

Antibody Mediated Intercommunication of Germinal Centers


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Overview of GC Model
2.2. Simulation of Multiple GCs
2.3. Antibody Feedback and GC–GC Interactions
3. Results
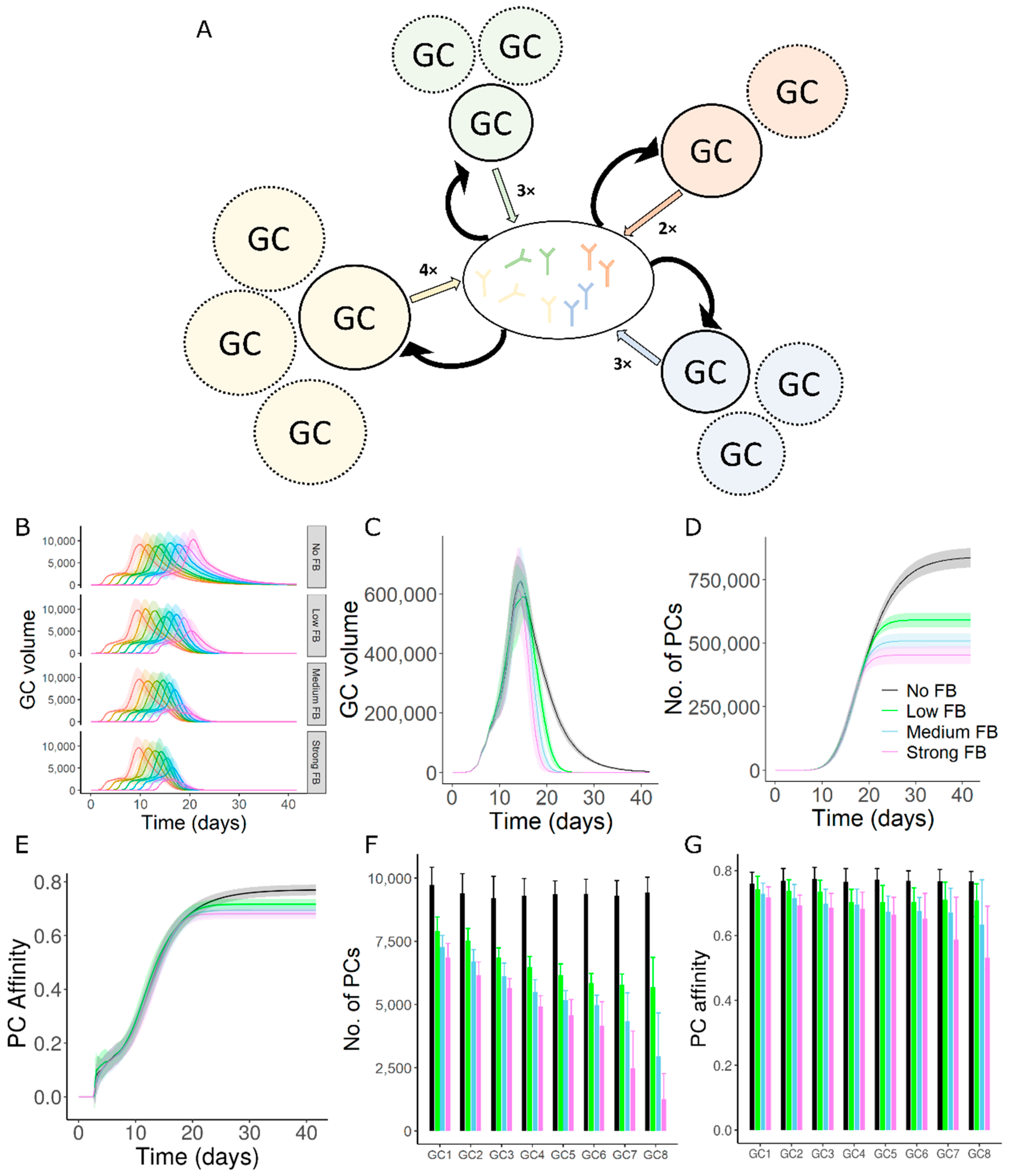
3.1. GC–GC Interactions Decrease the Longevity of GC Responses
3.2. Late Formed GCs Are Highly Sensitive to Antibody Feedback
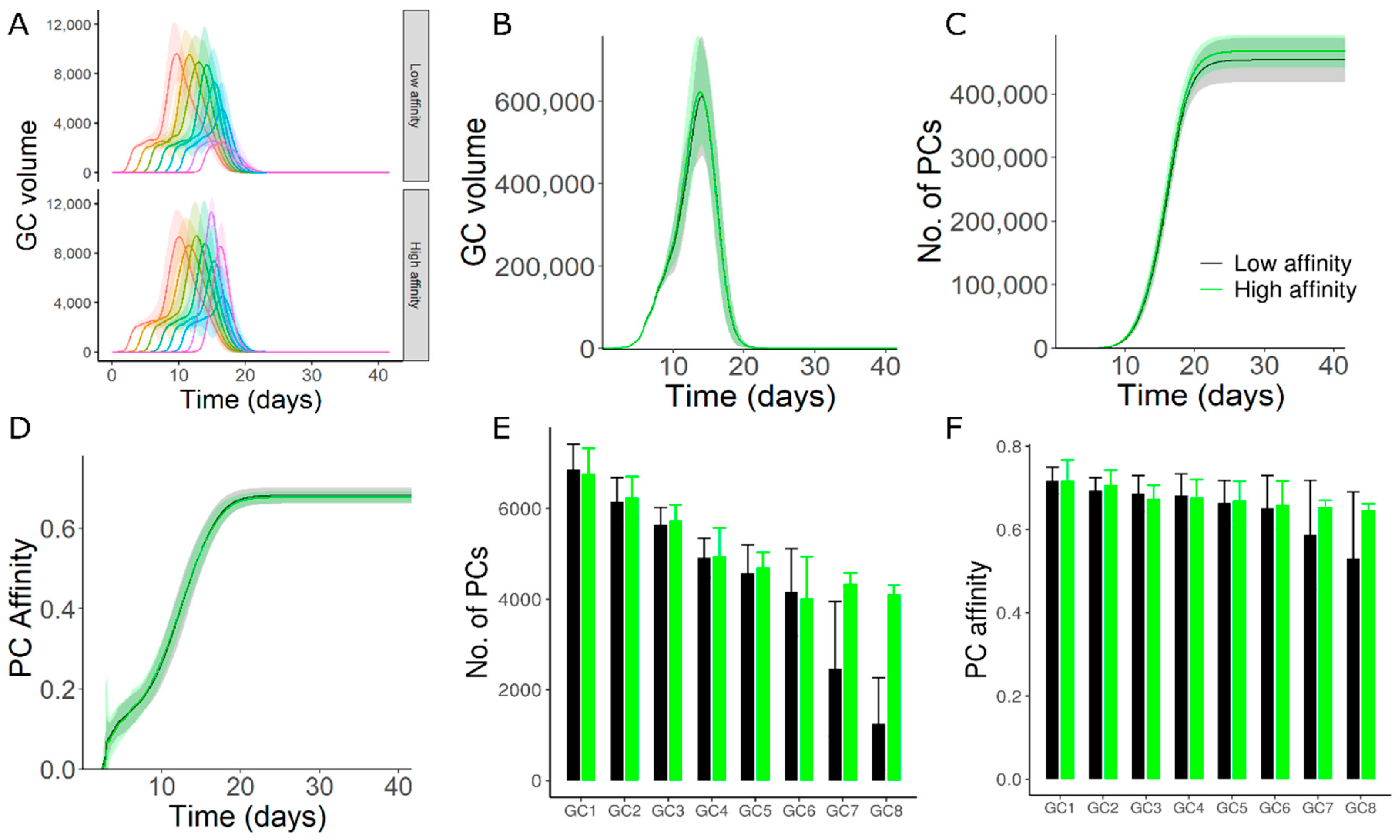
3.3. High Affinity Founder Cells Can Counteract Antibody Mediated Inhibition
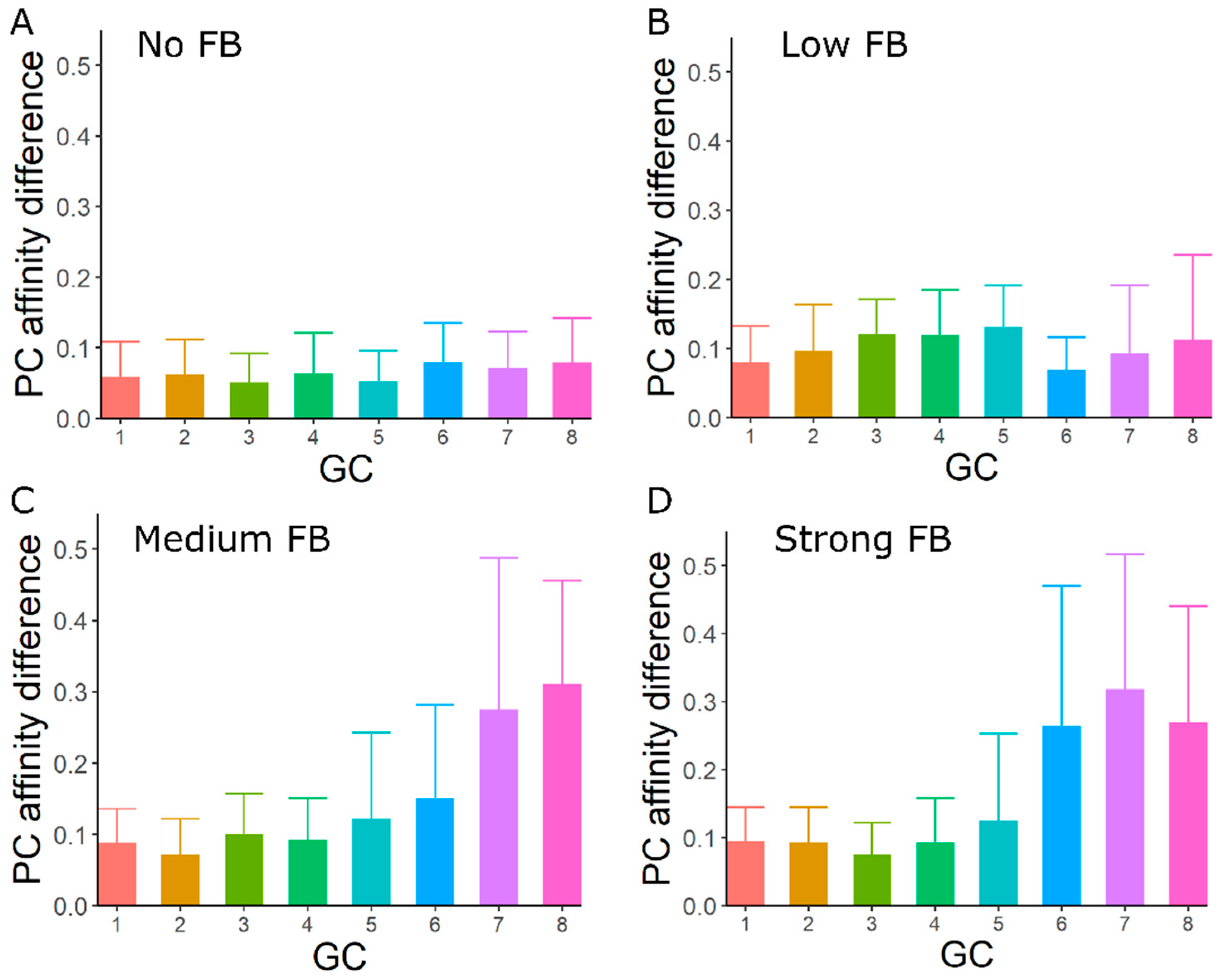
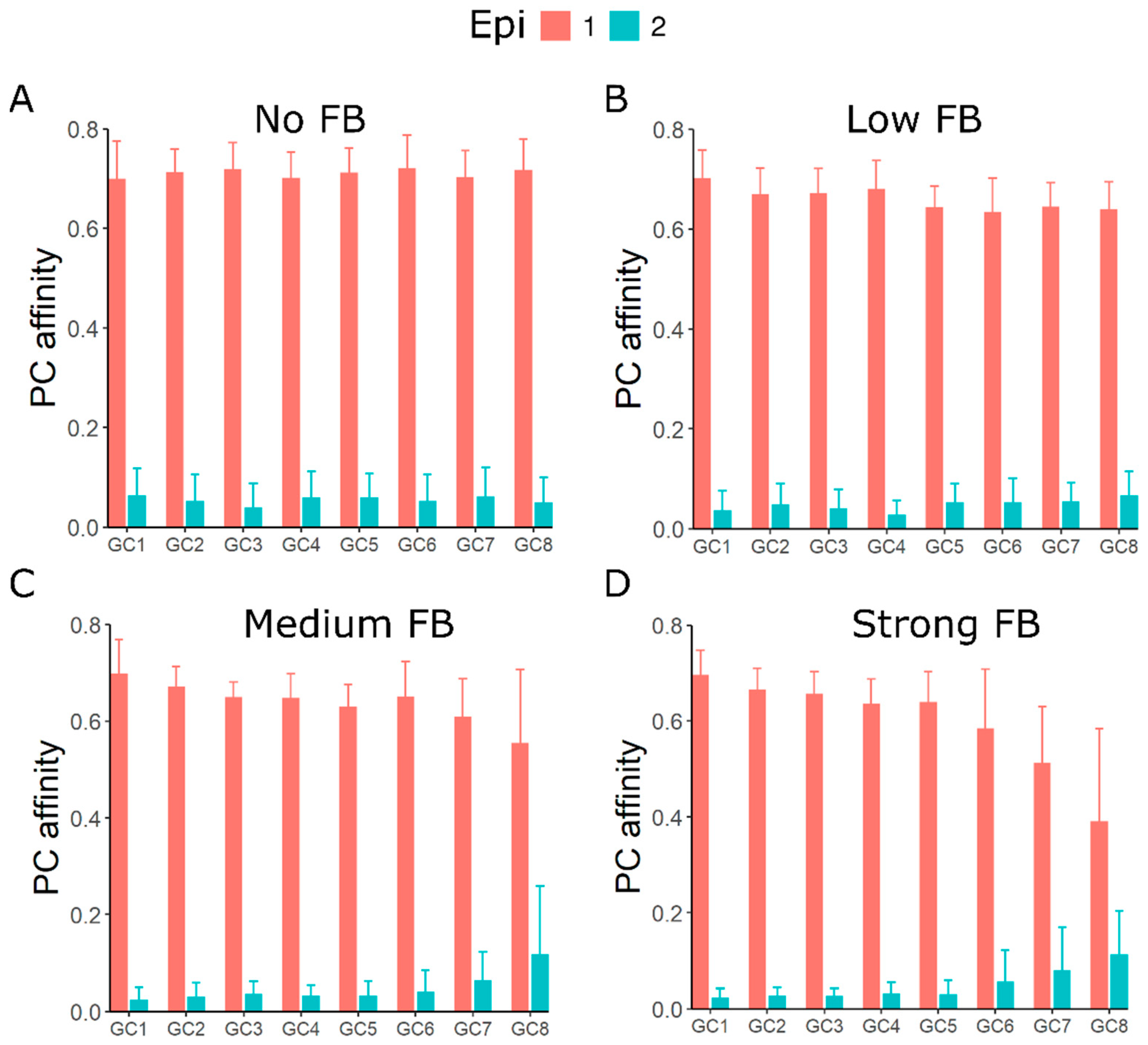
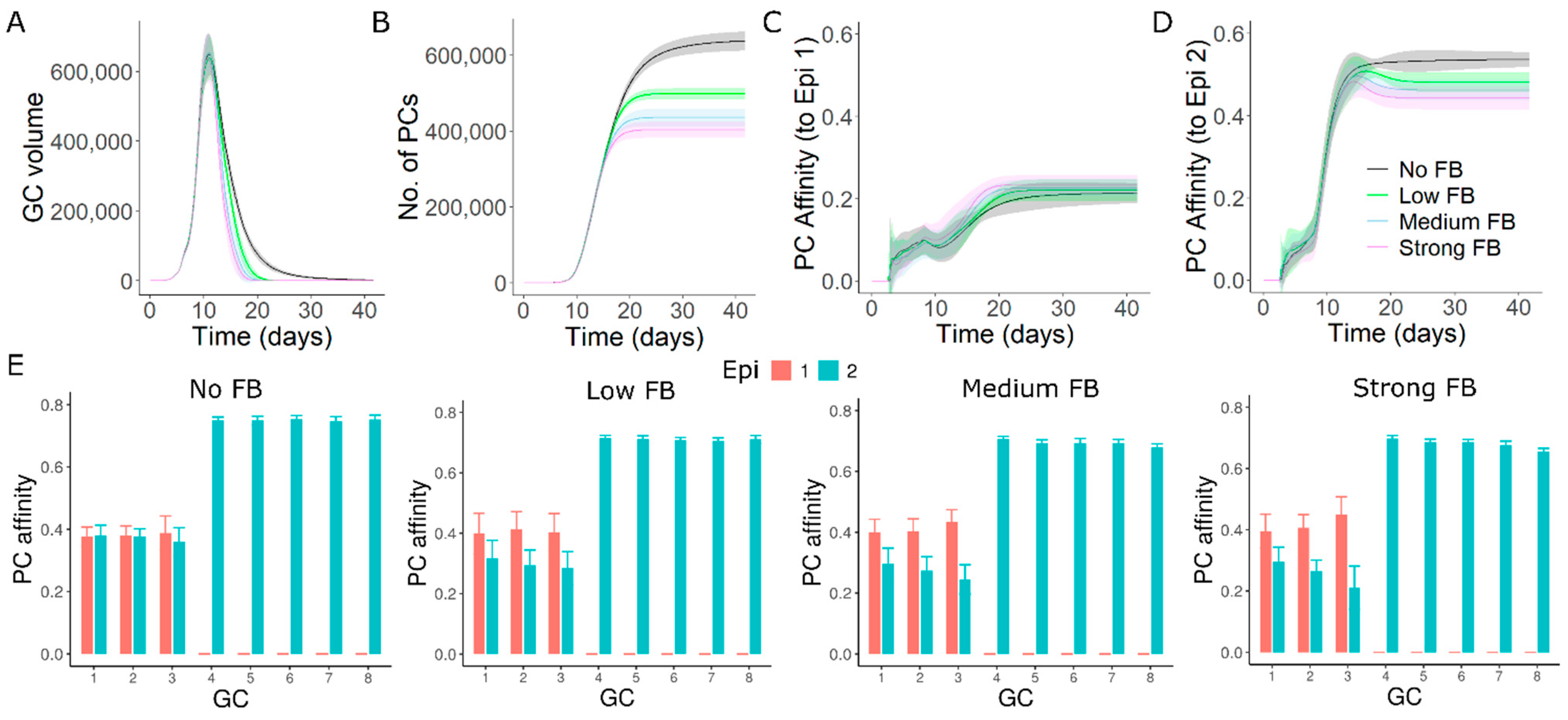
3.4. GC–GC Interactions Induce Diversity in Epitope Recognition among Individual GCs
3.5. GC–GC Interactions Induce Better Affinity Maturation of Rare Epitope in Late Formed GCs
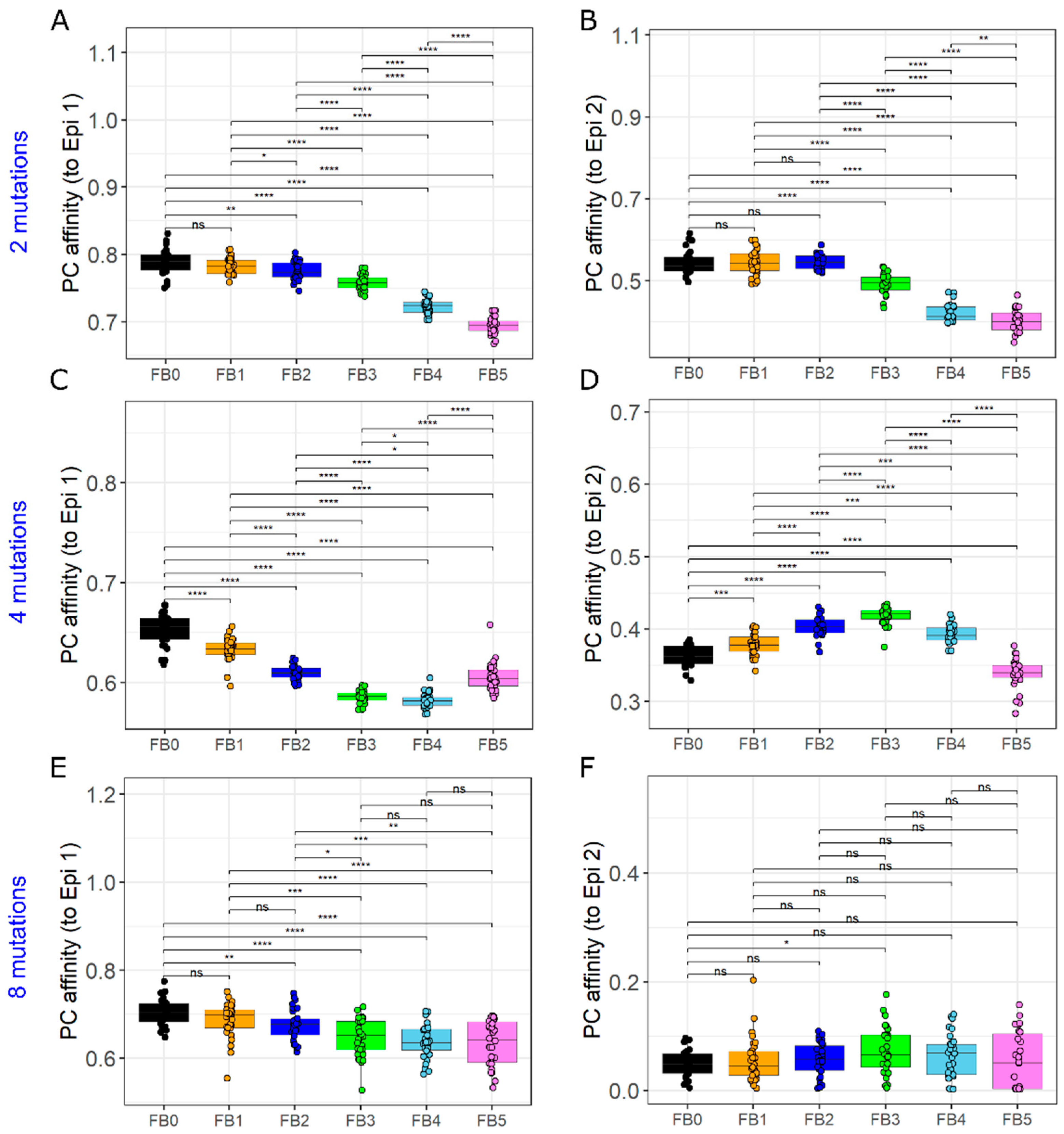
3.6. The GC Founder Cell Composition Determines the Impact of Antibody-Mediated GC–GC Interactions
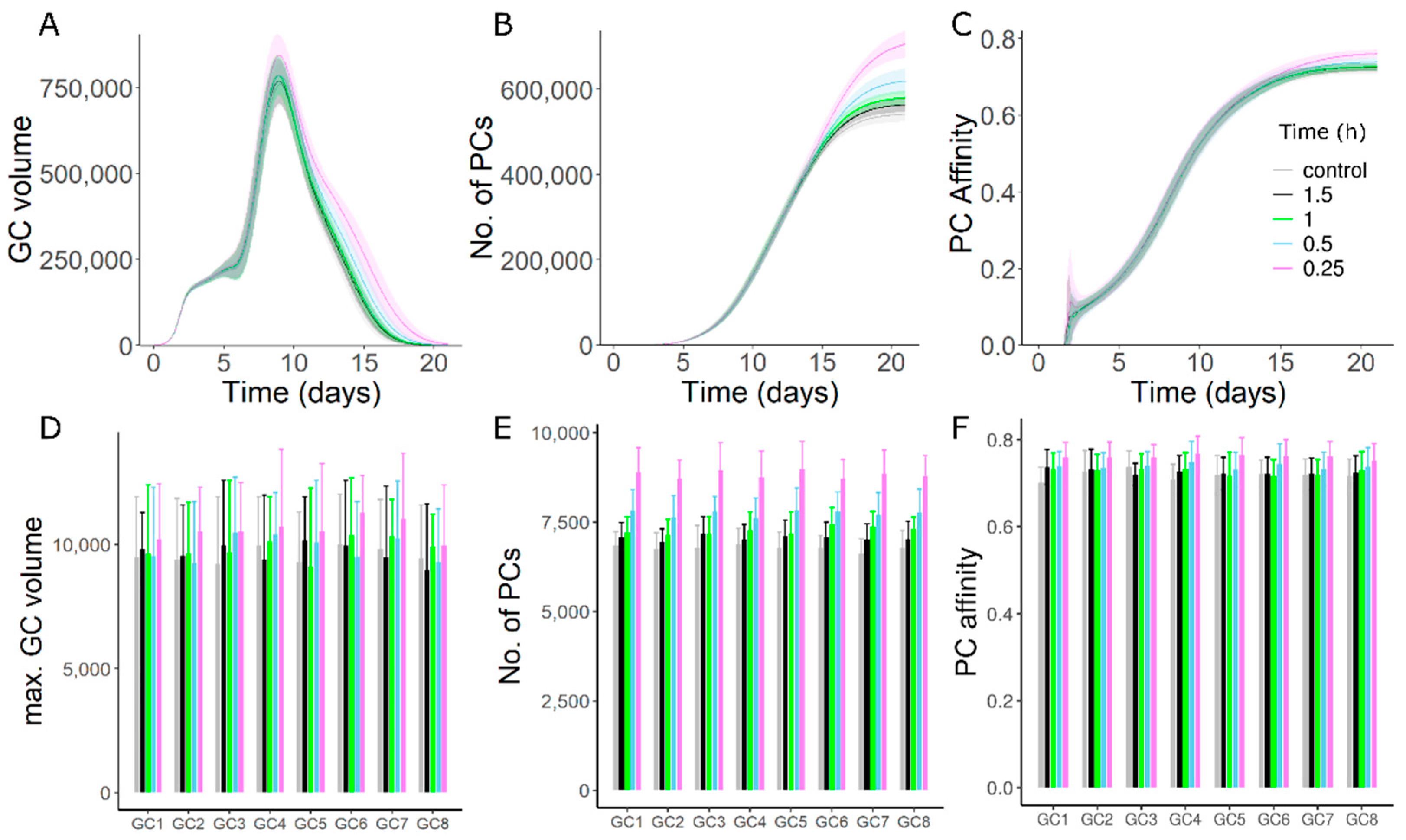
3.7. Persistent Addition of Antigen Can Overcome the Effects of GC–GC Interactions
3.8. GC–GC Interactions Modulate Responses to Antigenic Variants
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- McHeyzer-Williams, M.G.; Ahmed, R. B cell memory and the long-lived plasma cell. Curr. Opin. Immunol. 1999, 11, 172–179. [Google Scholar] [CrossRef]
- Allen, C.D.C.; Ansel, K.M.; Low, C.; Lesley, R.; Tamamura, H.; Fujii, N.; Cyster, J.G. Germinal center dark and light zone organization is mediated by CXCR4 and CXCR5. Nat. Immunol. 2004, 5, 943–952. [Google Scholar] [CrossRef] [PubMed]
