
Autophagy Induced by Toll-like Receptor Ligands Regulates Antigen Extraction and Presentation by B Cells

 ,
,  ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cells and Mice
2.2. Antibodies and Reagents
2.3. Preparation of Antigen-Coated Beads and Antigen-Coated Coverslip
2.4. Immobilized Surface Antigens
2.5. Antigen Extraction
2.6. Antigen Presentation
2.7. Activation of B Cells on Antigen-Coated Plates and Western Blot Densitometric Analysis
2.8. CRISPR Targeting
2.9. Microscopy
2.10. Image Analysis
2.10.1. Lysosome and GEF-H1 Recruitment
2.10.2. Polarity Analysis
2.10.3. Distribution of Lysosomes within Z Sections
2.11. Flow Cytometry
2.12. Statistical Analysis
3. Results
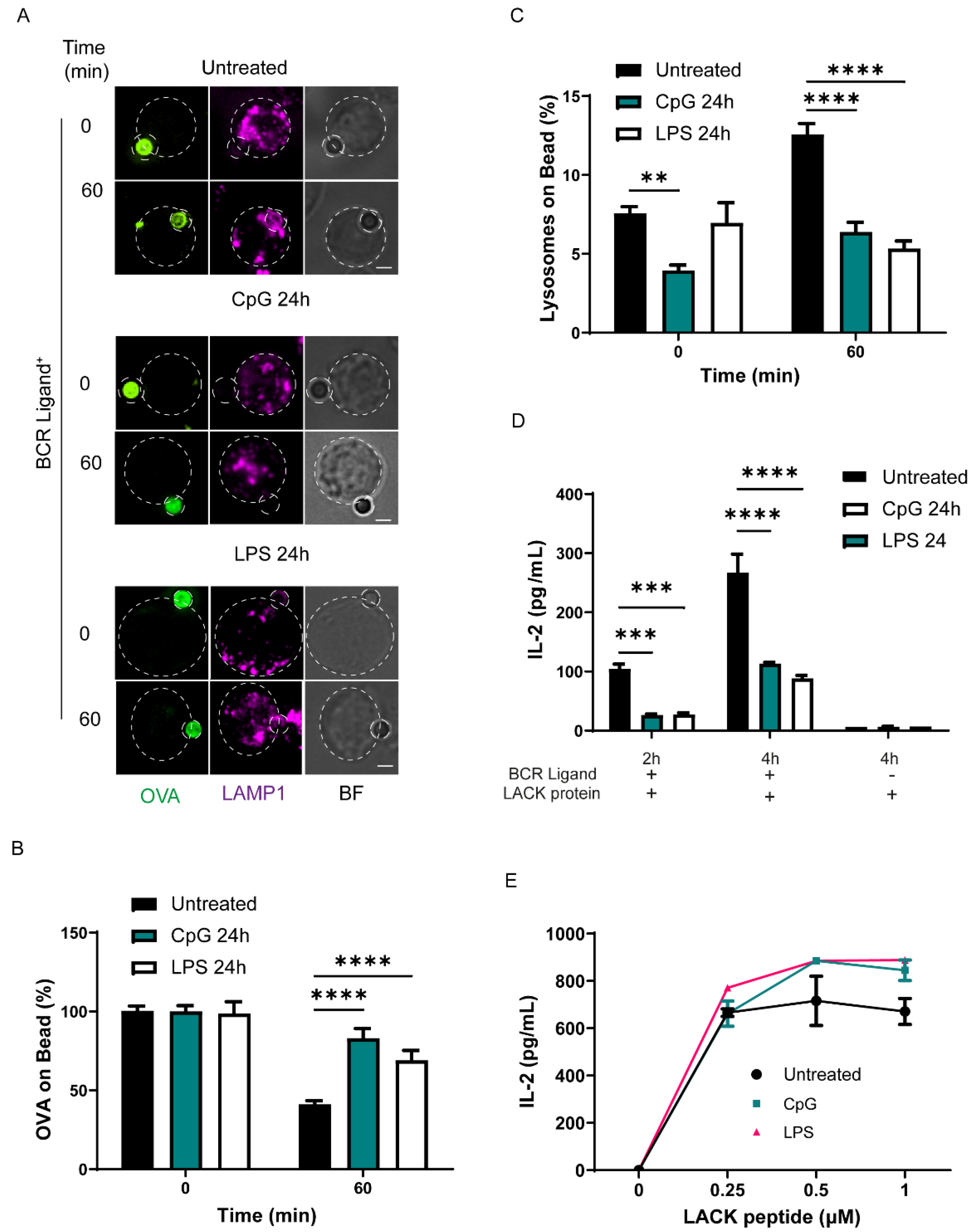
3.1. TLR Stimulation Decreases the Capacity of B Cells to Extract and Present Immobilized Antigens by Decreasing Lysosome Recruitment to the Immune Synapse
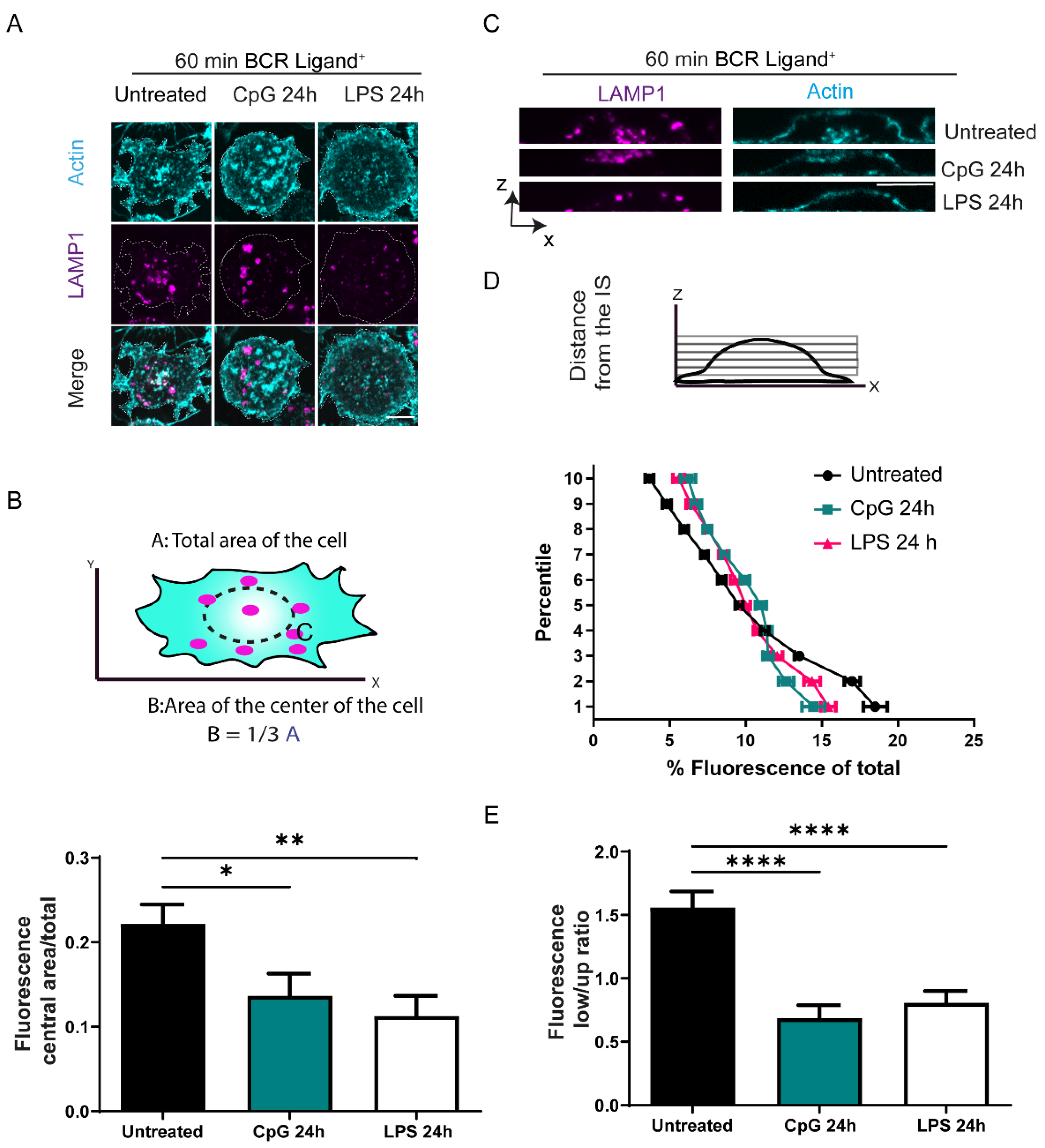
3.2. TLR Stimulation Regulates the Distribution of Lysosomes at the Immune Synapse
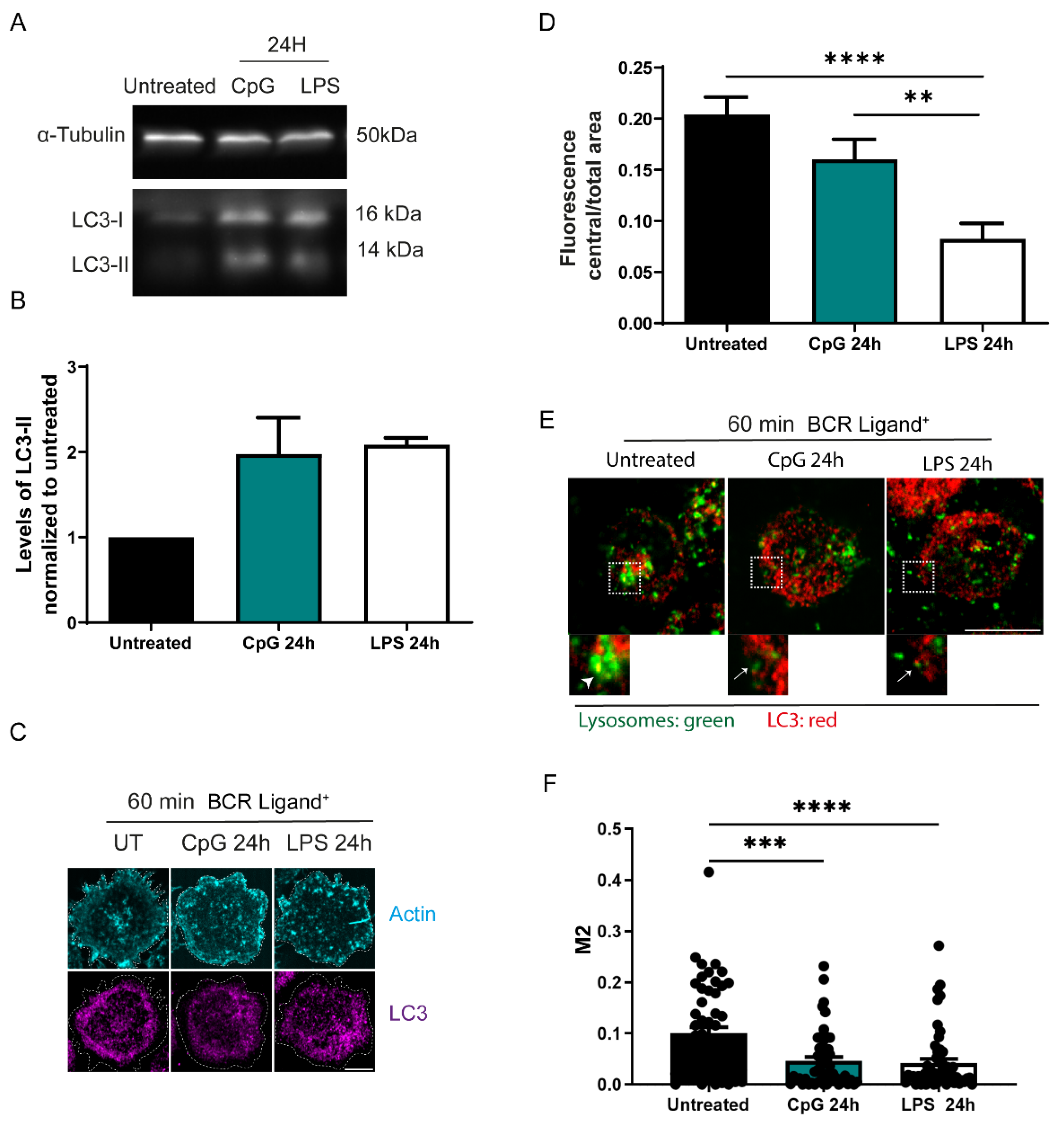
3.3. Autophagy Is Increased in B Cells Stimulated with TLR Ligands
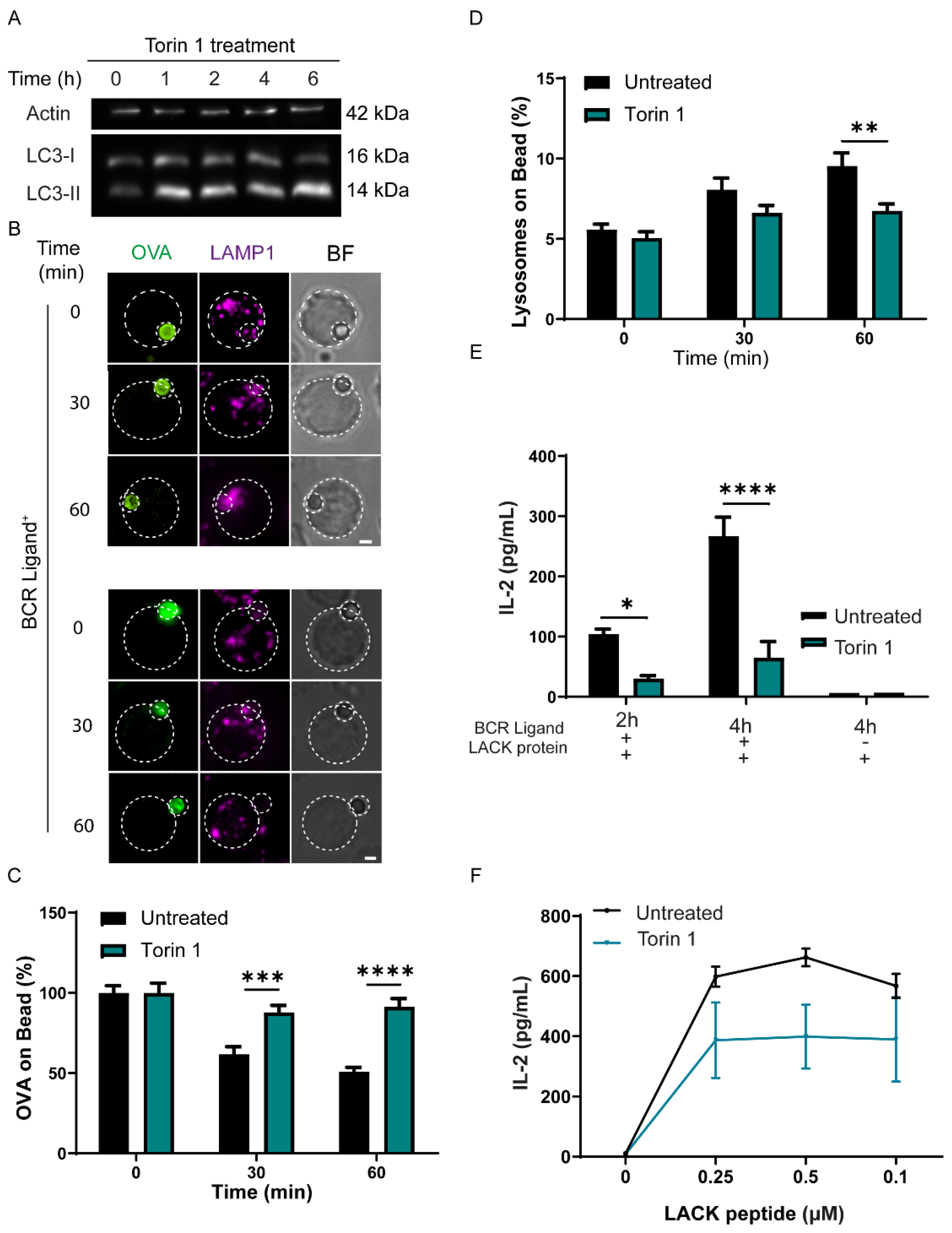
3.4. Induction of Autophagy Decreases the Capacity of B Cells to Extract and Present Immobilized Antigen
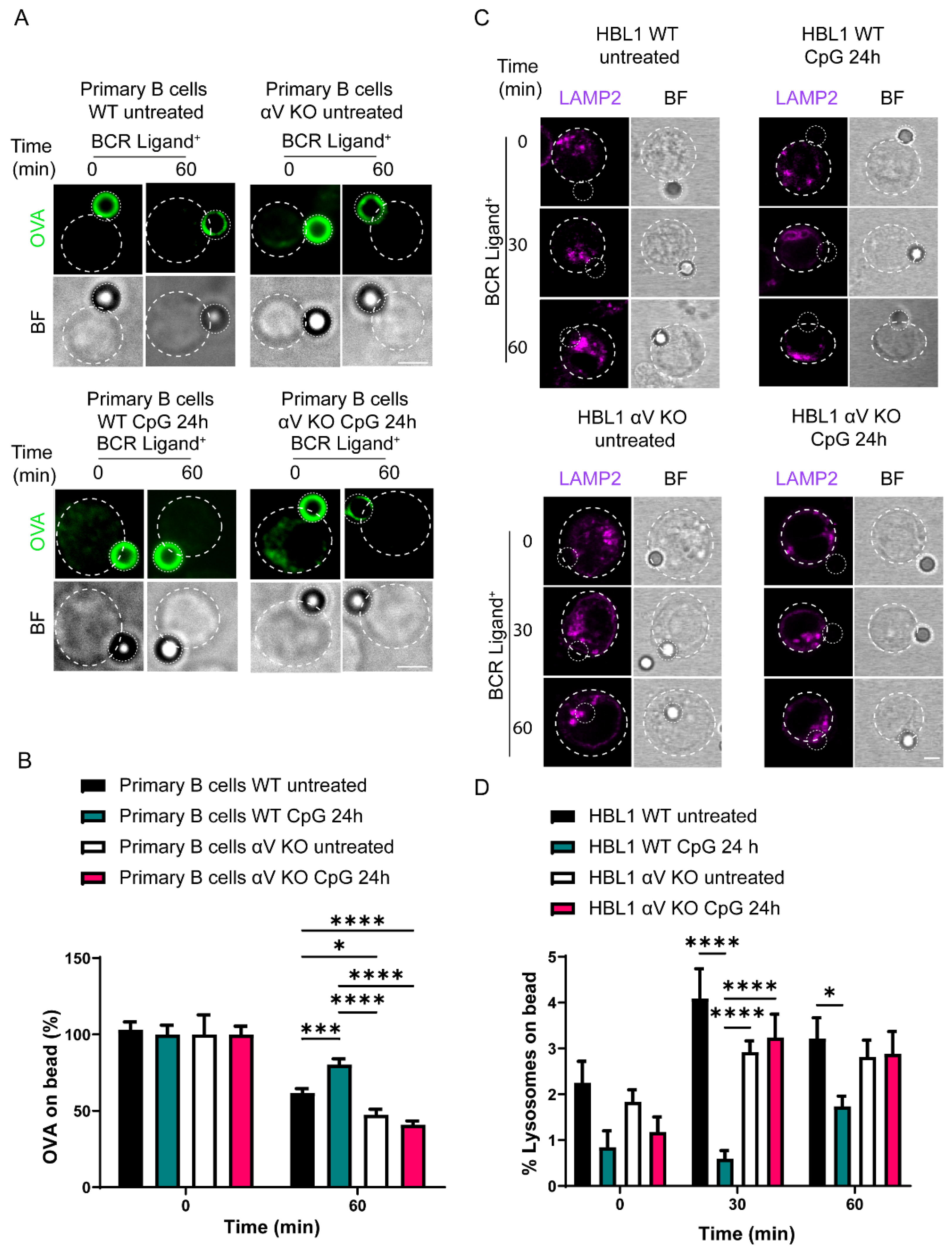
3.5. Antigen Extraction in B Cells Is Regulated by TLR-Induced Autophagy via Integrin αV
3.6. TLR-Induced Autophagy Controls Levels of GEF-H1, a Regulator of Lysosome Trafficking in B Cells
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Avalos, A.M.; Ploegh, H.L. Early BCR Events and Antigen Capture, Processing, and Loading on MHC Class II on B Cells. Front. Immunol. 2014, 5, 92. [Google Scholar] [CrossRef] [PubMed]
- Mitchison, N.A. T-Cell-B-Cell Cooperation. Nat. Rev. Immunol. 2004, 4, 308–312. [Google Scholar] [CrossRef] [PubMed]
- Harwood, N.E.; Batista, F.D. The Cytoskeleton Coordinates the Early Events of B-Cell Activation. Cold Spring Harb. Perspect. Biol. 2011, 3, a002360. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mattila, P.K.; Feest, C.; Depoil, D.; Treanor, B.; Montaner, B.; Otipoby, K.L.; Carter, R.; Justement, L.B.; Bruckbauer, A.; Batista, F.D. The Actin and Tetraspanin Networks Organize Receptor Nanoclusters to Regulate B Cell Receptor-Mediated Signaling. Immunity 2013, 38, 461–474. [Google Scholar] [CrossRef] [Green Version]
