
Tumor-Derived Extracellular Vesicles Induce CCL18 Production by Mast Cells: A Possible Link to Angiogenesis


Abstract
:1. Introduction
2. Materials and Methods
2.1. Antibodies and Reagents
2.2. Cell Culture
2.3. Isolation of Tumor-Derived Microvesicles (TMVs)
2.4. Mast Cell Activation
2.5. RNA Isolation
2.6. High-Throughput Sequencing and Data Analysis
2.7. Real-Time PCR
2.8. Human Cytokine Assay
2.9. SDS-PAGE and Immunoblotting
2.10. Cell Proliferation
2.11. Wound Healing Assay
2.12. In Vitro Angiogenesis Assay by Tube Formation
2.13. Statistical Analysis
3. Results
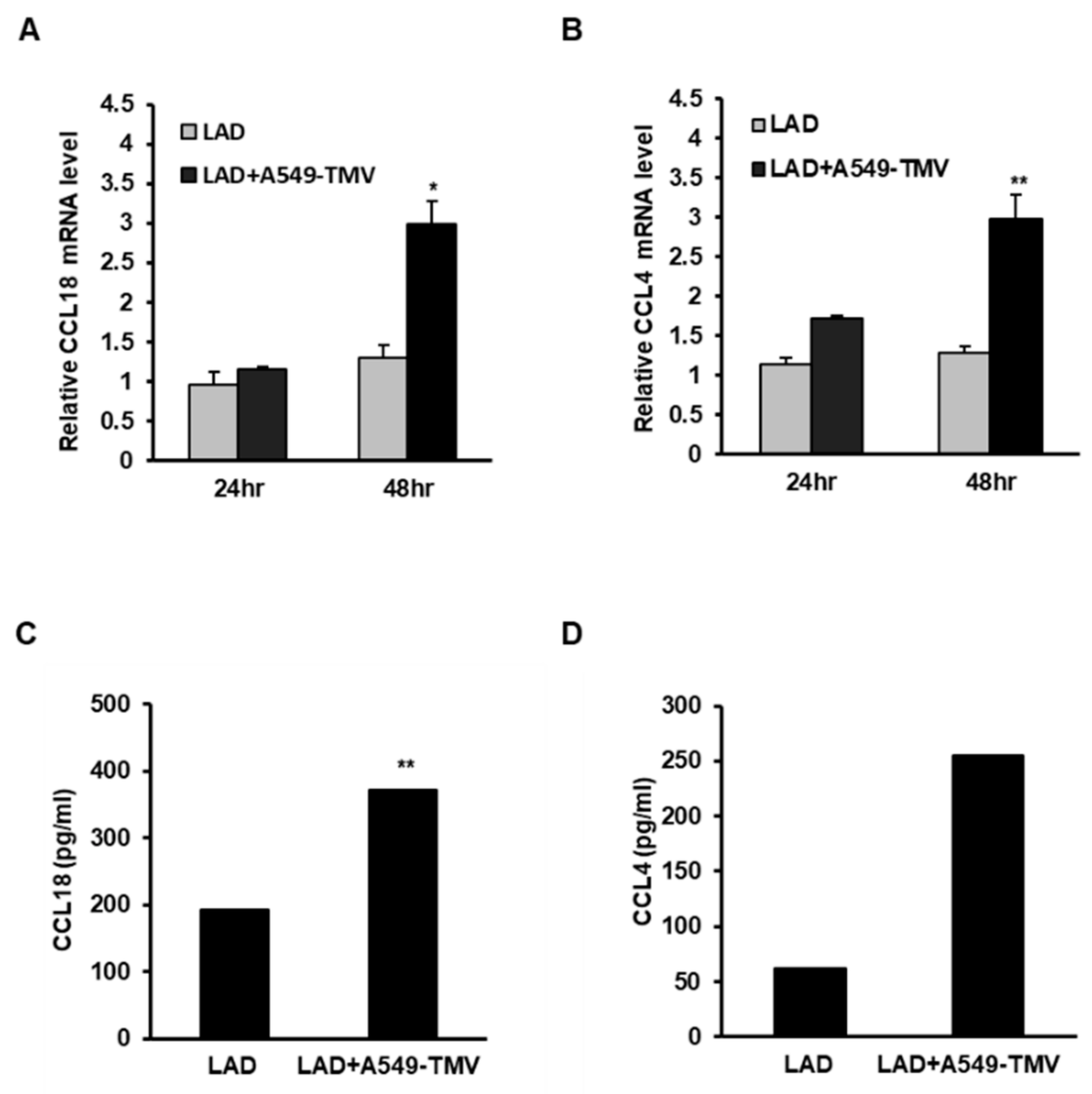
3.1. TMVs Derived from Lung Cancer Cells Induced CCL18 Release from Mast Cells
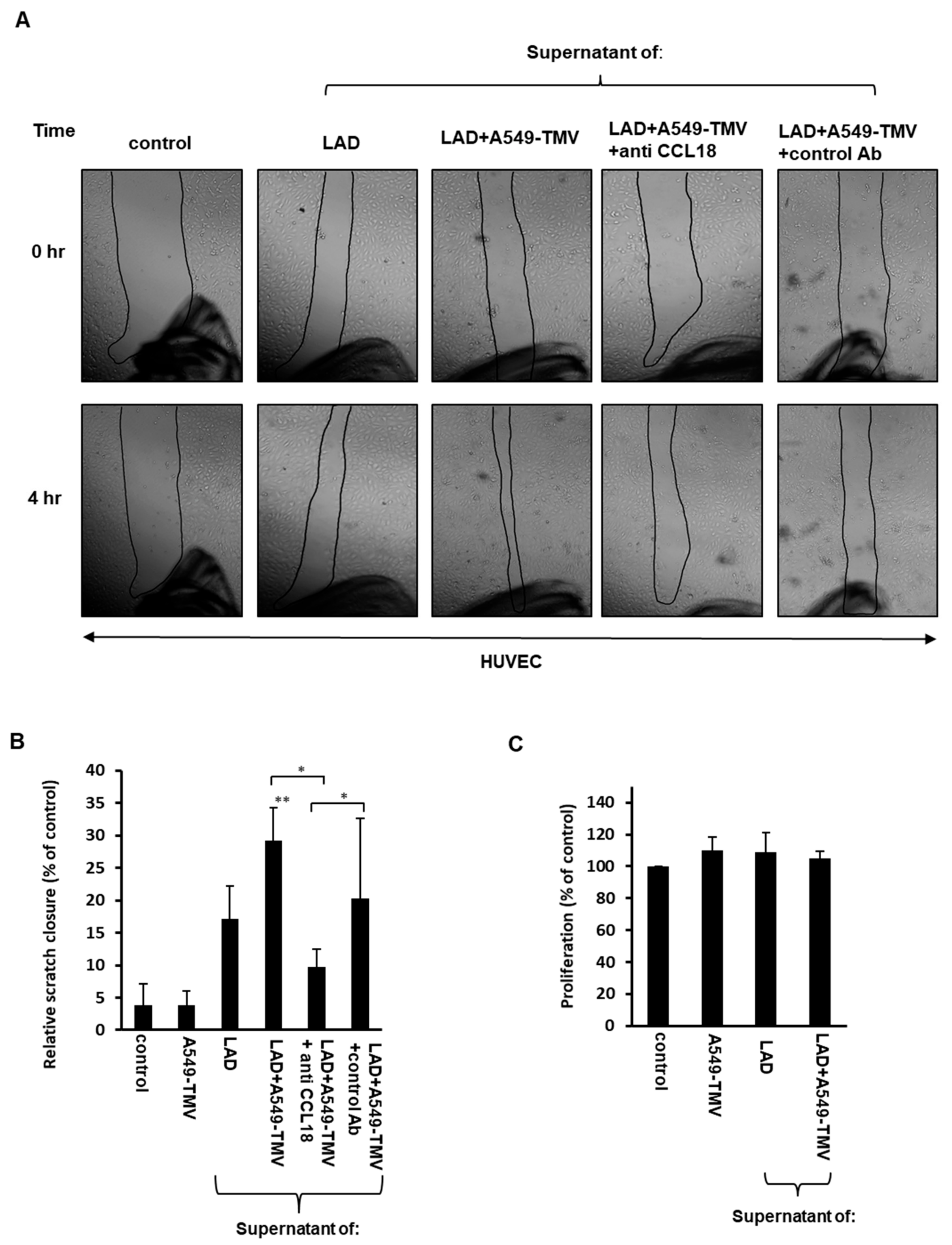
3.2. CCL18 Derived from TMV-Stimulated Mast Cells Induced HUVEC Migration
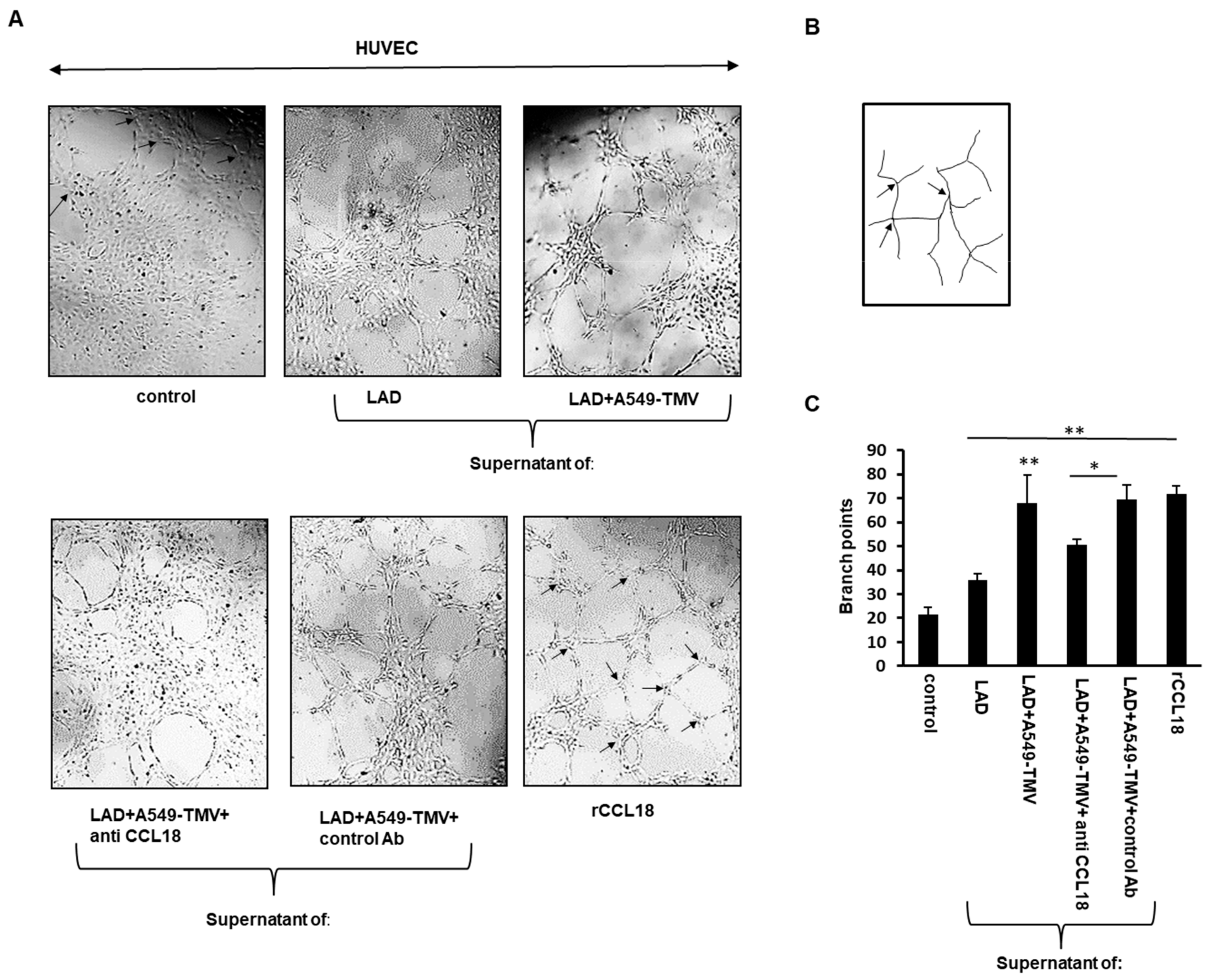
3.3. CCL18 Derived from TMV-Stimulated Mast Cells Induced HUVEC Tube Formation
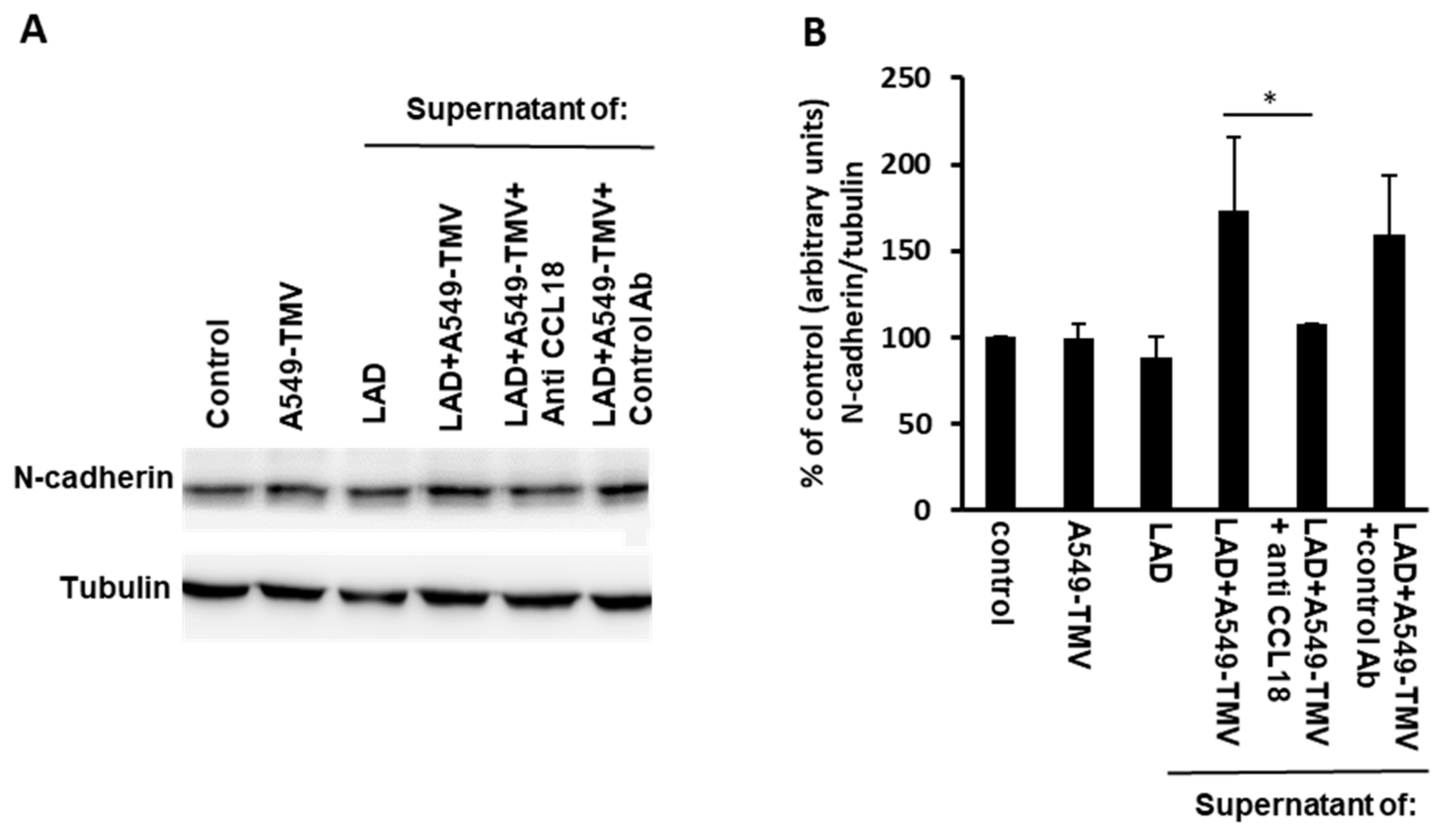
3.4. CCL18 Derived from TMV-Stimulated MCs Enhanced Endothelial–Mesenchymal Transition in HUVECs
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| CCL2 | CC chemokine ligand 2 |
| CCL4 | CC chemokine ligand 4 |
| CCL18 | CC chemokine ligand 18 |
| EndMT | Endothelial-to-mesenchymal transition |
| EV | Extracellular vesicle |
| HUVEC | Human umbilical cord endothelial cell |
| MC | Mast cell |
| MCP-1 | Monocyte chemoattractant protein 1 |
| NSCLC | Non-small-cell lung cancer |
| TAM | Tumor-associated macrophage |
| TMV | Tumor-derived microvesicle |
| TME | Tumor microenvironment |
References
- Kalesnikoff, J.; Galli, S.J. New developments in mast cell biology. Nat. Immunol. 2008, 9, 1215–1223. [Google Scholar] [CrossRef]
