
TRIM32 Deficiency Impairs the Generation of Pyramidal Neurons in Developing Cerebral Cortex

 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Mice
2.2. Genotyping Detection of TRIM32 Mice
2.3. Calculate the Embryonic Age of Mice
2.4. Antibodies
2.5. Immunofluorescence Staining and Image Analysis
2.6. Edu Pulse Chase
2.6.1. The Ability of NPCs Proliferation
2.6.2. The Ability of NPCs to Differentiate into TBR1 Positive Neurons
2.7. Sample Preparation and RNA-seq Analysis
2.8. Statistical Analysis
3. Results
3.1. The Expression Pattern of TRIM32 in the Cortex during Embryo
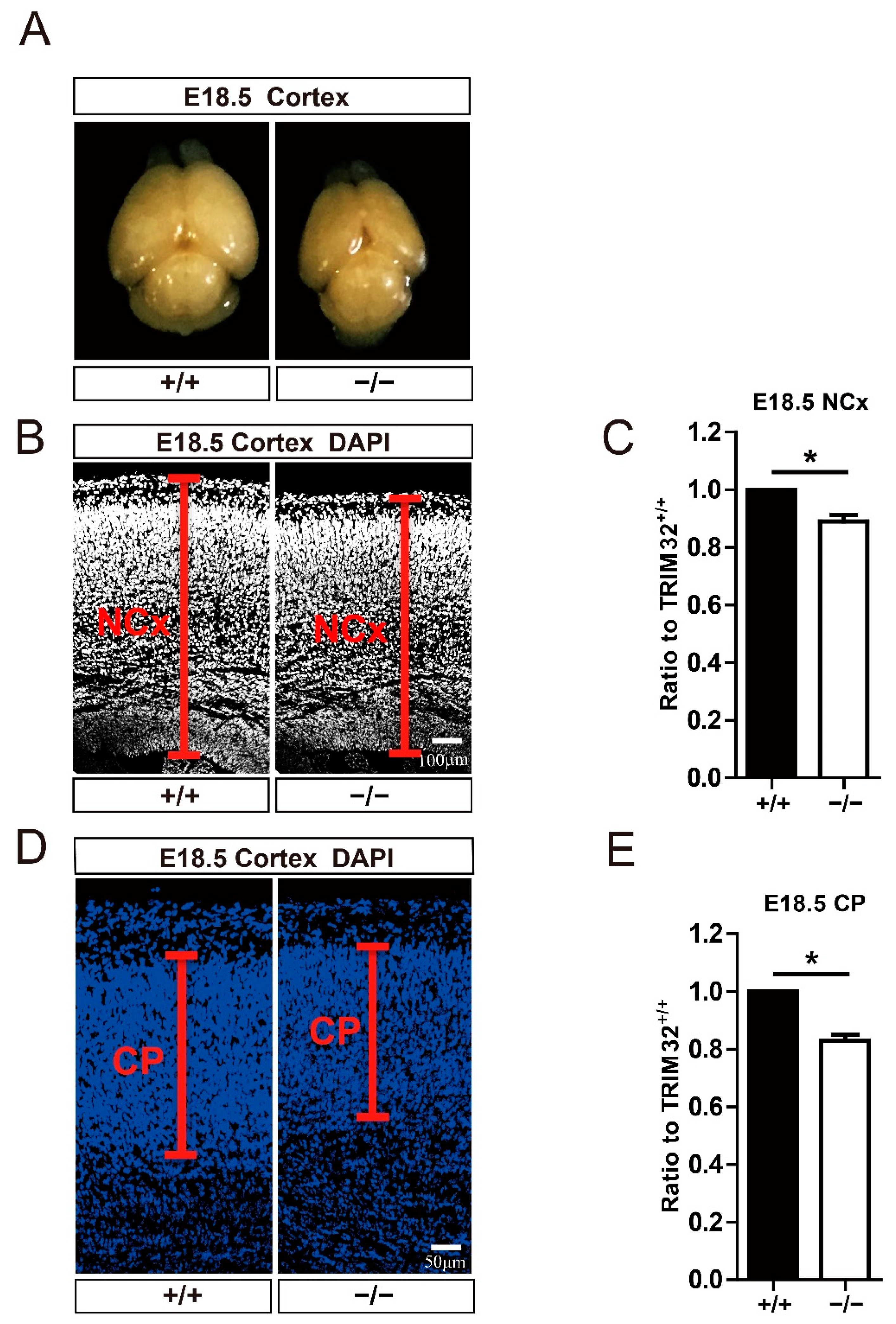
3.2. TRIM32 Deficiency Mice Exhibit Reduced Size of Brain
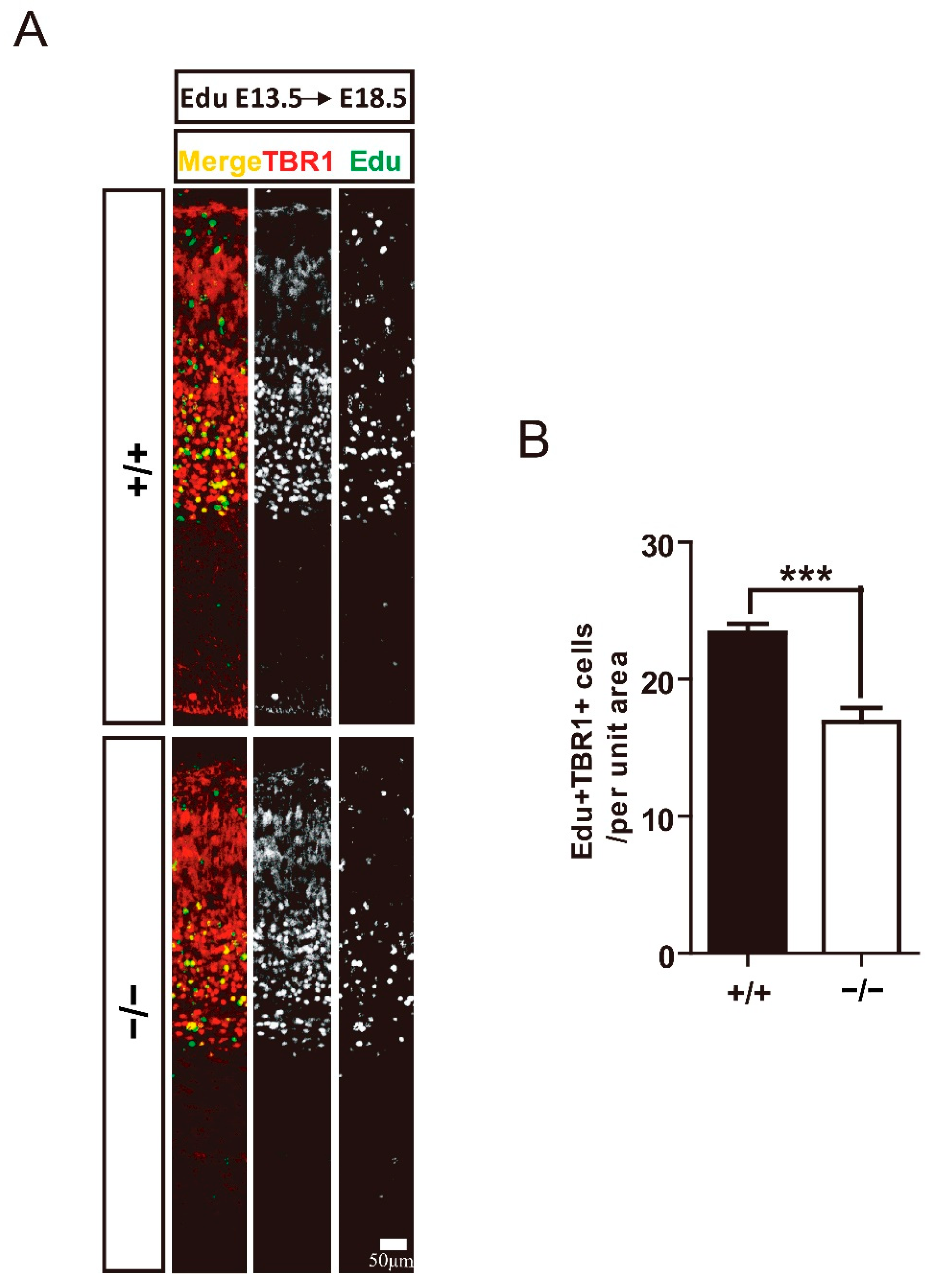
3.3. TRIM32 Deficiency Results in Reduced Generation of Cortical Neurons in Developing Cortex
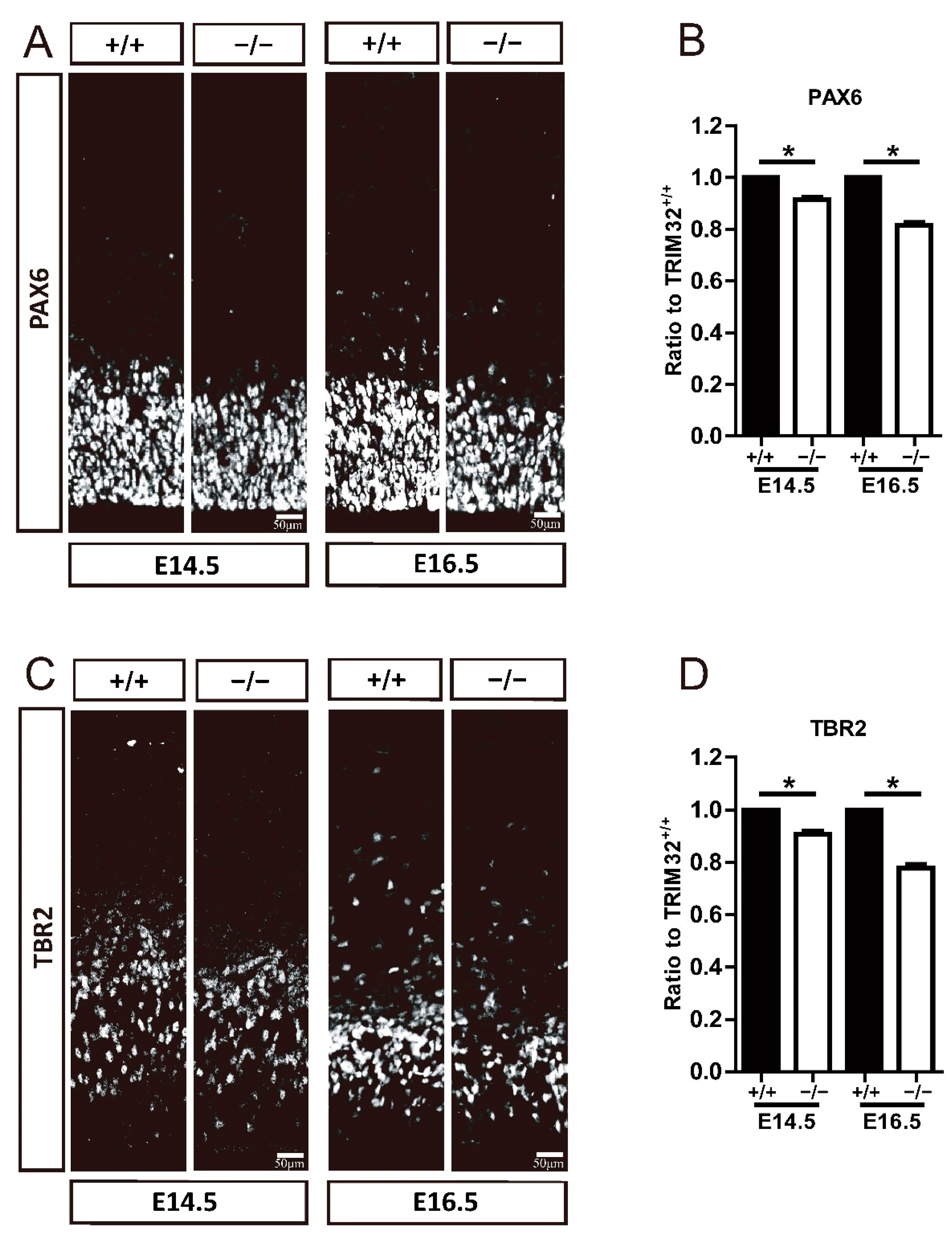