- Yuseff, M.I.; Reversat, A.; Lankar, D.; Diaz, J.; Fanget, I.; Pierobon, P.; Randrian, V.; Larochette, N.; Vascotto, F.; Desdouets, C.; et al. Polarized Secretion of Lysosomes at the B Cell Synapse Couples Antigen Extraction to Processing and Presentation. Immunity 2011, 35, 361–374. [Google Scholar] [CrossRef] [Green Version]
- Spillane, K.M.; Tolar, P. Mechanics of Antigen Extraction in the B Cell Synapse. Mol. Immunol. 2018, 101, 319–328. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Natkanski, E.; Lee, W.-Y.; Mistry, B.; Casal, A.; Molloy, J.E.; Tolar, P. B Cells Use Mechanical Energy to Discriminate Antigen Affinities. Science 2013, 340, 1587–1590. [Google Scholar] [CrossRef] [Green Version]
- Yuseff, M.-I.; Pierobon, P.; Reversat, A.; Lennon-Duménil, A.-M. How B Cells Capture, Process and Present Antigens: A Crucial Role for Cell Polarity. Nat. Rev. Immunol. 2013, 13, 475–486. [Google Scholar] [CrossRef]
- Sáez, J.J.; Diaz, J.; Ibañez, J.; Bozo, J.P.; Cabrera Reyes, F.; Alamo, M.; Gobert, F.-X.; Obino, D.; Bono, M.R.; Lennon-Duménil, A.-M.; et al. The Exocyst Controls Lysosome Secretion and Antigen Extraction at the Immune Synapse of B Cells. J. Cell Biol. 2019, 218, 2247–2264. [Google Scholar] [CrossRef]
- Obino, D.; Fetler, L.; Soza, A.; Malbec, O.; Saez, J.J.; Labarca, M.; Oyanadel, C.; del Valle Batalla, F.; Goles, N.; Chikina, A.; et al. Galectin-8 Favors the Presentation of Surface-Tethered Antigens by Stabilizing the B Cell Immune Synapse. Cell Rep. 2018, 25, 3110–3122.e6. [Google Scholar] [CrossRef]
- Hou, B.; Saudan, P.; Ott, G.; Wheeler, M.L.; Ji, M.; Kuzmich, L.; Lee, L.M.; Coffman, R.L.; Bachmann, M.F.; DeFranco, A.L. Selective Utilization of Toll-like Receptor and Myd88 Signaling in B Cells for Enhancement of the Antiviral Germinal Center Response. Immunity 2011, 34, 375–384. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lau, C.M.; Broughton, C.; Tabor, A.S.; Akira, S.; Flavell, R.A.; Mamula, M.J.; Christensen, S.R.; Shlomchik, M.J.; Viglianti, G.A.; Rifkin, I.R.; et al. RNA-Associated Autoantigens Activate B Cells by Combined B Cell Antigen Receptor/Toll-like Receptor 7 Engagement. J. Exp. Med. 2005, 202, 1171–1177. [Google Scholar] [CrossRef] [PubMed]
- Akkaya, M.; Akkaya, B.; Kim, A.S.; Miozzo, P.; Sohn, H.; Pena, M.; Roesler, A.S.; Theall, B.P.; Henke, T.; Kabat, J.; et al. Toll-like Receptor 9 Antagonizes Antibody Affinity Maturation Article. Nat. Immunol. 2018, 19, 255–266. [Google Scholar] [CrossRef] [PubMed]