- Liu, J.; Zhang, Y.; Zhao, J.; Yang, Z.; Li, D.; Katirai, F.; Huang, B. Mast cell: Insight into remodeling a tumor microenvironment. Cancer Metastasis Rev. 2011, 30, 177–184. [Google Scholar] [CrossRef] [PubMed]
- Rigoni, A.; Colombo, M.P.; Pucillo, C. The Role of Mast Cells in Molding the Tumor Microenvironment. Cancer Microenviron. 2015, 8, 167–176. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Varricchi, G.; Galdiero, M.R.; Loffredo, S.; Marone, G.; Iannone, R.; Marone, G.; Granata, F. Are Mast Cells MASTers in Cancer? Front. Immunol. 2017, 8, 424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dyduch, G.; Kaczmarczyk, K.; Okon, K. Mast cells and cancer: Enemies or allies? Pol. J. Pathol. Off. J. Pol. Soc. Pathol. 2012, 63, 1–7. [Google Scholar]
- Aponte-López, A.; Muñoz-Cruz, S. Mast Cells in the Tumor Microenvironment. Adv. Exp. Med. Biol. 2020, 1273, 159–173. [Google Scholar] [CrossRef]
- Detmar, M. Tumor angiogenesis. J. Investig. Dermatol. Symp. Proc. 2000, 5, 20–23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ribatti, D. Genetic and epigenetic mechanisms in the early development of the vascular system. J. Anat. 2006, 208, 139–152. [Google Scholar] [CrossRef] [PubMed]
- Ribatti, D.; Crivellato, E. Mast cells, angiogenesis, and tumour growth. Biochim. Biophys. Acta 2012, 1822, 2–8. [Google Scholar] [CrossRef] [Green Version]
- Sullivan, R.; Maresh, G.; Zhang, X.; Salomon, C.; Hooper, J.; Margolin, D.; Li, L. The Emerging Roles of Extracellular Vesicles As Communication Vehicles within the Tumor Microenvironment and Beyond. Front. Endocrinol. 2017, 8, 194. [Google Scholar] [CrossRef] [Green Version]
