


S100A8-Mediated NLRP3 Inflammasome-Dependent Pyroptosis in Macrophages Facilitates Liver Fibrosis Progression


Abstract
:1. Introduction
2. Materials and Methods
2.1. Human Samples
2.2. CCl4-Induced Liver Fibrosis Mouse Models
2.3. Mouse Serum and Liver Samples Preparation
2.4. Analysis of Liver Function, Liver Pathology, and Fibrosis
2.5. Immunohistochemical Staining
2.6. Immunofluorescence Staining
2.7. Enzyme-Linked Immunosorbent Assay (ELISA)
2.8. Preparation of Recombinant Proteins
2.9. Cell Culture and In Vitro Treatment
2.10. RNA Extraction and Quantitative Real-Time PCR
2.11. Western Blot
2.12. Flow Cytometry
2.13. Statistical Analysis
3. Results
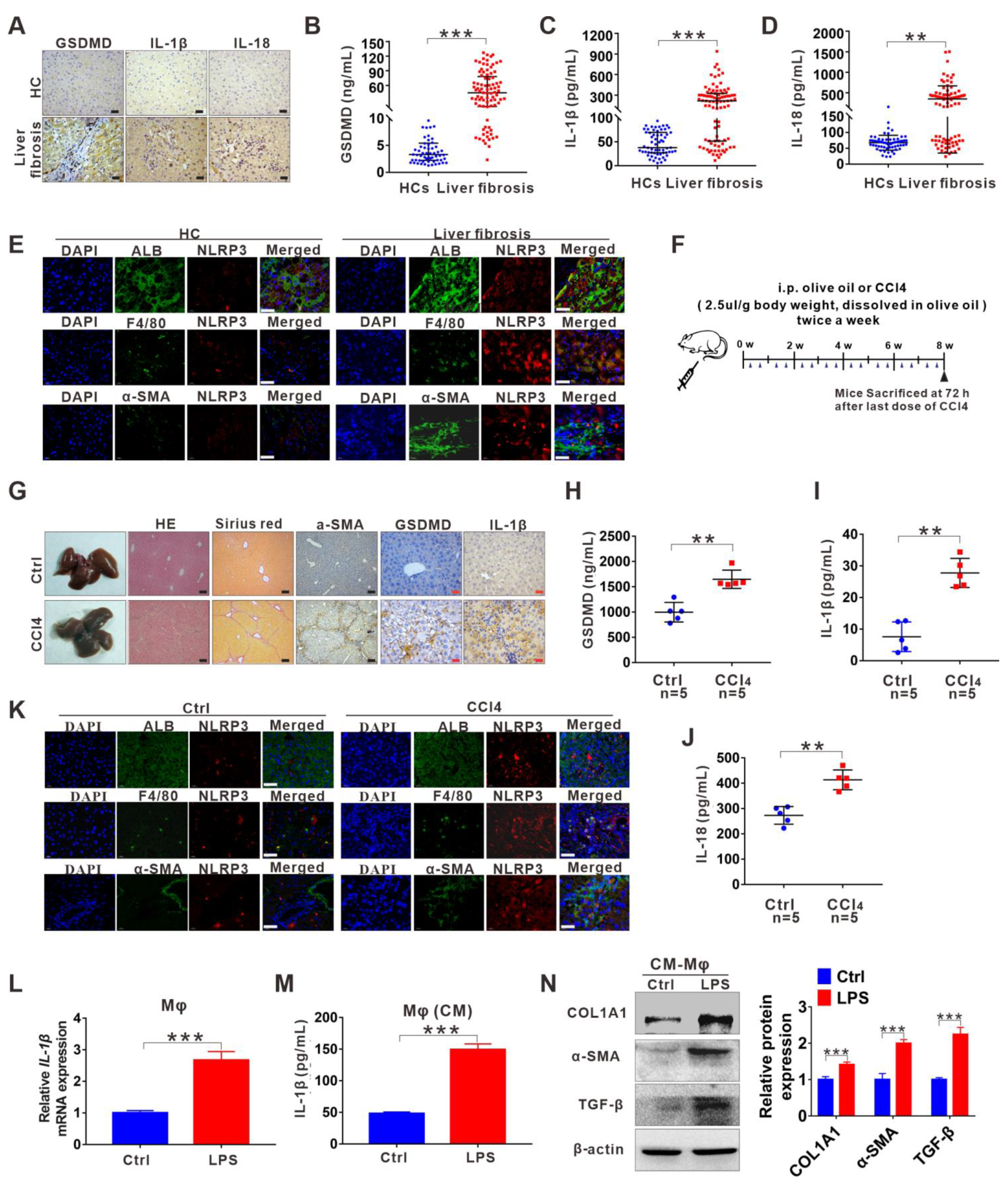
3.1. NLRP3 Inflammasome-Dependent Pyroptosis Occurs in Liver Fibrosis
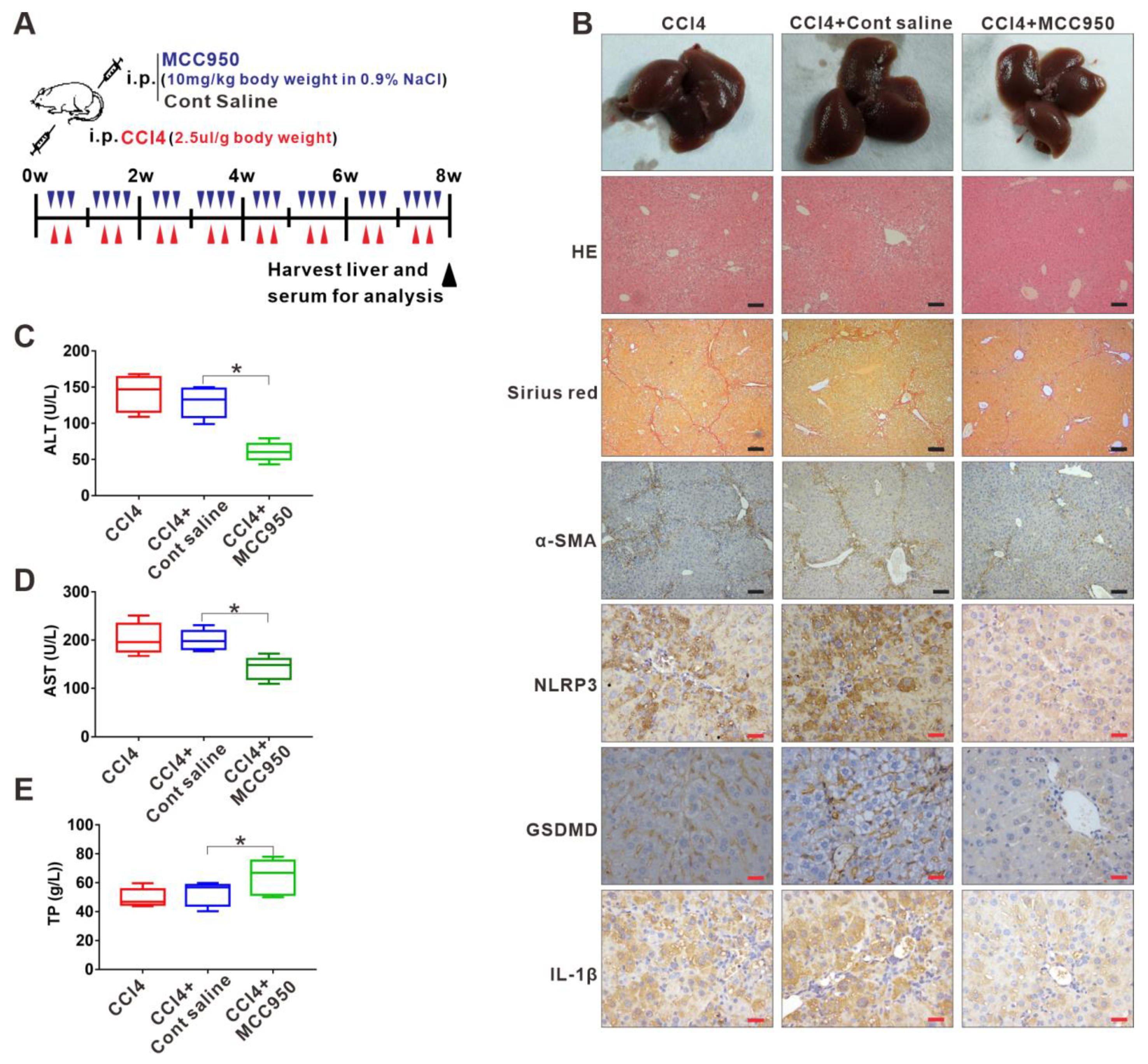
3.2. Inhibition of NLRP3 Inflammasome-Dependent Pyroptosis Alleviates Liver Fibrosis Progression
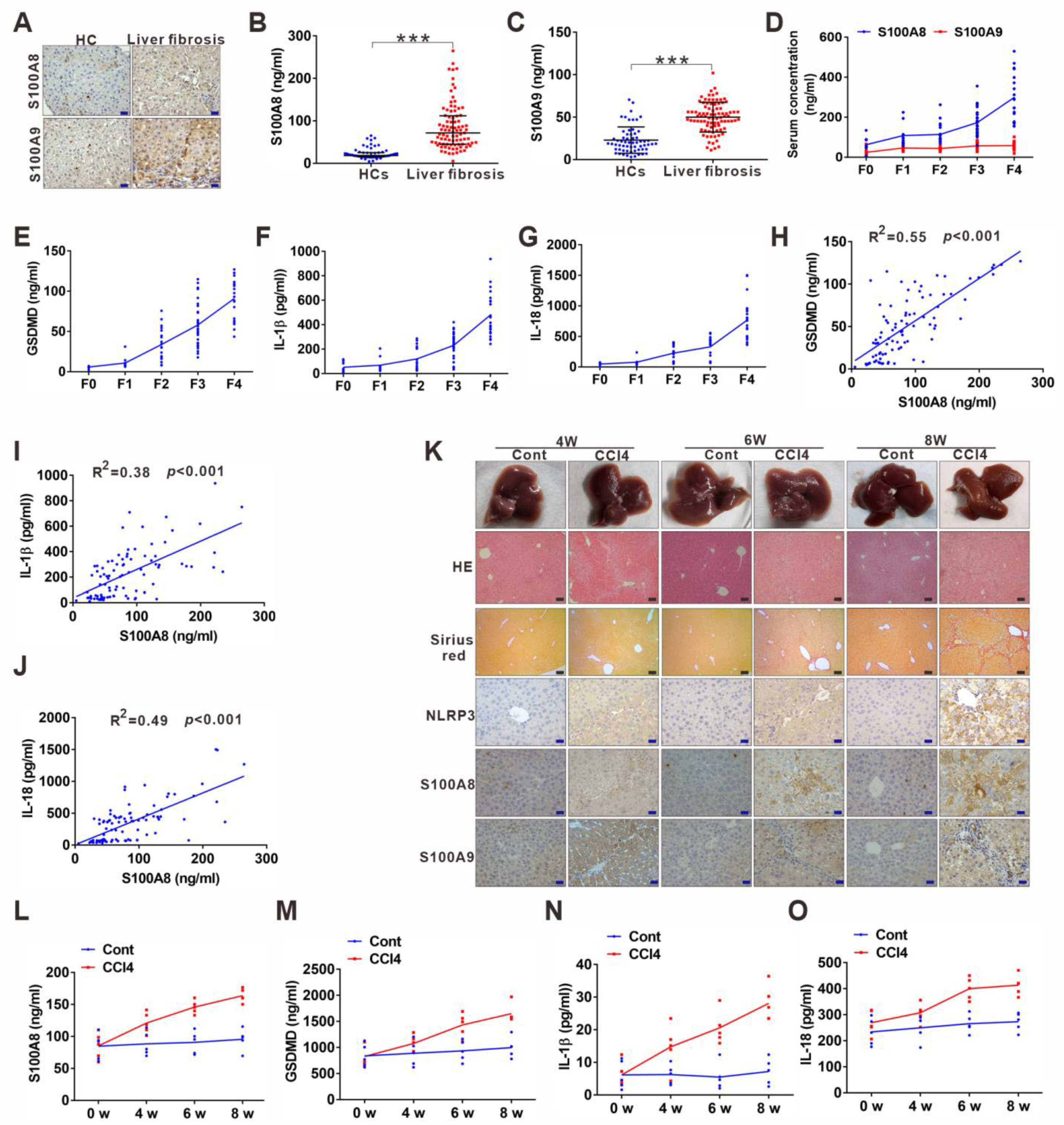
3.3. DAMP S100A8 along with NLRP3 Inflammasome-Dependent Pyroptosis Is Positively Related to the Progression of Liver Fibrosis
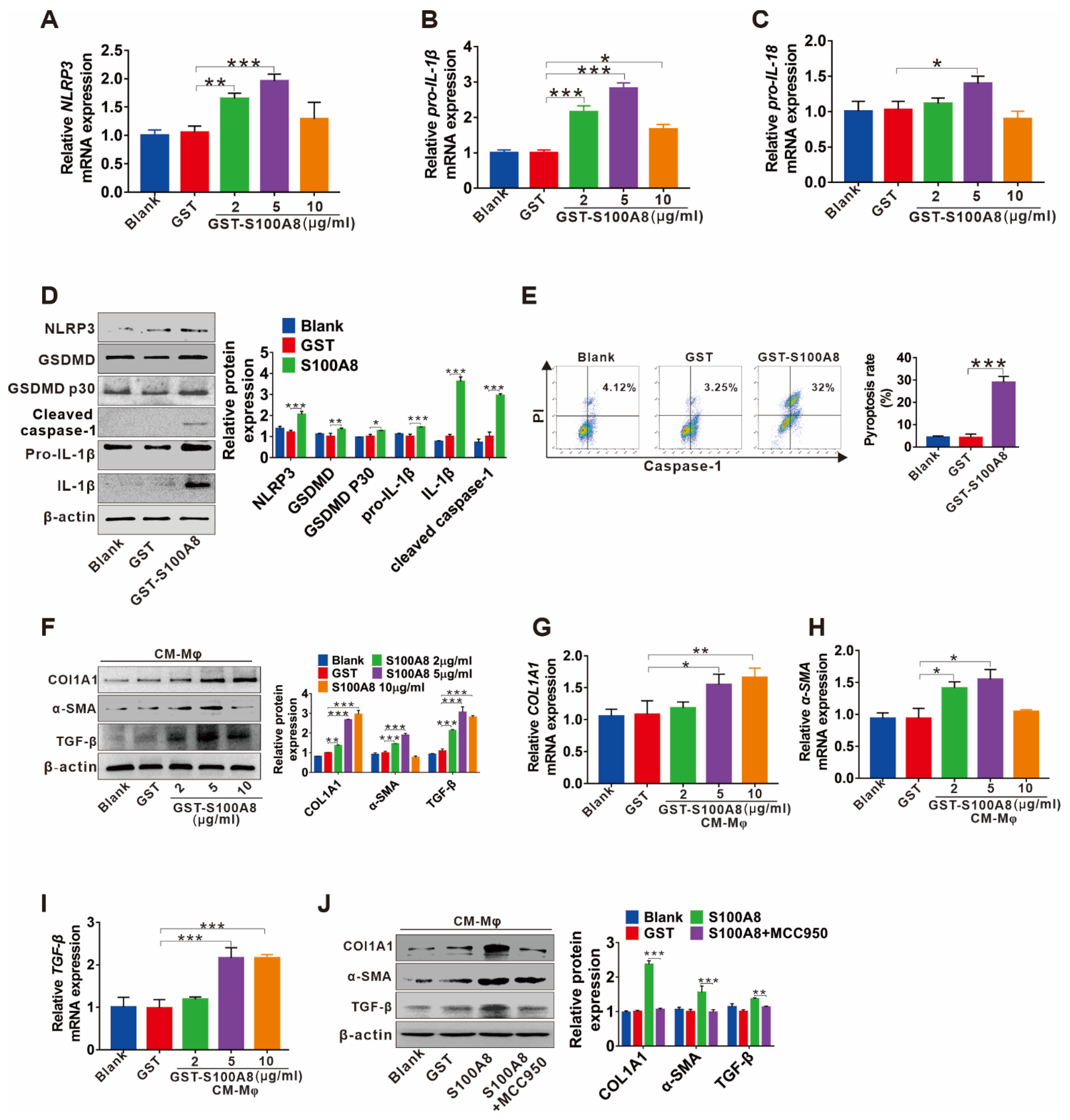
3.4. S100A8-Mediated NLRP3 Inflammasome-Dependent Pyroptotic Macrophage Death Amplifies the Activation of Human Hepatic Stellate Cells
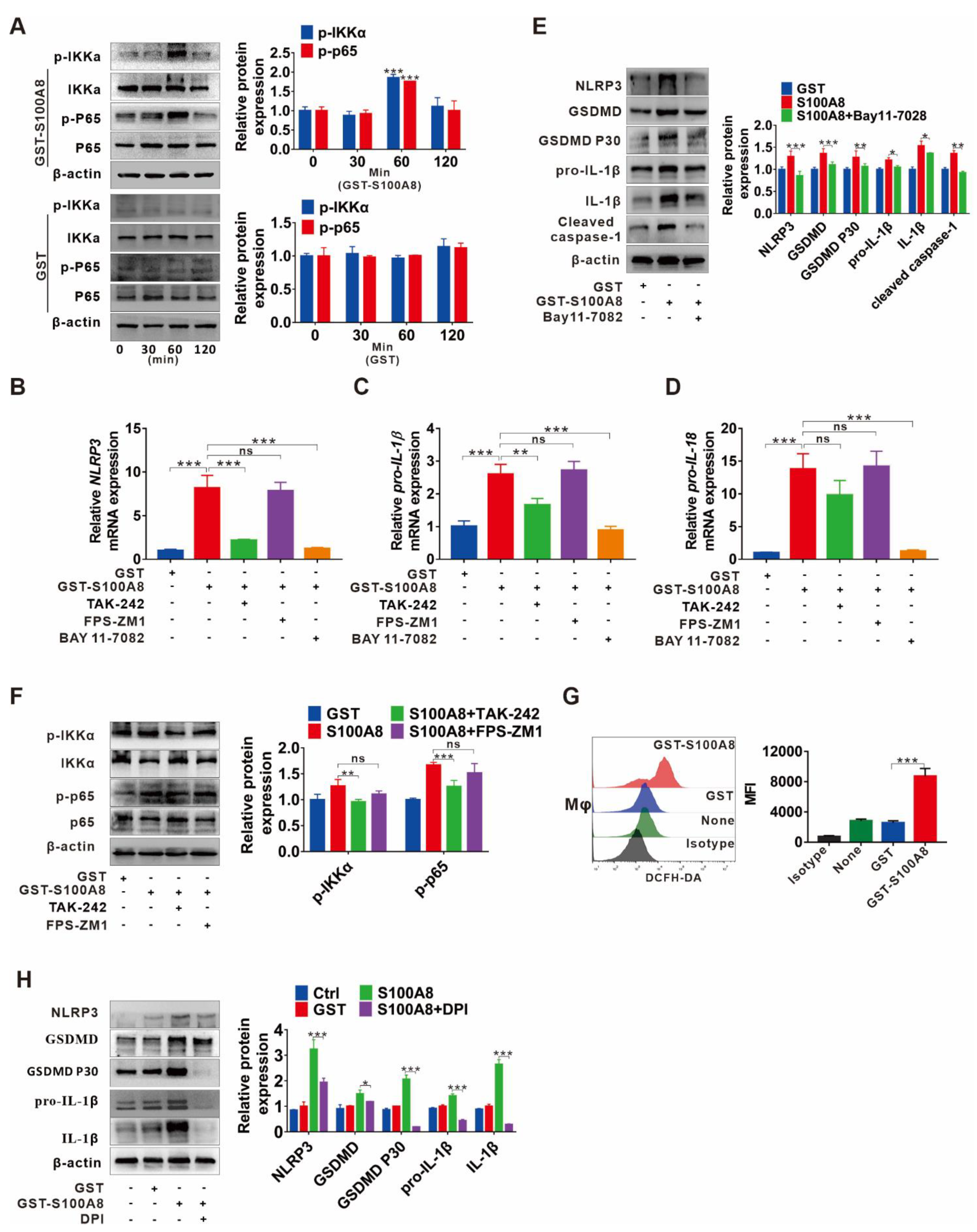
3.5. TLR4/NF-κB Signaling Cascade and ROS Abundance Are Responsible for S100A8-Induced NLRP3 Inflammasome-Dependent Pyroptotic Death in Macrophages
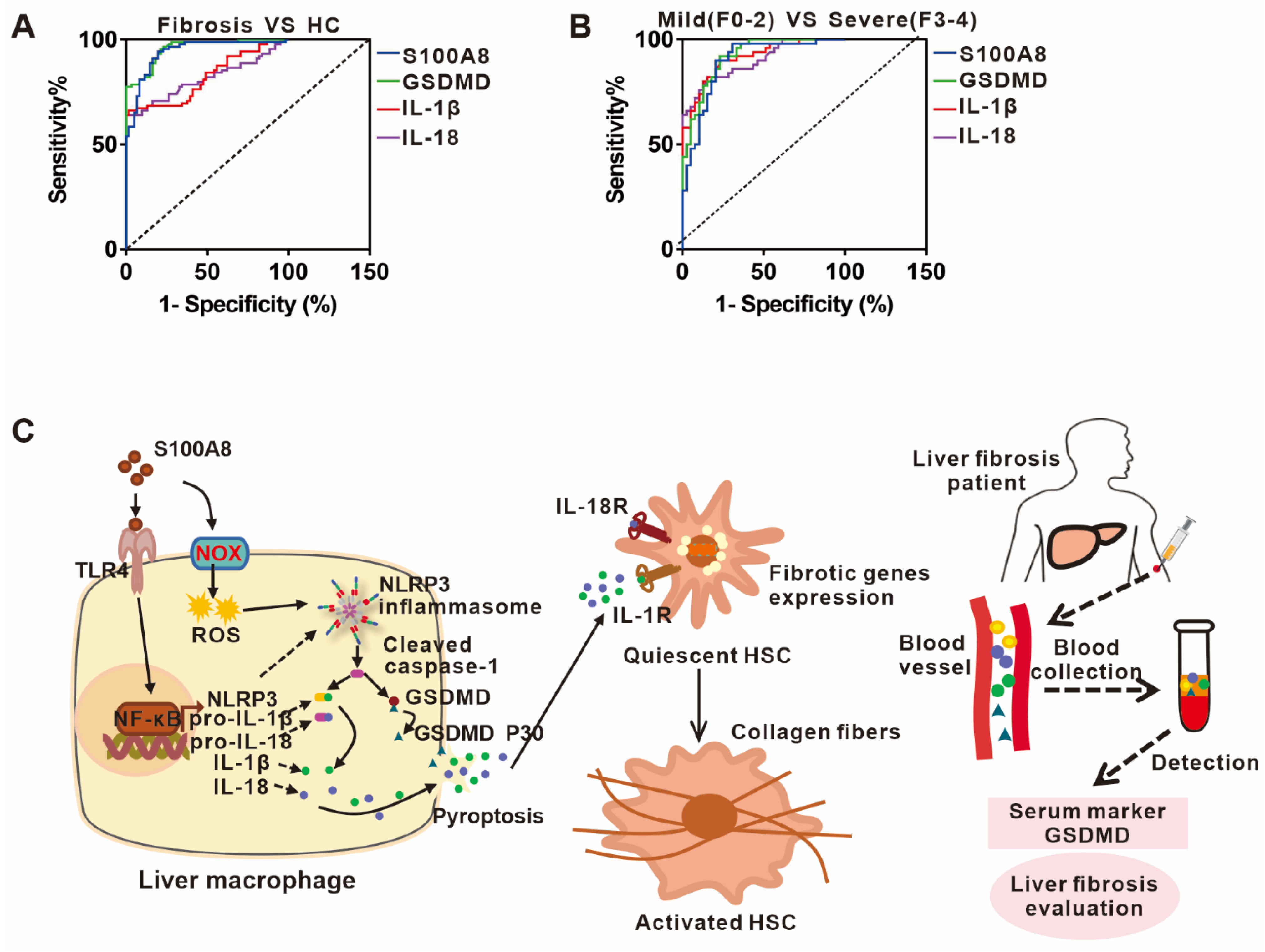
3.6. The Potential Predictive Powers of S100A8, GSDMD, IL-1β, and IL-18 for the Occurrence and Severity of Liver Fibrosis
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Hernandez-Gea, V.; Friedman, S.L. Pathogenesis of liver fibrosis. Annu. Rev. Pathol. 2011, 6, 425–456. [Google Scholar] [CrossRef] [PubMed]
- Iredale, J.P. Models of liver fibrosis: Exploring the dynamic nature of inflammation and repair in a solid organ. J. Clin. Investig. 2007, 117, 539–548. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lo, R.C.; Kim, H. Histopathological evaluation of liver fibrosis and cirrhosis regression. Clin. Mol. Hepatol. 2017, 23, 302–307. [Google Scholar] [CrossRef] [Green Version]