- Camacho, S.A.; Kosco-Vilbois, M.H.; Berek, C. The dynamic structure of the germinal center. Immunol. Today 1998, 19, 511–514. [Google Scholar] [CrossRef]
- Nossal, G.J. V The molecular and cellular basis of affinity maturation in the antibody response. Cell 1992, 68, 1–2. [Google Scholar] [CrossRef]
- Kosco-Vilbois, M.H. Are follicular dendritic cells really good for nothing? Nat. Rev. Immunol. 2003, 3, 764–769. [Google Scholar] [CrossRef] [PubMed]
- Linterman, M.A.; Beaton, L.; Yu, D.; Ramiscal, R.R.; Srivastava, M.; Hogan, J.J.; Verma, N.K.; Smyth, M.J.; Rigby, R.J.; Vinuesa, C.G. IL-21 acts directly on B cells to regulate Bcl-6 expression and germinal center responses. J. Exp. Med. 2010, 207, 353–363. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Victora, G.D.; Schwickert, T.A.; Fooksman, D.R.; Kamphorst, A.O.; Meyer-Hermann, M.; Dustin, M.L.; Nussenzweig, M.C. Germinal Center Dynamics Revealed by Multiphoton Microscopy with a Photoactivatable Fluorescent Reporter. Cell 2010, 143, 592–605. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Depoil, D.; Zaru, R.; Guiraud, M.; Chauveau, A.; Harriague, J.; Bismuth, G.; Utzny, C.; Müller, S.; Valitutti, S. Immunological Synapses Are Versatile Structures Enabling Selective T Cell Polarization. Immunity 2005, 22, 185–194. [Google Scholar] [CrossRef] [Green Version]
- Mayer, C.T.; Gazumyan, A.; Kara, E.E.; Gitlin, A.D.; Golijanin, J.; Viant, C.; Pai, J.; Oliveira, T.Y.; Wang, Q.; Escolano, A.; et al. The microanatomic segregation of selection by apoptosis in the germinal center. Science. 2017, 358, eaao2602. [Google Scholar] [CrossRef] [Green Version]