- Bian, X.; Xiao, Y.T.; Wu, T.; Yao, M.; Du, L.; Ren, S.; Wang, J. Microvesicles and chemokines in tumor microenvironment: Mediators of intercellular communications in tumor progression. Mol. Cancer 2019, 18, 50. [Google Scholar] [CrossRef] [PubMed]
- Salamon, P.; Mekori, Y.A.; Shefler, I. Lung cancer-derived extracellular vesicles: A possible mediator of mast cell activation in the tumor microenvironment. Cancer Immunol. Immunother. 2020, 69, 373–381. [Google Scholar] [CrossRef] [PubMed]
- Shi, L.; Zhang, B.; Sun, X.; Zhang, X.; Lv, S.; Li, H.; Wang, X.; Zhao, C.; Zhang, H.; Xie, X.; et al. CC chemokine ligand 18(CCL18) promotes migration and invasion of lung cancer cells by binding to Nir1 through Nir1-ELMO1/DOC180 signaling pathway. Mol. Carcinog. 2016, 55, 2051–2062. [Google Scholar] [CrossRef] [PubMed]
- Kirshenbaum, A.S.; Akin, C.; Wu, Y.; Rottem, M.; Goff, J.P.; Beaven, M.A.; Rao, V.K.; Metcalfe, D.D. Characterization of novel stem cell factor responsive human mast cell lines LAD 1 and 2 established from a patient with mast cell sarcoma/leukemia; activation following aggregation of FcepsilonRI or FcgammaRI. Leuk. Res. 2003, 27, 677–682. [Google Scholar] [CrossRef]
- Shefler, I.; Salamon, P.; Reshef, T.; Mor, A.; Mekori, Y.A. T cell-induced mast cell activation: A role for microparticles released from activated T cells. J. Immunol. 2010, 185, 4206–4212. [Google Scholar] [CrossRef] [PubMed]
- Shefler, I.; Pasmanik-Chor, M.; Kidron, D.; Mekori, Y.A.; Hershko, A.Y. T cell-derived microvesicles induce mast cell production of IL-24: Relevance to inflammatory skin diseases. J. Allergy Clin. Immunol. 2014, 133, 217–224.e3. [Google Scholar] [CrossRef]