- Schuppan, D.; Kim, Y.O. Evolving therapies for liver fibrosis. J. Clin. Investig. 2013, 123, 1887–1901. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Strowig, T.; Henao-Mejia, J.; Elinav, E.; Flavell, R. Inflammasomes in health and disease. Nature 2012, 481, 278–286. [Google Scholar] [CrossRef]
- Sharma, B.R.; Kanneganti, T.-D. NLRP3 inflammasome in cancer and metabolic diseases. Nat. Immunol. 2021, 22, 550–559. [Google Scholar] [CrossRef]
- He, Y.; Hara, H.; Núñez, G. Mechanism and Regulation of NLRP3 Inflammasome Activation. Trends Biochem. Sci. 2016, 41, 1012–1021. [Google Scholar] [CrossRef] [Green Version]
- Tian, R.; Zhu, Y.; Yao, J.; Meng, X.; Wang, J.; Xie, H.; Wang, R. NLRP3 participates in the regulation of EMT in bleomycin-induced pulmonary fibrosis. Exp. Cell Res. 2017, 357, 328–334. [Google Scholar] [CrossRef]
- Wu, M.; Han, W.; Song, S.; Du, Y.; Liu, C.; Chen, N.; Wu, H.; Shi, Y.; Duan, H. NLRP3 deficiency ameliorates renal inflammation and fibrosis in diabetic mice. Mol. Cell. Endocrinol. 2018, 478, 115–125. [Google Scholar] [CrossRef]
- Pinar, A.A.; Scott, T.E.; Huuskes, B.M.; Cáceres, F.E.T.; Kemp-Harper, B.K.; Samuel, C.S. Targeting the NLRP3 inflammasome to treat cardiovascular fibrosis. Pharmacol. Ther. 2020, 209, 107511. [Google Scholar] [CrossRef]
- Wree, A.; Eguchi, A.; McGeough, M.D.; Pena, C.A.; Johnson, C.D.; Canbay, A.; Hoffman, H.M.; Feldstein, A.E. NLRP3 inflammasome activation results in hepatocyte pyroptosis, liver inflammation, and fibrosis in mice. Hepatology 2013, 59, 898–910. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wree, A.; McGeough, M.D.; Peña, C.A.; Schlattjan, M.; Li, H.; Inzaugarat, M.E.; Messer, K.; Canbay, A.; Hoffman, H.M.; Feldstein, A.E. NLRP3 inflammasome activation is required for fibrosis development in NAFLD. Klin. Wochenschr. 2014, 92, 1069–1082. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mridha, A.R.; Wree, A.; Robertson, A.A.; Yeh, M.M.; Johnson, C.D.; Van Rooyen, D.M.; Haczeyni, F.; Teoh, N.C.-H.; Savard, C.; Ioannou, G.N.; et al. NLRP3 inflammasome blockade reduces liver inflammation and fibrosis in experimental NASH in mice. J. Hepatol. 2017, 66, 1037–1046. [Google Scholar] [CrossRef]