- van Eijk, M.; Medema, J.P.; de Groot, C. Cutting Edge: Cellular Fas-Associated Death Domain-Like IL-1-Converting Enzyme-Inhibitory Protein Protects Germinal Center B Cells from Apoptosis During Germinal Center Reactions. J. Immunol. 2001, 166, 6473–6476. [Google Scholar] [CrossRef]
- Shapiro-Shelef, M.; Lin, K.-I.; McHeyzer-Williams, L.J.; Liao, J.; McHeyzer-Williams, M.G.; Calame, K. Blimp-1 Is Required for the Formation of Immunoglobulin Secreting Plasma Cells and Pre-Plasma Memory B Cells. Immunity 2003, 19, 607–620. [Google Scholar] [CrossRef] [Green Version]
- Higgins, B.W.; McHeyzer-Williams, L.J.; McHeyzer-Williams, M.G. Programming Isotype-Specific Plasma Cell Function. Trends Immunol. 2019, 40, 345–357. [Google Scholar] [CrossRef] [PubMed]
- Weisel, F.J.; Zuccarino-Catania, G.V.; Chikina, M.; Shlomchik, M.J. A temporal switch in the germinal center determines differential output of memory B and plasma cells. Immunity 2016, 44, 116–130. [Google Scholar] [CrossRef] [Green Version]
- MacLennan, I.C.M. Germinal Centers. Annu. Rev. Immunol. 1994, 12, 117–139. [Google Scholar] [CrossRef] [PubMed]
- Meyer-Hermann, M.; Deutsch, A.; Or-Guil, M. Recycling Probability and Dynamical Properties of Germinal Center Reactions. J. Theor. Biol. 2001, 210, 265–285. [Google Scholar] [CrossRef] [Green Version]
- Allen, C.D.C.; Okada, T.; Tang, H.L.; Cyster, J.G. Imaging of Germinal Center Selection Events During Affinity Maturation. Science. 2007, 315, 528–531. [Google Scholar] [CrossRef]