- Rashid, G.; Benchetrit, S.; Fishman, D.; Bernheim, J. Effect of advanced glycation end-products on gene expression and synthesis of TNF-alpha and endothelial nitric oxide synthase by endothelial cells. Kidney Int. 2004, 66, 1099–1106. [Google Scholar] [CrossRef] [Green Version]
- Hashimshony, T.; Senderovich, N.; Avital, G.; Klochendler, A.; de Leeuw, Y.; Anavy, L.; Gennert, D.; Li, S.; Livak, K.J.; Rozenblatt-Rosen, O.; et al. CEL-Seq2: Sensitive highly-multiplexed single-cell RNA-Seq. Genome Biol. 2016, 17, 77. [Google Scholar] [CrossRef] [Green Version]
- Baram, D.; Vaday, G.G.; Salamon, P.; Drucker, I.; Hershkoviz, R.; Mekori, Y.A. Human mast cells release metalloproteinase-9 on contact with activated T cells: Juxtacrine regulation by TNF-alpha. J. Immunol. 2001, 167, 4008–4016. [Google Scholar] [CrossRef] [Green Version]
- Distler, O.; Neidhart, M.; Gay, R.E.; Gay, S. The molecular control of angiogenesis. Int. Rev. Immunol. 2002, 21, 33–49. [Google Scholar] [CrossRef]
- Lin, L.; Chen, Y.S.; Yao, Y.D.; Chen, J.Q.; Chen, J.N.; Huang, S.Y.; Zeng, Y.J.; Yao, H.R.; Zeng, S.H.; Fu, Y.S.; et al. CCL18 from tumor-associated macrophages promotes angiogenesis in breast cancer. Oncotarget 2015, 6, 34758–34773. [Google Scholar] [CrossRef] [Green Version]
- Lamouille, S.; Xu, J.; Derynck, R. Molecular mechanisms of epithelial-mesenchymal transition. Nat. Rev. Mol. Cell Biol. 2014, 15, 178–196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gonzalez, D.M.; Medici, D. Signaling mechanisms of the epithelial-mesenchymal transition. Sci. Signal. 