- Reiter, F.P.; Wimmer, R.; Wottke, L.; Artmann, R.; Nagel, J.M.; Carranza, M.O.; Mayr, D.; Rust, C.; Fickert, P.; Trauner, M.; et al. Role of interleukin-1 and its antagonism of hepatic stellate cell proliferation and liver fibrosis in the Abcb4(-/-) mouse model. World J. Hepatol. 2016, 8, 401–410. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Zhan, X.; Wu, R.; You, Y.; Chen, W.; Duan, L. Assessment of Pyroptosis-Related Indicators as Potential Biomarkers and Their Association with Severity in Patients with Liver Cirrhosis. J. Inflamm. Res. 2021, ume 14, 3185–3196. [Google Scholar] [CrossRef]
- Youssef, P.; Roth, J.; Frosch, M.; Costello, P.; Fitzgerald, O.; Sorg, C. Expression of myeloid related proteins (MRP) 8 and 14 and the MRP8/14 heterodimer in rheumatoid arthritis synovial membrane. J. Rheumatol. 1999, 26, 2523–2528. [Google Scholar]
- Sands, B.E. Biomarkers of Inflammation in Inflammatory Bowel Disease. Gastroenterology 2015, 149, 1275–1285.e2. [Google Scholar] [CrossRef]
- Achouiti, A.; Vogl, T.; Endeman, H.; Mortensen, B.L.; Laterre, P.F.; Wittebole, X.; van Zoelen, M.A.; Zhang, Y.; Hoogerwerf, J.J.; Florquin, S.; et al. Myeloid-related protein-8/14 facilitates bacterial growth during pneumococcal pneumonia. Thorax 2014, 69, 1034–1042. [Google Scholar] [CrossRef] [Green Version]
- Sallman, D.A.; Cluzeau, T.; Basiorka, A.A.; List, A. Unraveling the Pathogenesis of MDS: The NLRP3 inflammasome and Py-roptosis Drive the MDS Phenotype. Front. Oncol. 2016, 6, 151. [Google Scholar] [CrossRef] [Green Version]
- Kim, K.; Kim, H.J.; Binas, B.; Kang, J.H.; Chung, I.Y. Inflammatory mediators ATP and S100A12 activate the NLRP3 inflammasome to induce MUC5AC production in airway epithelial cells. Biochem. Biophys. Res. Commun. 2018, 503, 657–664. [Google Scholar] [CrossRef]