- Acharya, M.; Edkins, A.L.; Ozanne, B.W.; Cushley, W. SDF-1 and PDGF Enhance Avβ5-Mediated ERK Activation and Adhesion-Independent Growth of Human Pre-B Cell Lines. Leukemia 2009, 23, 1807–1817. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leifer, C.A.; Medvedev, A.E. Molecular Mechanisms of Regulation of Toll-like Receptor Signaling. J. Leukoc. Biol. 2016, 100, 927–941. [Google Scholar] [CrossRef] [PubMed]
- Acharya, M.; Sokolovska, A.; Tam, J.M.; Conway, K.L.; Stefani, C.; Raso, F.; Mukhopadhyay, S.; Feliu, M.; Paul, E.; Savill, J.; et al. Av Integrins Combine with LC3 and Atg5 to Regulate Toll-like Receptor Signalling in B Cells. Nat. Commun. 2016, 7, 10917. [Google Scholar] [CrossRef] [Green Version]
- Freeman, S.A.; Lei, V.; Dang-Lawson, M.; Mizuno, K.; Roskelley, C.D.; Gold, M.R. Cofilin-Mediated F-Actin Severing Is Regulated by the Rap GTPase and Controls the Cytoskeletal Dynamics That Drive Lymphocyte Spreading and BCR Microcluster Formation. J. Immunol. 2011, 187, 5887–5900. [Google Scholar] [CrossRef] [Green Version]
- Cuervo, A.M. Autophagy: Many Paths to the Same End. Mol. Cell Biochem. 2004, 263, 55–72. [Google Scholar] [CrossRef]
- Galluzzi, L.; Green, D.R. Autophagy-Independent Functions of the Autophagy Machinery. Cell 2019, 177, 1682–1699. [Google Scholar] [CrossRef]
- Martinez, J.; Malireddi, R.K.S.; Lu, Q.; Cunha, L.D.; Pelletier, S.; Gingras, S.; Orchard, R.; Guan, J.L.; Tan, H.; Peng, J.; et al. Molecular Characterization of LC3-Associated Phagocytosis Reveals Distinct Roles for Rubicon, NOX2 and Autophagy Proteins. Nat. Cell Biol. 2015, 17, 893–906. [Google Scholar] [CrossRef]
- Chen, M.; Kodali, S.; Jang, A.; Kuai, L.; Wang, J. Requirement for Autophagy in the Long-Term Persistence but Not Initial Formation of Memory B Cells. J. Immunol. 2015, 194, 2607–2615. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clarke, A.J.; Riffelmacher, T.; Braas, D.; Cornall, R.J.; Simon, A.K. B1a B Cells Require Autophagy for Metabolic Homeostasis and Self-Renewal. J. Exp. Med. 2018, 215, 399–413. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pengo, N.; Scolari, M.; Oliva, L.; Milan, E.; Mainoldi, F.; Raimondi, A.; Fagioli, C.; Merlini, A.; Mariani, E.; Pasqualetto, E.; et al. Plasma Cells Require Autophagy for Sustainable Immunoglobulin Production. Nat. Immunol. 2013, 14, 298–305. [Google Scholar] [CrossRef] [PubMed]