- Figge, M.T.; Garin, A.; Gunzer, M.; Kosco-Vilbois, M.; Toellner, K.-M.; Meyer-Hermann, M. Deriving a germinal center lymphocyte migration model from two-photon data. J. Exp. Med. 2008, 205, 3019–3029. [Google Scholar] [CrossRef]
- Eisen, H.N.; Siskind, G.W. Variations in Affinities of Antibodies during the Immune Response*. Biochemistry 1964, 3, 996–1008. [Google Scholar] [CrossRef]
- Hanna, M.G.; Congdon, C.C.; Wust, C.J. Effect of Antigen Dose on Lymphatic Tissue Germinal Center Changes. Proc. Soc. Exp. Biol. Med. 1966, 121, 286–290. [Google Scholar] [CrossRef]
- Meyer-Hermann, M.E.; Maini, P.K.; Iber, D. An analysis of B cell selection mechanisms in germinal centers. Math. Med. Biol. A J. IMA 2006, 23, 255–277. [Google Scholar] [CrossRef] [PubMed]
- Arulraj, T.; Binder, S.C.; Robert, P.A.; Meyer-Hermann, M. Synchronous Germinal Center Onset Impacts the Efficiency of Antibody Responses. Front. Immunol. 2019, 10, 2116. [Google Scholar] [CrossRef]
- Molari, M.; Eyer, K.; Baudry, J.; Cocco, S.; Monasson, R. Quantitative modeling of the effect of antigen dosage on B-cell affinity distributions in maturating germinal centers. Elife 2020, 9, e55678. [Google Scholar] [CrossRef] [PubMed]
- Cirelli, K.M.; Carnathan, D.G.; Nogal, B.; Martin, J.T.; Rodriguez, O.L.; Upadhyay, A.A.; Enemuo, C.A.; Gebru, E.H.; Choe, Y.; Viviano, F.; et al. Slow Delivery Immunization Enhances HIV Neutralizing Antibody and Germinal Center Responses via Modulation of Immunodominance. Cell 2019, 177, 1153–1171.e28. [Google Scholar] [CrossRef]
- Cirelli, K.M.; Crotty, S. Germinal center enhancement by extended antigen availability. Curr. Opin. Immunol. 2017, 47, 64–69. [Google Scholar] [CrossRef] [PubMed]