- Wu, R.; Zhang, Y.; Xiang, Y.; Tang, Y.; Cui, F.; Cao, J.; Zhou, L.; You, Y.; Duan, L. Association between serum S100A9 levels and liver necroin-flammation in chronic hepatitis B. J. Transl. Med. 2018, 16, 83. [Google Scholar] [CrossRef] [PubMed]
- Batts, K.P.; Ludwig, J. Chronic hepatitis. An update on terminology and reporting. Am. J. Surg. Pathol. 1995, 19, 1409–1417. [Google Scholar] [CrossRef] [PubMed]
- Duan, L.; Wu, R.; Ye, L.; Wang, H.; Yang, X.; Zhang, Y.; Chen, X.; Zuo, G.; Zhang, Y.; Weng, Y.; et al. S100A8 and S100A9 are associated with colorectal carcinoma pro-gression and contribute to colorectal carcinoma cell survival and migration via Wnt/beta-catenin pathway. PLoS ONE 2013, 8, e62092. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vogl, T.; Tenbrock, K.; Ludwig, S.; Leukert, N.; Ehrhardt, C.; van Zoelen, M.A.; Nacken, W.; Foell, D.; van der Pol, l.T.; Sorg, C.; et al. Mrp8 and Mrp14 are endogenous activators of Toll- like receptor 4, promoting lethal, endotoxin-induced shock. Nat. Med. 2007, 13, 1042–1049. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Yuan, Y.; Huang, Z.-X.; Chen, H.; Lan, R.; Wang, Z.; Lai, K.; Chen, H.; Chen, Z.; Zou, Z.; et al. GSDME-mediated pyroptosis promotes inflammation and fibrosis in obstructive nephropathy. Cell Death Differ. 2021, 28, 2333–2350. [Google Scholar] [CrossRef]
- Li, F.J.; Surolia, R.; Li, H.; Wang, Z.; Liu, G.; Kulkarni, T.; Massicano, A.V.F.; Mobley, J.A.; Mondal, S.; de Andrade, J.A.; et al. Citrullinated vimentin mediates development and progression of lung fibrosis. Sci. Transl. Med. 2021, 13. [Google Scholar] [CrossRef]
- Hofmann, M.A.; Drury, S.; Fu, C.; Qu, W.; Taguchi, A.; Lu, Y.; Avila, C.; Kambham, N.; Bierhaus, A.; Nawroth, P.; et al. RAGE mediates a novel proinflammatory axis: A central cell surface receptor for S100/calgranulin polypeptides. Cell 1999, 97, 889–901. [Google Scholar] [CrossRef] [Green Version]
- Jiang, J.X.; Chen, X.; Serizawa, N.; Szyndralewiez, C.; Page, P.; Schröder, K.; Brandes, R.P.; Devaraj, S.; Török, N.J. Liver fibrosis and hepatocyte apoptosis are attenuated by GKT137831, a novel NOX4/NOX1 inhibitor in vivo. Free Radic. Biol. Med. 2012, 53, 289–296. [Google Scholar] [CrossRef] [Green Version]