- Conway, K.L.; Kuballa, P.; Khor, B.; Zhang, M.; Shi, H.N.; Virgin, H.W.; Xavier, R.J. ATG5 Regulates Plasma Cell Differentiation. Autophagy 2013, 9, 528–537. [Google Scholar] [CrossRef] [Green Version]
- Arbogast, F.; Arnold, J.; Hammann, P.; Kuhn, L.; Chicher, J.; Murera, D.; Weishaar, J.; Muller, S.; Fauny, J.D.; Gros, F. ATG5 Is Required for B Cell Polarization and Presentation of Particulate Antigens. Autophagy 2018, 15, 280–294. [Google Scholar] [CrossRef]
- Martinez-Martin, N.; Maldonado, P.; Gasparrini, F.; Frederico, B.; Aggarwal, S.; Gaya, M.; Tsui, C.; Burbage, M.; Keppler, S.J.; Montaner, B.; et al. A Switch from Canonical to Noncanonical Autophagy Shapes B Cell Responses. Science 2017, 355, 641–647. [Google Scholar] [CrossRef] [Green Version]
- Sanjuan, M.A.; Dillon, C.P.; Tait, S.W.G.; Moshiach, S.; Dorsey, F.; Connell, S.; Komatsu, M.; Tanaka, K.; Cleveland, J.L.; Withoff, S.; et al. Toll-like Receptor Signalling in Macrophages Links the Autophagy Pathway to Phagocytosis. Nature 2007, 450, 1253–1257. [Google Scholar] [CrossRef]
- Jones, B.; Tite, J.P.; Janeway, C.A. Different Phenotypic Variants of the Mouse B Cell Tumor A20/2J Are Selected by Antigen- and Mitogen-Triggered Cytotoxicity of L3T4-Positive, I-A-Restricted T Cell Clones. J. Immunol. 1986, 136, 348–356. [Google Scholar]
- Lankar, D.; Vincent-Schneider, H.; Briken, V.; Yokozeki, T.; Raposo, G.; Bonnerot, C. Dynamics of Major Histocompatibility Complex Class II Compartments during B Cell Receptor-Mediated Cell Activation. J. Exp. Med. 2002, 195, 461–472. [Google Scholar] [CrossRef] [Green Version]
- Malherbe, L.; Filippi, C.; Julia, V.; Foucras, G.; Moro, M.; Appel, H.; Wucherpfennig, K.; Guéry, J.C.; Glaichenhaus, N. Selective Activation and Expansion of High-Affinity CD4+ T Cells in Resistant Mice upon Infection with Leishmania Major. Immunity 2000, 13, 771–782. [Google Scholar] [CrossRef] [Green Version]
- le Roux, D.; Lankar, D.; Yuseff, M.I.; Vascotto, F.; Yokozeki, T.; Faure-André, G.; Mougneau, E.; Glaichenhaus, N.; Manoury, B.; Bonnerot, C.; et al. Syk-Dependent Actin Dynamics Regulate Endocytic Trafficking and Processing of Antigens Internalized through the B-Cell Receptor. Mol. Biol. Cell 2007, 18, 3451–3462. [Google Scholar] [CrossRef] [PubMed]