- Tam, H.H.; Melo, M.B.; Kang, M.; Pelet, J.M.; Ruda, V.M.; Foley, M.H.; Hu, J.K.; Kumari, S.; Crampton, J.; Baldeon, A.D.; et al. Sustained antigen availability during germinal center initiation enhances antibody responses to vaccination. Proc. Natl. Acad. Sci. 2016, 113, E6639–E6648. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heesters, B.A.; Chatterjee, P.; Kim, Y.-A.; Gonzalez, S.F.; Kuligowski, M.P.; Kirchhausen, T.; Carroll, M.C. Endocytosis and recycling of immune complexes by follicular dendritic cells enhances B cell antigen binding and activation. Immunity 2013, 38, 1164–1175. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heesters, B.A.; Lindqvist, M.; Vagefi, P.A.; Scully, E.P.; Schildberg, F.A.; Altfeld, M.; Walker, B.D.; Kaufmann, D.E.; Carroll, M.C. Follicular dendritic cells retain infectious HIV in cycling endosomes. PLoS Pathog. 2015, 11, e1005285. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arulraj, T.; Binder, S.C.; Meyer-Hermann, M. Rate of Immune Complex Cycling in Follicular Dendritic Cells Determines the Extent of Protecting Antigen Integrity and Availability to Germinal Center B Cells. J. Immunol. 2021, 206, ji2001355. [Google Scholar] [CrossRef]
- Zhang, Y.; Meyer-Hermann, M.; George, L.A.; Figge, M.T.; Khan, M.; Goodall, M.; Young, S.P.; Reynolds, A.; Falciani, F.; Waisman, A.; et al. Germinal center B cells govern their own fate via antibody feedback. J. Exp. Med. 2013, 210, 457–464. [Google Scholar] [CrossRef] [Green Version]
- Meyer-Hermann, M. Injection of Antibodies against Immunodominant Epitopes Tunes Germinal Centers to Generate Broadly Neutralizing Antibodies. Cell Rep. 2019, 29, 1066–1073. [Google Scholar] [CrossRef]