- Simard, J.C.; Cesaro, A.; Chapeton-Montes, J.; Tardif, M.; Antoine, F.; Girard, D.; Tessier, P.A. S100A8 and S100A9 Induce Cytokine Ex-pression and Regulate the NLRP3 Inflammasome via ROS-Dependent Activation of NF-kappa B. PLoS ONE 2013, 8, e72138. [Google Scholar] [CrossRef] [Green Version]
- Benedyk, M.; Sopalla, C.; Nacken, W.; Bode, G.; Melkonyan, H.; Banfi, B.; Kerkhoff, C. HaCaT keratinocytes overexpressing the S100 proteins S100A8 and S100A9 show increased NADPH oxidase and NF-kappa B activities. J. Investig. Dermatol. 2007, 127, 2001–2011. [Google Scholar] [CrossRef] [Green Version]
- Liang, F.Q.; Zhang, F.; Zhang, L.L.; Wei, W. The advances in pyroptosis initiated by inflammasome in inflammatory and im-mune diseases. Inflamm. Res. 2020, 69, 159–166. [Google Scholar] [CrossRef] [PubMed]
- Alegre, F.; Pelegrin, P.; Feldstein, A.E. Inflammasomes in Liver Fibrosis. Semin. Liver Dis. 2017, 37, 119–127. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, C.; He, L.; Zhang, J.; Ma, H.; Yuan, X.; Hu, G.; Tao, L.; Zhang, J.; Meng, J. Fluorofenidone attenuates pulmonary inflammation and fibrosis via inhibiting the activation of NALP3 inflammasome and IL-1/IL-1R1/MyD88/NF-B pathway. J. Cell. Mol. Med. 2016, 20, 2064–2077. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Artlett, C.M.; Sassi-Gaha, S.; Rieger, J.L.; Boesteanu, A.C.; Feghali-Bostwick, C.A.; Katsikis, P. The inflammasome activating caspase 1 mediates fibrosis and myofibroblast differentiation in systemic sclerosis. Arthritis Care Res. 2011, 63, 3563–3574. [Google Scholar] [CrossRef] [PubMed]
- Pan, J.; Ou, Z.; Cai, C.; Li, P.; Gong, J.; Ruan, X.Z.; He, K. Fatty acid activates NLRP3 inflammasomes in mouse Kupffer cells through mitochondrial DNA release. Cell. Immunol. 2018, 332, 111–120. [Google Scholar] [CrossRef] [PubMed]