- Vascotto, F.; Lankar, D.; Faure-André, G.; Vargas, P.; Diaz, J.; le Roux, D.; Yuseff, M.I.; Sibarita, J.B.; Boes, M.; Raposo, G.; et al. The Actin-Based Motor Protein Myosin II Regulates MHC Class II Trafficking and BCR-Driven Antigen Presentation. J. Cell Biol. 2007, 176, 1007–1019. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lacy-Hulbert, A.; Smith, A.M.; Tissire, H.; Barry, M.; Crowley, D.; Bronson, R.T.; Roes, J.T.; Savill, J.S.; Hynes, R.O. Ulcerative Colitis and Autoimmunity Induced by Loss of Myeloid Alphav Integrins. Proc. Natl. Acad. Sci. USA 2007, 104, 15823–15828. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reversat, A.; Yuseff, M.-I.; Lankar, D.; Malbec, O.; Obino, D.; Maurin, M.; Penmatcha, N.V.G.; Amoroso, A.; Sengmanivong, L.; Gundersen, G.G.; et al. Polarity Protein Par3 Controls B-Cell Receptor Dynamics and Antigen Extraction at the Immune Synapse. Mol. Biol. Cell 2015, 26, 1273–1285. [Google Scholar] [CrossRef] [PubMed]
- Muir, V.; Sagadiev, S.; Liu, S.; Holder, U.; Armendariz, A.M.; Suchland, E.; Meitlis, I.; Camp, N.; Giltiay, N.; Tam, J.M.; et al. Transcriptomic Analysis of Pathways Associated with ITGAV/Alpha(v) Integrin-Dependent Autophagy in Human B Cells. Autophagy 2022, 1–17. [Google Scholar] [CrossRef]
- Schindelin, J.; Arganda-Carreras, I.; Frise, E.; Kaynig, V.; Longair, M.; Pietzsch, T.; Preibisch, S.; Rueden, C.; Saalfeld, S.; Schmid, B.; et al. Fiji: An Open-Source Platform for Biological-Image Analysis. Nat. Methods 2012, 9, 676–682. [Google Scholar] [CrossRef] [Green Version]
- Ulloa, R.; Corrales, O.; Cabrera-Reyes, F.; Jara-Wilde, J.; Saez, J.J.; Rivas, C.; Lagos, J.; Härtel, S.; Quiroga, C.; Yuseff, M.I.; et al. B Cells Adapt Their Nuclear Morphology to Organize the Immune Synapse and Facilitate Antigen Extraction. Front. Immunol. 2022, 12, 801164. [Google Scholar] [CrossRef]
- Bekeredjian-Ding, I.; Jego, G. Toll-like Receptors-Sentries in the B-Cell Response. Immunology 2009, 128, 311–323. [Google Scholar] [CrossRef]
- Chaturvedi, A.; Dorward, D.; Pierce, S.K. The B Cell Receptor Governs the Subcellular Location of Toll-like Receptor 9 Leading to Hyperresponses to DNA-Containing Antigens. Immunity 2008, 28, 799–809. [Google Scholar] [CrossRef] [Green Version]
- Ibañez-Vega, J.; Fuentes, D.; Lagos, J.; Cancino, J.; Yuseff, M.I. Studying Organelle Dynamics in B Cells during Immune Synapse Formation. J. Vis. Exp. 2019, 2019, e59621. [Google Scholar] [CrossRef] [Green Version]
- Obino, D.; Diaz, J.; Saez, J.J.; Ibanez-Vega, J.; Saez, P.J.; Alamo, M.; Lankar, D.; Yuseff, M.-I. Vamp-7 Dependent Secretion at the Immune Synapse Regulates Antigen Extraction and Presentation in B Lymphocytes Dorian. Mol. Biol. Cell 2017, 28, 890–897. [Google Scholar] [CrossRef] [PubMed]