- Meyer-Hermann, M.; Mohr, E.; Pelletier, N.; Zhang, Y.; Victora, G.D.; Toellner, K.-M. A theory of germinal center B cell selection, division, and exit. Cell Rep. 2012, 2, 162–174. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meyer-Hermann, M. Overcoming the dichotomy of quantity and quality in antibody responses. J. Immunol. 2014, 193, 1401828. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Binder, S.C.; Meyer-Hermann, M. Implications of intravital imaging of murine germinal centers on the control of B cell selection and division. Front. Immunol. 2016, 7, 593. [Google Scholar] [CrossRef] [Green Version]
- Arulraj, T.; Binder, S.C.; Meyer-Hermann, M. In Silico Analysis of the Longevity and Timeline of Individual Germinal Center Reactions in a Primary Immune Response. Cells 2021, 10, 1736. [Google Scholar] [CrossRef] [PubMed]
- Perelson, A.S.; Oster, G.F. Theoretical studies of clonal selection: Minimal antibody repertoire size and reliability of self-non-self discrimination. J. Theor. Biol. 1979, 81, 645–670. [Google Scholar] [CrossRef]
- Wedemayer, G.J.; Patten, P.A.; Wang, L.H.; Schultz, P.G.; Stevens, R.C. Structural insights into the evolution of an antibody combining site. Science. 1997, 276, 1665–1669. [Google Scholar] [CrossRef]
- Küppers, R.; Zhao, M.; Hansmann, M.L.; Rajewsky, K. Tracing B cell development in human germinal centres by molecular analysis of single cells picked from histological sections. EMBO J. 1993, 12, 4955–4967. [Google Scholar] [CrossRef]