- Mangan, M.S.J.; Olhava, E.J.; Roush, W.R.; Seidel, H.M.; Glick, G.D.; Latz, E. Targeting the NLRP3 inflammasome in in-flammatory diseases. Nat. Rev. Drug Discov. 2018, 17, 688. [Google Scholar] [CrossRef] [Green Version]
- Medzhitov, R. Origin and physiological roles of inflammation. Nature 2008, 454, 428–435. [Google Scholar] [CrossRef]
- Leimkühler, N.B.; Gleitz, H.F.; Ronghui, L.; Snoeren, I.A.; Fuchs, S.N.; Nagai, J.S.; Banjanin, B.; Lam, K.H.; Vogl, T.; Kuppe, C.; et al. Heterogeneous bone-marrow stromal pro-genitors drive myelofibrosis via a druggable alarmin axis. Cell Stem Cell 2021, 28, 637–652.e8. [Google Scholar] [CrossRef]
- Tammaro, A.; Florquin, S.; Brok, M.; Claessen, N.; Butter, L.M.; Teske, G.J.D.; de Boer, O.J.; Vogl, T.; Leemans, J.C.; Dessing, M.C. S100A8/A9 promotes parenchymal damage and renal fibrosis in obstructive nephropathy. Clin. Exp. Immunol. 2018, 193, 361–375. [Google Scholar] [CrossRef] [Green Version]
- Wang, S.; Song, R.; Wang, Z.; Jing, Z.; Wang, S.; Ma, J. S100A8/A9 in Inflammation. Front. Immunol. 2018, 9, 1298. [Google Scholar] [CrossRef] [Green Version]
- Gebhardt, C.; Németh, J.; Angel, P.; Hess, J. S100A8 and S100A9 in inflammation and cancer. Biochem. Pharmacol. 2006, 72, 1622–1631. [Google Scholar] [CrossRef] [PubMed]
- Tan, X.X.; Zheng, X.H.; Huang, Z.N.; Lin, J.Q.; Xie, C.L.; Lin, Y. Involvement of S100A8/A9-TLR4-NLRP3 Inflammasome Pathway in Contrast-Induced Acute Kidney Injury. Cell Physiol. Biochem. 2017, 43, 209–222. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.P.; Di Luqin, W.Y.; Tang, N.; Ai, X.M.; Yao, X.X. Mechanism of interleukin-1b-induced proliferation in rat hepatic stellate cells from different levels of signal transduction. Apmis 2014, 122, 392–398. [Google Scholar]
- Mihm, S. Danger-Associated Molecular Patterns (DAMPs): Molecular Triggers for Sterile Inflammation in the Liver. Int. J. Mol. Sci. 2018, 19, 3104. [Google Scholar] [CrossRef] [Green Version]
- Xue, Y.; Tuipulotu, D.E.; Tan, W.H.; Kay, C.; Man, S.M. Emerging Activators and Regulators of Inflammasomes and Pyroptosis. Trends Immunol. 2019, 40, 1035–1052. [Google Scholar] [CrossRef] [PubMed]