2014, 7, re8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maacha, S.; Bhat, A.A.; Jimenez, L.; Raza, A.; Haris, M.; Uddin, S.; Grivel, J.C. Extracellular vesicles-mediated intercellular communication: Roles in the tumor microenvironment and anti-cancer drug resistance. Mol. Cancer 2019, 18, 55. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kogure, A.; Kosaka, N.; Ochiya, T. Cross-talk between cancer cells and their neighbors via miRNA in extracellular vesicles: An emerging player in cancer metastasis. J. Biomed. Sci. 2019, 26, 7. [Google Scholar] [CrossRef]
- Wysoczynski, M.; Ratajczak, M.Z. Lung cancer secreted microvesicles: Underappreciated modulators of microenvironment in expanding tumors. Int. J. Cancer 2009, 125, 1595–1603. [Google Scholar] [CrossRef] [Green Version]
- Ploenes, T.; Scholtes, B.; Krohn, A.; Burger, M.; Passlick, B.; Müller-Quernheim, J.; Zissel, G. CC-chemokine ligand 18 induces epithelial to mesenchymal transition in lung cancer A549 cells and elevates the invasive potential. PLoS ONE 2013, 8, e53068. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Q.; Tang, Y.; Yu, H.; Yin, Q.; Li, M.; Shi, L.; Zhang, W.; Li, D.; Li, L. CCL18 from tumor-cells promotes epithelial ovarian cancer metastasis via mTOR signaling pathway. Mol. Carcinog. 2016, 55, 1688–1699. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Yao, Y.; Gong, C.; Yu, F.; Su, S.; Chen, J.; Liu, B.; Deng, H.; Wang, F.; Lin, L.; et al. CCL18 from tumor-associated macrophages promotes breast cancer metastasis via PITPNM3. Cancer Cell 2011, 19, 541–555. [Google Scholar] [CrossRef] [Green Version]
- She, L.; Qin, Y.; Wang, J.; Liu, C.; Zhu, G.; Li, G.; Wei, M.; Chen, C.; Liu, G.; Zhang, D.; et al. Tumor-associated macrophages derived CCL18 promotes metastasis in squamous cell carcinoma of the head and neck. Cancer Cell Int. 2018, 18, 120. [Google Scholar] [CrossRef]