- Korolchuk, V.I.; Saiki, S.; Lichtenberg, M.; Siddiqi, F.H.; Roberts, E.A.; Imarisio, S.; Jahreiss, L.; Sarkar, S.; Futter, M.; Menzies, F.M.; et al. Lysosomal Positioning Coordinates Cellular Nutrient Responses. Nat. Cell Biol. 2011, 13, 453–462. [Google Scholar] [CrossRef] [PubMed]
- Pathak, R.; DerMardirossian, C. GEF-H1: Orchestrating the Interplay between Cytoskeleton and Vesicle Trafficking. Small GTPases 2013, 4, 174–179. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoshida, T.; Tsujioka, M.; Honda, S.; Tanaka, M.; Shimizu, S. Autophagy Suppresses Cell Migration by Degrading GEF-H1, a RhoA GEF. Oncotarget 2016, 7, 34420–34429. [Google Scholar] [CrossRef]
- Defranco, A.L.; Rookhuizen, D.C.; Hou, B. Contribution of Toll-like Receptor Signaling to Germinal Center Antibody Responses. Immunol. Rev. 2012, 247, 64–72. [Google Scholar] [CrossRef] [Green Version]
- Rawlings, D.J.; Schwartz, M.A.; Jackson, S.W.; Meyer-Bahlburg, A. Integration of B Cell Responses through Toll-like Receptors and Antigen Receptors. Nat. Rev. Immunol. 2012, 12, 282–294. [Google Scholar] [CrossRef]
- Crotzer, V.L.; Blum, J.S. Autophagy and Its Role in MHC-Mediated Antigen Presentation. J. Immunol. 2009, 182, 3335. [Google Scholar] [CrossRef] [Green Version]
- English, L.; Chemali, M.; Duron, J.; Rondeau, C.; Laplante, A.; Gingras, D.; Alexander, D.; Leib, D.; Norbury, C.; Lippé, R.; et al. Autophagy Enhances the Presentation of Endogenous Viral Antigens on MHC Class I Molecules during HSV-1 Infection. Nat. Immunol. 2009, 10, 480–487. [Google Scholar] [CrossRef] [Green Version]
- Paludan, C.; Schmid, D.; Landthaler, M.; Vockerodt, M.; Kube, D.; Tuschl, T.; Münz, C. Endogenous MHC Class II Processing of a Viral Nuclear Antigen after Autophagy. Science 2005, 307, 593–596. [Google Scholar] [CrossRef]
- Muniz-Feliciano, L.; Doggett, T.A.; Zhou, Z.; Ferguson, T.A. RUBCN/Rubicon and EGFR Regulate Lysosomal Degradative Processes in the Retinal Pigment Epithelium (RPE) of the Eye. Autophagy 2017, 13, 2072–2085. [Google Scholar] [CrossRef] [Green Version]
- Nedjic, J.; Aichinger, M.; Emmerich, J.; Mizushima, N.; Klein, L. Autophagy in Thymic Epithelium Shapes the T-Cell Repertoire and Is Essential for Tolerance. Nature 2008, 455, 396–400. [Google Scholar] [CrossRef]