- Artlett, C.M.; Thacker, J.D. Molecular Activation of the NLRP3 Inflammasome in Fibrosis: Common Threads Linking Divergent Fibrogenic Diseases. Antioxidants Redox Signal. 2015, 22, 1162–1175. [Google Scholar] [CrossRef]
- Morgan, M.J.; Liu, Z.G. Crosstalk of reactive oxygen species and NF-kappa B signaling. Cell Res. 2011, 21, 103–115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- West, A.P.; Brodsky, I.E.; Rahner, C.; Woo, D.K.; Erdjument-Bromage, H.; Tempst, P.; Walsh, M.C.; Choi, Y.; Shadel, G.S.; Ghosh, S. TLR signalling augments macrophage bactericidal activity through mitochondrial ROS. Nature 2011, 472, 476–480. [Google Scholar] [CrossRef] [Green Version]
- Yang, X.; Okamura, D.M.; Lu, X.; Chen, Y.; Moorhead, J.; Varghese, Z.; Ruan, X.Z. CD36 in chronic kidney disease: Novel insights and therapeutic opportunities. Nat. Rev. Nephrol. 2017, 13, 769–781. [Google Scholar] [CrossRef]
- Shu, H.; Peng, Y.; Hang, W.; Nie, J.; Zhou, N.; Wang, D.W. The role of CD36 in cardiovascular disease. Cardiovasc. Res. 2020, 118, 115–129. [Google Scholar] [CrossRef]
- Chen, Y.; Zhang, J.; Cui, W.; Silverstein, R.L. CD36, a signaling receptor and fatty acid transporter that regulates immune cell metabolism and fate. J. Exp. Med. 2022, 219, e20211314. [Google Scholar] [CrossRef] [PubMed]
- Patel, K.; Bedossa, P.; Castera, L. Diagnosis of Liver Fibrosis: Present and Future. Semin. Liver Dis. 2015, 35, 166–183. [Google Scholar] [CrossRef] [PubMed]
- Saadeh, S.; Cammell, G.; M.D., W.D.C.; Younossi, Z.; Barnes, D.; Easley, K. The role of liver biopsy in chronic hepatitis C. Hepatology 2001, 33, 196–200. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vilar-Gomez, E.; Chalasani, N. Non-invasive assessment of non-alcoholic fatty liver disease: Clinical prediction rules and blood-based biomarkers. J. Hepatol. 2018, 68, 305–315. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameters | Liver fibrosis | HCs | |
|---|---|---|---|
| Serum/tissue Specimen (n = 89) | Serum specimen (n = 60) | Tissue Specimen (n = 5) | |
| Gender | |||
| Male n (%) | 47 (52.81) | 34 (56.66) | 3 (60) |