- Pivarcsi, A.; Gombert, M.; Dieu-Nosjean, M.C.; Lauerma, A.; Kubitza, R.; Meller, S.; Rieker, J.; Muller, A.; Da Cunha, L.; Haahtela, A.; et al. CC chemokine ligand 18, an atopic dermatitis-associated and dendritic cell-derived chemokine, is regulated by staphylococcal products and allergen exposure. J. Immunol. 2004, 173, 5810–5817. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiao, H.; He, M.; Xie, G.; Liu, Y.; Zhao, Y.; Ye, X.; Li, X.; Zhang, M. The release of tryptase from mast cells promote tumor cell metastasis via exosomes. BMC Cancer 2019, 19, 1015. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dongre, A.; Weinberg, R.A. New insights into the mechanisms of epithelial-mesenchymal transition and implications for cancer. Nat. Rev. Mol. Cell Biol. 2019, 20, 69–84. [Google Scholar] [CrossRef]
- Garside, V.C.; Chang, A.C.; Karsan, A.; Hoodless, P.A. Co-ordinating Notch, BMP, and TGF-β signaling during heart valve development. Cell. Mol. Life Sci. 2013, 70, 2899–2917. [Google Scholar] [CrossRef] [PubMed]
- van Meeteren, L.A.; ten Dijke, P. Regulation of endothelial cell plasticity by TGF-β. Cell Tissue Res. 2012, 347, 177–186. [Google Scholar] [CrossRef] [Green Version]
- Coultas, L.; Chawengsaksophak, K.; Rossant, J. Endothelial cells and VEGF in vascular development. Nature 2005, 438, 937–945. [Google Scholar] [CrossRef]
- Zeisberg, E.M.; Potenta, S.; Xie, L.; Zeisberg, M.; Kalluri, R. Discovery of endothelial to mesenchymal transition as a source for carcinoma-associated fibroblasts. Cancer Res. 2007, 67, 10123–10128. [Google Scholar] [CrossRef] [PubMed] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| ToppGene | p-Value GOrilla | String | |