- Berek, C.; Milstein, C. Mutation Drift and Repertoire Shift in the Maturation of the Immune Response. Immunol. Rev. 1987, 96, 23–41. [Google Scholar] [CrossRef]
- Thaunat, O.; Granja, A.G.; Barral, P.; Filby, A.; Montaner, B.; Collinson, L.; Martinez-Martin, N.; Harwood, N.E.; Bruckbauer, A.; Batista, F.D. Asymmetric Segregation of Polarized Antigen on B Cell Division Shapes Presentation Capacity. Science. 2012, 335, 475–479. [Google Scholar] [CrossRef]
- Rao, S.P.; Vora, K.A.; Manser, T. Differential Expression of the Inhibitory IgG Fc Receptor FcγRIIB on Germinal Center Cells: Implications for Selection of High-Affinity B Cells. J. Immunol. 2002, 169, 1859–1868. [Google Scholar] [CrossRef]
- Chapman, M.E.; Hu, L.; Plato, C.F.; Kohan, D.E. Bioimpedance spectroscopy for the estimation of body fluid volumes in mice. Am. J. Physiol. Physiol. 2010, 299, F280–F283. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Randall, T.D.; Parkhouse, R.M.; Corley, R.B. J chain synthesis and secretion of hexameric IgM is differentially regulated by lipopolysaccharide and interleukin 5. Proc. Natl. Acad. Sci. 1992, 89, 962–966. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Uvarovskii, A. Mathematical Modelling of Lymphocyte-mediated Immune Responses. Technische Universität Carolo-Wilhelmina zu Braunschweig: Braunschweig, Germany, 2017. [Google Scholar] [CrossRef]
- ISO/IEC. ‘ISO International Standard ISO/IEC 14882:2014(E)–Programming Language C++’. Geneva, Switzerland: International Organization for Standardization (ISO). 2015. Available online: https://isocpp.org/std/the-standard (accessed on 8 July 2021).