- Ireland, J.M.; Unanue, E.R. Autophagy in Antigen-Presenting Cells Results in Presentation of Citrullinated Peptides to CD4 T Cells. J. Exp. Med. 2011, 208, 2625–2632. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Münz, C. Autophagy beyond Intracellular MHC Class II Antigen Presentation. Trends Immunol. 2016, 37, 755–763. [Google Scholar] [CrossRef] [PubMed]
- Dengjel, J.; Schoor, O.; Fischer, R.; Reich, M.; Kraus, M.; Muller, M.; Kreymborg, K.; Altenberend, F.; Brandenburg, J.; Kalbacher, H.; et al. Autophagy Promotes MHC Class II Presentation of Peptides from Intracellular Source Proteins. Proc. Natl. Acad. Sci. USA 2005, 102, 7922–7927. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmid, D.; Pypaert, M.; Münz, C. Antigen-Loading Compartments for Major Histocompatibility Complex Class II Molecules Continuously Receive Input from Autophagosomes. Immunity 2007, 26, 79–92. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, H.K.; Mattei, L.M.; Steinberg, B.E.; Alberts, P.; Lee, Y.H.; Chervonsky, A.; Mizushima, N.; Grinstein, S.; Iwasaki, A. In Vivo Requirement for Atg5 in Antigen Presentation by Dendritic Cells. Immunity 2010, 32, 227–239. [Google Scholar] [CrossRef] [Green Version]
- Subramani, S.; Malhotra, V. Non-Autophagic Roles of Autophagy-Related Proteins. EMBO Rep. 2013, 14, 143–151. [Google Scholar] [CrossRef]
- Saric, A.; Hipolito, V.E.B.; Kay, J.G.; Canton, J.; Antonescu, C.N.; Botelho, R.J. MTOR Controls Lysosome Tubulation and Antigen Presentation in Macrophages and Dendritic Cells. Mol. Biol. Cell 2016, 27, 321–333. [Google Scholar] [CrossRef]
- Hernández-Pérez, S.; Vainio, M.; Kuokkanen, E.; Šuštar, V.; Petrov, P.; Forstén, S.; Paavola, V.; Rajala, J.; Awoniyi, L.O.; Sarapulov, A.V.; et al. B Cells Rapidly Target Antigen and Surface-Derived MHCII into Peripheral Degradative Compartments. J. Cell Sci. 2020, 133, jcs235192. [Google Scholar] [CrossRef]
- Karlsson, L. DM and DO Shape the Repertoire of Peptide–MHC-Class-II Complexes. Curr. Opin. Immunol. 2005, 17, 65–70. [Google Scholar] [CrossRef]
- Nolte, M.A.; Nolte-’t Hoen, E.N.M.; Margadant, C. Integrins Control Vesicular Trafficking; New Tricks for Old Dogs. Trends Biochem Sci. 2021, 46, 124–137. [Google Scholar] [CrossRef] [PubMed]