|---|---|---|---|
| Up-regulated genes Cellular response to stimulus | |||
| Cellular response to stimulus | 2.19 × 10−6 | 1.54 × 10−6 | |
| Chemokine-mediated signaling pathway | 7.89 × 10−5 | 1.22 × 10−4 | |
| Cellular response to organic substance | 1.32 × 10−4 | ||
| Cytokine–cytokine receptor interaction | 4.59 × 10−5 | 0.0076 | |
| Interleukin-10 signaling | 2.38 × 10−4 | ||
| Inflammation mediated by chemokine and cytokine signaling pathway | 1.22 × 10−3 | ||
| NF-kappa B signaling pathway | 1.66 × 10−3 | 0.00024 | |
| TNF signaling pathway | 0.0017 | ||
| IL-17 signaling pathway | 0.0126 | ||
| Toll-like receptor signaling pathway | 1.22 × 10−4 | ||
| Cell chemotaxis | 5.60 × 10−4 | ||
| Lymphocyte migration | 5.00 × 10−4 | ||
| Monocyte chemotaxis | 5.00 × 10−4 | ||
| Lymphocyte migration chemotaxis | 5.00 × 10−4 | ||
| Positive regulation of ERK1 and ERK2 cascade | 5.42 × 10−5 | 5.00 × 10−4 | |
| Apoptosis | 9.77 × 10−3 | 0.0119 | |
| Down-regulated genes | |||
| Cell cycle | |||
| Cell cycle, mitotic | 7.54 × 10−107 | 1.73 × 10−5 | 3.70 × 10−28 |
| Cell cycle checkpoints | 9.19 × 10−93 | 3.27 × 10−6 | |
| MicroRNAs in cancer | 6.72 × 10−26 | 8.59 × 10−5 | |
| DNA replication | 3.85 × 10−27 | 2.27 × 10−16 | |
| p53 signaling pathway | 1.40 × 10−5 | ||
| Cell division | 1.41 × 10−9 | 6.65 × 10−4 |
| Gene Symbol | Description | Fold Change | p-Value |
|---|---|---|---|
| EGR1 | Early growth response 1 | 4.74 | 4.72 × 10−23 |
| EGR3 | Early growth response 3 | 3.67 | 9.11 × 10−16 |
| CCL4 | CC chemokine ligand 4 | 2.82 | 1.80 × 10−10 |