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2018; Available online: https://www.r-project.org/ (accessed on 8 July 2021).
- Wickham, H. ggplot2: Elegant Graphics for Data Analysis; 2016. Available online: https://ggplot2.tidyverse.org (accessed on 8 July 2021).
- Arulraj, T.; Binder, S.C.; Meyer-Hermann, M. Investigating the Mechanism of Germinal Center Shutdown. Front. Immunol. 2022, 13, 922318. [Google Scholar] [CrossRef]
- Le, D.; Miller, J.D.; Ganusov, V. V Mathematical modeling provides kinetic details of the human immune response to vaccination. Front. Cell. Infect. Microbiol. 2015, 4, 177. [Google Scholar] [CrossRef] [PubMed]
- Mesin, L.; Ersching, J.; Victora, G.D. Germinal Center B Cell Dynamics. Immunity 2016, 45, 471–482. [Google Scholar] [CrossRef] [Green Version]
- Dogan, I.; Bertocci, B.; Vilmont, V.; Delbos, F.; Mégret, J.; Storck, S.; Reynaud, C.-A.; Weill, J.-C. Multiple layers of B cell memory with different effector functions. Nat. Immunol. 2009, 10, 1292–1299. [Google Scholar] [CrossRef]
- Mesin, L.; Schiepers, A.; Ersching, J.; Barbulescu, A.; Cavazzoni, C.B.; Angelini, A.; Okada, T.; Kurosaki, T.; Victora, G.D. Restricted Clonality and Limited Germinal Center Reentry Characterize Memory B Cell Reactivation by Boosting. Cell 2020, 180, 92–106.e11. [Google Scholar] [CrossRef] [Green Version]
- Shulman, Z.; Gitlin, A.D.; Targ, S.; Jankovic, M.; Pasqual, G.; Nussenzweig, M.C.; Victora, G.D. T follicular helper cell dynamics in germinal centers. Science. 2013, 341, 673–677. [Google Scholar] [CrossRef] [Green Version]
- McNamara, H.A.; Idris, A.H.; Sutton, H.J.; Vistein, R.; Flynn, B.J.; Cai, Y.; Wiehe, K.; Lyke, K.E.; Chatterjee, D.; KC, N.; et al. Antibody Feedback Limits the Expansion of B Cell Responses to Malaria Vaccination but Drives Diversification of the Humoral Response. Cell Host Microbe 2020, 28, 572–585.e7. [Google Scholar] [CrossRef]
- Bergström, J.J.E.; Xu, H.; Heyman, B. Epitope-Specific Suppression of IgG Responses by Passively Administered Specific IgG: Evidence of Epitope Masking. Front. Immunol. 2017, 8, 238. [Google Scholar] [CrossRef] [Green Version]
- He, J.-S.; Meyer-Hermann, M.; Xiangying, D.; Zuan, L.Y.; Jones, L.A.; Ramakrishna, L.; de Vries, V.C.; Dolpady, J.; Aina, H.; Joseph, S.; et al. The distinctive germinal center phase of IgE+ B lymphocytes limits their contribution to the classical memory response. J. Exp. Med. 2013, 210, 2755–2771. [Google Scholar] [CrossRef] [Green Version]
- Roco, J.A.; Mesin, L.; Binder, S.C.; Nefzger, C.; Gonzalez-Figueroa, P.; Canete, P.F.; Ellyard, J.; Shen, Q.; Robert, P.A.; Cappello, J.; et al. Class-Switch Recombination Occurs Infrequently in Germinal Centers. Immunity 2019, 51, 337–350.e7. [Google Scholar] [CrossRef]
- Heyman, B. Regulation of Antibody Responses via Antibodies, Complement, and Fc Receptors. Annu. Rev. Immunol. 2000, 18, 709–737. [Google Scholar] [CrossRef] [PubMed]
- Gruber, D.R.; Richards, A.L.; Howie, H.L.; Hay, A.M.; Lebedev, J.N.; Wang, X.; Zimring, J.C.; Hudson, K.E. Passively transferred IgG enhances humoral immunity to a red blood cell alloantigen in mice. Blood Adv. 2020, 4, 1526–1537. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mackay, M.; Stanevsky, A.; Wang, T.; Aranow, C.; Li, M.; Koenig, S.; Ravetch, J.V.; Diamond, B. Selective dysregulation of the FcγIIB receptor on memory B cells in SLE. J. Exp. Med. 2006, 203, 2157–2164. [Google Scholar] [CrossRef] [PubMed]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Arulraj, T.; Binder, S.C.; Meyer-Hermann, M. Antibody Mediated Intercommunication of Germinal Centers. Cells 2022, 11, 3680. https://doi.org/10.3390/cells11223680
Arulraj T, Binder SC, Meyer-Hermann M. Antibody Mediated Intercommunication of Germinal Centers. Cells. 2022; 11(22):3680. https://doi.org/10.3390/cells11223680
Chicago/Turabian StyleArulraj, Theinmozhi, Sebastian C. Binder, and Michael Meyer-Hermann. 2022. "Antibody Mediated Intercommunication of Germinal Centers" Cells 11, no. 22: 3680. https://doi.org/10.3390/cells11223680
APA StyleArulraj, T., Binder, S. C., & Meyer-Hermann, M. (2022). Antibody Mediated Intercommunication of Germinal Centers. Cells, 11(22), 3680. https://doi.org/10.3390/cells11223680