- Kenific, C.M.; Stehbens, S.J.; Goldsmith, J.; Leidal, A.M.; Faure, N.; Ye, J.; Wittmann, T.; Debnath, J. NBR 1 Enables Autophagy-Dependent Focal Adhesion Turnover. J. Cell Biol. 2016, 212, 577–590. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharifi, M.N.; Mowers, E.E.; Drake, L.E.; Collier, C.; Chen, H.; Zamora, M.; Mui, S.; Macleod, K.F. Autophagy Promotes Focal Adhesion Disassembly and Cell Motility of Metastatic Tumor Cells through the Direct Interaction of Paxillin with LC3. Cell Rep. 2016, 15, 1660–1672. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rafiq, N.B.M.; Nishimura, Y.; Plotnikov, S. v.; Thiagarajan, V.; Zhang, Z.; Shi, S.; Natarajan, M.; Viasnoff, V.; Kanchanawong, P.; Jones, G.E.; et al. A Mechano-Signalling Network Linking Microtubules, Myosin IIA Filaments and Integrin-Based Adhesions. Nat. Mater. 2019, 18, 638–649. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carrasco, Y.R.; Fleire, S.J.; Cameron, T.; Dustin, M.L.; Batista, F.D. LFA-1/ICAM-1 Interaction Lowers the Threshold of B Cell Activation by Facilitating B Cell Adhesion and Synapse Formation. Immunity 2004, 20, 589–599. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.C.; Yim, Y.I.; Wu, X.; Jaumouillé, V.; Cameron, A.; Waterman, C.M.; Kehrl, J.H.; Hammer, J.A. A B Cell Actomyosin Arc Network Couples Integrin Co-Stimulation to Mechanical Force-Dependent Immune Synapse Formation. eLife 2022, 11, e72805. [Google Scholar] [CrossRef]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lagos, J.; Sagadiev, S.; Diaz, J.; Bozo, J.P.; Guzman, F.; Stefani, C.; Zanlungo, S.; Acharya, M.; Yuseff, M.I. Autophagy Induced by Toll-like Receptor Ligands Regulates Antigen Extraction and Presentation by B Cells. Cells 2022, 11, 3883. https://doi.org/10.3390/cells11233883
Lagos J, Sagadiev S, Diaz J, Bozo JP, Guzman F, Stefani C, Zanlungo S, Acharya M, Yuseff MI. Autophagy Induced by Toll-like Receptor Ligands Regulates Antigen Extraction and Presentation by B Cells. Cells. 2022; 11(23):3883. https://doi.org/10.3390/cells11233883
Chicago/Turabian StyleLagos, Jonathan, Sara Sagadiev, Jheimmy Diaz, Juan Pablo Bozo, Fanny Guzman, Caroline Stefani, Silvana Zanlungo, Mridu Acharya, and Maria Isabel Yuseff. 2022. "Autophagy Induced by Toll-like Receptor Ligands Regulates Antigen Extraction and Presentation by B Cells" Cells 11, no. 23: 3883. https://doi.org/10.3390/cells11233883
APA StyleLagos, J., Sagadiev, S., Diaz, J., Bozo, J. P., Guzman, F., Stefani, C., Zanlungo, S., Acharya, M., & Yuseff, M. I. (2022). Autophagy Induced by Toll-like Receptor Ligands Regulates Antigen Extraction and Presentation by B Cells. Cells, 11(23), 3883. https://doi.org/10.3390/cells11233883