| CCL18 | CC chemokine ligand 18 | 2.59 | 2.72 × 10−20 |
| FOSB | FosB proto-oncogene, AP-1 transcription factor subunit | 2.53 | 1.13 × 10−8 |
| ATF4 | Activating transcription factor 4 | 2.52 | 1.35 × 10−86 |
| CCL4L2 | C-C motif chemokine ligand 4 like 2 | 2.51 | 8.19 × 10−9 |
| CCL3L1 | C-C motif chemokine ligand 3 like 1 | 2.43 | 3.20 × 10−8 |
| BMP7 | Bone morphogenetic protein 7 | 2.01 | 9.57 × 10−9 |
| CCL3 | C-C motif chemokine ligand 3 | 2.0 | 4.79 × 10−6 |
| TNFRSF12A | TNF receptor superfamily member 12A | 1.93 | 2.18 × 10−6 |
| PTGS2 | Prostaglandin-endoperoxide synthase 2 | 1.92 | 5.47 × 10−5 |
| TNF | Tumor necrosis factor | 1.89 | 6.20 × 10−6 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shefler, I.; Salamon, P.; Zitman-Gal, T.; Mekori, Y.A. Tumor-Derived Extracellular Vesicles Induce CCL18 Production by Mast Cells: A Possible Link to Angiogenesis. Cells 2022, 11, 353. https://doi.org/10.3390/cells11030353
Shefler I, Salamon P, Zitman-Gal T, Mekori YA. Tumor-Derived Extracellular Vesicles Induce CCL18 Production by Mast Cells: A Possible Link to Angiogenesis. Cells. 2022; 11(3):353. https://doi.org/10.3390/cells11030353
Chicago/Turabian StyleShefler, Irit, Pazit Salamon, Tali Zitman-Gal, and Yoseph A. Mekori. 2022. "Tumor-Derived Extracellular Vesicles Induce CCL18 Production by Mast Cells: A Possible Link to Angiogenesis" Cells 11, no. 3: 353. https://doi.org/10.3390/cells11030353
APA StyleShefler, I., Salamon, P., Zitman-Gal, T., & Mekori, Y. A. (2022). Tumor-Derived Extracellular Vesicles Induce CCL18 Production by Mast Cells: A Possible Link to Angiogenesis. Cells, 11(3), 353. https://doi.org/10.3390/cells11030353