 11816–11837. [Google Scholar] [CrossRef]
- Lin, C.W.; Peng, Y.J.; Lin, Y.Y.; Mersmann, H.J.; Ding, S.T. LRRK2 Regulates CPT1A to Promote beta-Oxidation in HepG2 Cells. Molecules 2020, 25, 4122. [Google Scholar] [CrossRef] [PubMed]
- Moody, L.; Xu, G.B.; Chen, H.; Pan, Y.-X. Epigenetic regulation of carnitine palmitoyltransferase 1 (Cpt1a) by high fat diet. Biochim. Biophys. Acta (BBA)-Gene Regul. Mech. 2018, 1862, 141–152. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameter | Pre-Op | Post-Op |
|---|---|---|
| Number of patients | 14 | 14 |
| Age in years (mean ± SD) | 46 ± 12.1 | 46 ± 12.1 |
| Female/male (number) | 12/2 | 12/2 |
| BMI (kg/m2) (mean ± SD) | 51.8 ± 9.8 | 38.1 ± 9.1 |
| NAS (mean ± SD) | 3.1 ± 2.3 | 1 ± 1.2 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Breternitz, W.; Sandkühler, F.; Grohmann, F.; Hampe, J.; Brosch, M.; Herrmann, A.; Schafmayer, C.; Meinhardt, C.; Schreiber, S.; Arlt, A.; et al. The HSF1-CPT1a Pathway Is Differentially Regulated in NAFLD Progression. Cells 2022, 11, 3504. https://doi.org/10.3390/cells11213504
Breternitz W, Sandkühler F, Grohmann F, Hampe J, Brosch M, Herrmann A, Schafmayer C, Meinhardt C, Schreiber S, Arlt A, et al. The HSF1-CPT1a Pathway Is Differentially Regulated in NAFLD Progression. Cells. 2022; 11(21):3504. https://doi.org/10.3390/cells11213504
Chicago/Turabian StyleBreternitz, Wiebke, Friedrich Sandkühler, Frauke Grohmann, Jochen Hampe, Mario Brosch, Alexander Herrmann, Clemens Schafmayer, Christian Meinhardt, Stefan Schreiber, Alexander Arlt, and et al. 2022. "The HSF1-CPT1a Pathway Is Differentially Regulated in NAFLD Progression" Cells 11, no. 21: 3504. https://doi.org/10.3390/cells11213504
APA StyleBreternitz, W., Sandkühler, F., Grohmann, F., Hampe, J., Brosch, M., Herrmann, A., Schafmayer, C., Meinhardt, C., Schreiber, S., Arlt, A., & Geismann, C. (2022). The HSF1-CPT1a Pathway Is Differentially Regulated in NAFLD Progression. Cells, 11(21), 3504. https://doi.org/10.3390/cells11213504