| Fale n (%) | 42 (47.19) | 26 (43.33) | 2 (40) |
| Age (years) (IQR) | 57 (12.75) | 57 (14.5) | 59 (17.5) |
| Aetiology | |||
| Viral hepatitis n (%) | 42 (47.19) | NA | NA |
| Cholestatic /Autoimmune n (%) | 25 (28.08) | NA | NA |
| Alcohol n (%) | 16 (17.97) | NA | NA |
| Others n (%) | 6 (6.74) | NA | NA |
| Stage of fibrosis (F) | |||
| F0 n (%) | 9 (10.11) | NA | NA |
| F1 n (%) | 10 (11.23) | NA | NA |
| F2 n (%) | 20 (22.47) | NA | NA |
| F3 n (%) | 29 (32.58) | NA | NA |
| F4 n (%) | 21 (23.59) | NA | NA |
| Genes | Forward (5′-3′) | Reverse (5′-3′) |
|---|---|---|
| NLRP3 | CTTCTCTGATGAGGCCCAAG | GCAGCAAACTGGAAAGGAAG |
| pro-IL-1β | TCCAGGGACAGGATATGGAG | TCTTTCAACACGCAGGACAG |
| pro-IL-18 | AAGATGGCTGCTGAACCAGT | GAGGCCGATTTCCTTGGTCA |
| Col1a1 | AAGAGTGGAGAGTACTGGATT | GTTCTTGCTGATGTACCAGT |
| α-SMA | CGTGGGTGACGAAGCACAG | GGTGGGATGCTCTTCAGGG |
| TGF-β | GGCCAGATCCTGTCCAAGC | GTGGGTTTCCACCATTAGCAC |
| GAPDH | CCACTCCTCCACCTTTGAC | ACCCTGTTGCTGTAGCCA |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, Y.; Kong, X.; You, Y.; Xiang, L.; Zhang, Y.; Wu, R.; Zhou, L.; Duan, L. S100A8-Mediated NLRP3 Inflammasome-Dependent Pyroptosis in Macrophages Facilitates Liver Fibrosis Progression. Cells 2022, 11, 3579. https://doi.org/10.3390/cells11223579
Liu Y, Kong X, You Y, Xiang L, Zhang Y, Wu R, Zhou L, Duan L. S100A8-Mediated NLRP3 Inflammasome-Dependent Pyroptosis in Macrophages Facilitates Liver Fibrosis Progression. Cells. 2022; 11(22):3579. https://doi.org/10.3390/cells11223579
Chicago/Turabian StyleLiu, Yan, Xuehua Kong, Yan You, Linwei Xiang, Yan Zhang, Rui Wu, Lan Zhou, and Liang Duan. 2022. "S100A8-Mediated NLRP3 Inflammasome-Dependent Pyroptosis in Macrophages Facilitates Liver Fibrosis Progression" Cells 11, no. 22: 3579. https://doi.org/10.3390/cells11223579
APA StyleLiu, Y., Kong, X., You, Y., Xiang, L., Zhang, Y., Wu, R., Zhou, L., & Duan, L. (2022). S100A8-Mediated NLRP3 Inflammasome-Dependent Pyroptosis in Macrophages Facilitates Liver Fibrosis Progression. Cells, 11(22), 3579. https://doi.org/10.3390/cells11223579