3.4. TRIM32 Deficiency Causes Smaller Size of Neural Progenitor Pool
3.5. TRIM32 Deficiency Decreases Proliferation and Mitosis of Both RGCs and IPCs
3.6. TRIM32 Deficiency Does Not Affect Apoptosis in Developing Cortex
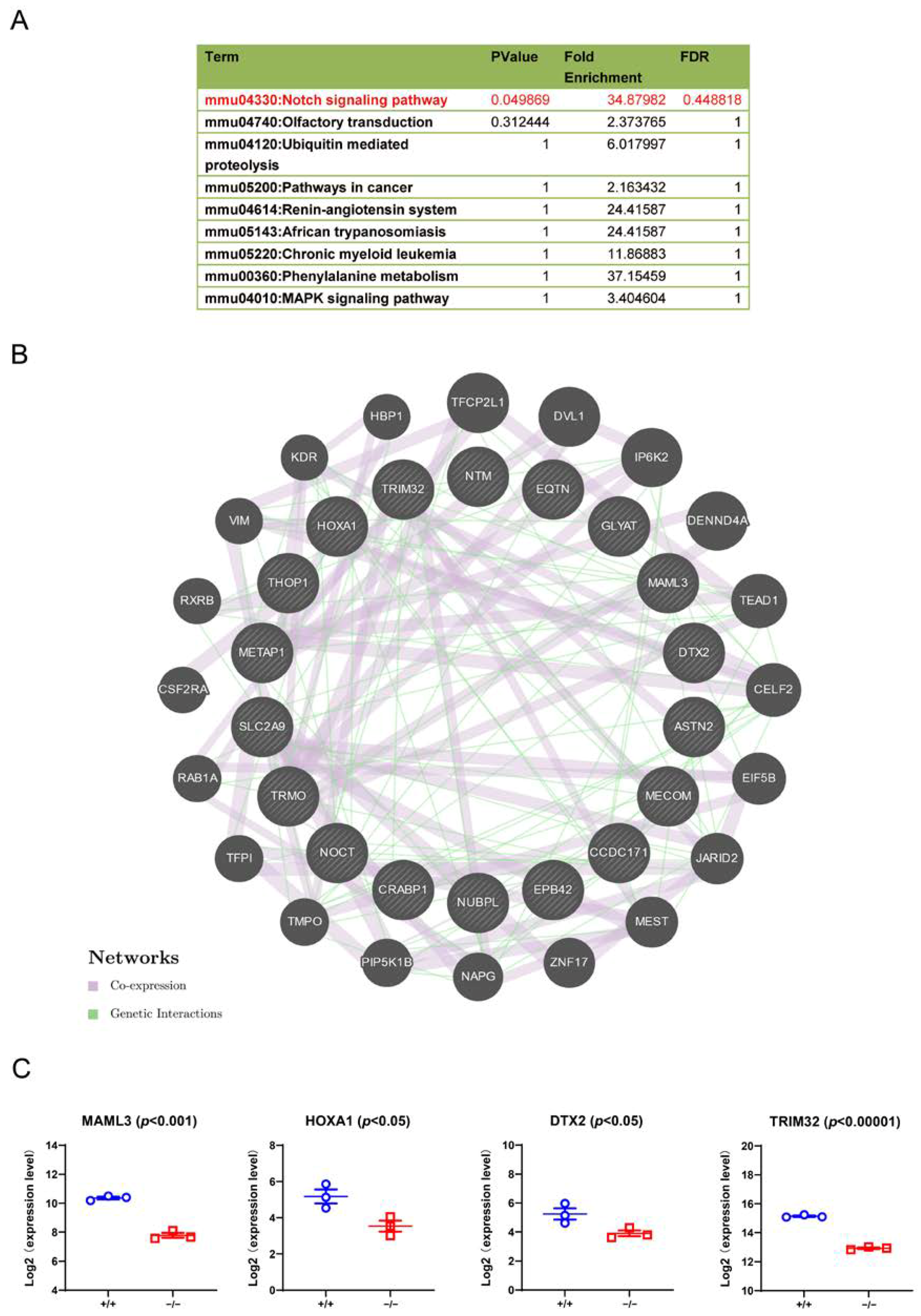
3.7. Downstream Signalling Regulated by TRIM32
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Lee, E.; Lee, J.; Kim, E. Excitation/Inhibition Imbalance in Animal Models of Autism Spectrum Disorders. Biol. Psychiatry 2017, 81, 838–847. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, Z.Q.; Li, D.; Huang, Y.; Chen, X.P.; Huang, W.; Liu, C.F.; Zhao, H.Q.; Xu, R.X.; Cheng, M.; Schachner, M.; et al. Caspr Controls the Temporal Specification of Neural Progenitor Cells through Notch Signaling in the Developing Mouse Cerebral Cortex. Cereb. Cortex 2017, 27, 1369–1385. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seuntjens, E.; Nityanandam, A.; Miquelajauregui, A.; Debruyn, J.; Stryjewska, A.; Goebbels, S.; Nave, K.A.; Huylebroeck, D.; Tarabykin, V. Sip1 regulates sequential fate decisions by feedback signaling from postmitotic neurons to progenitors. Nat. Neurosci. 2009, 12, 1373–1380. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, J.L.; Nedivi, E. Highly specific structural plasticity of inhibitory circuits in the adult neocortex. Neuroscientist 2013, 19, 384–393. [Google Scholar] [CrossRef] [Green Version]
- Borrell, V.; Gotz, M. Role of radial glial cells in cerebral cortex folding. Curr. Opin. Neurobiol. 2014, 27, 39–46. [Google Scholar] [CrossRef] [Green Version]
- Nadarajah, B.; Parnavelas, J.G. Modes of neuronal migration in the developing cerebral cortex. Nat. Rev. Neurosci. 2002, 3, 423–432. [Google Scholar] [CrossRef]
- Reymond, A.; Meroni, G.; Fantozzi, A.; Merla, G.; Cairo, S.; Luzi, L.; Riganelli, D.; Zanaria, E.; Messali, S.; Cainarca, S.; et al. The tripartite motif family identifies cell compartments. EMBO J. 2001, 20, 2140–2151. [Google Scholar] [CrossRef] [Green Version]
- Kudryashova, E.; Kudryashov, D.; Kramerova, I.; Spencer, M.J. Trim32 is a ubiquitin ligase mutated in limb girdle muscular dystrophy type 2H that binds to skeletal muscle myosin and ubiquitinates actin. J. Mol. Biol. 2005, 354, 413–424. [Google Scholar] [CrossRef]
- Hillje, A.L.; Beckmann, E.; Pavlou, M.A.S.; Jaeger, C.; Pacheco, M.P.; Sauter, T.; Schwamborn, J.C.; Lewejohann, L. The neural stem cell fate determinant TRIM32 regulates complex behavioral traits. Front. Cell. Neurosci. 2015, 9, 75. [Google Scholar] [CrossRef] [Green Version]
- Hillje, A.L.; Worlitzer, M.M.A.; Palm, T.; Schwamborn, J.C. Neural Stem Cells Maintain Their Stemness through Protein Kinase C zeta-Mediated Inhibition of TRIM32. Stem Cells 2011, 29, 1437–1447. [Google Scholar] [CrossRef]
- Schwamborn, J.C.; Berezikov, E.; Knoblich, J.A. The TRIM-NHL Protein TRIM32 Activates MicroRNAs and Prevents Self-Renewal in Mouse Neural Progenitors. Cell 2009, 136, 913–925. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lionel, A.C.; Tammimies, K.; Vaags, A.K.; Rosenfeld, J.A.; Ahn, J.W.; Merico, D.; Noor, A.; Runke, C.K.; Pillalamarri, V.K.; Carter, M.T.; et al. Disruption of the ASTN2/TRIM32 locus at 9q33.1 is a risk factor in males for autism spectrum disorders, ADHD and other neurodevelopmental phenotypes. Hum. Mol. Genet. 2014, 23, 2752–2768. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lionel, A.C.; Crosbie, J.; Barbosa, N.; Goodale, T.; Thiruvahindrapuram, B.; Rickaby, J.; Gazzellone, M.; Carson, A.R.; Howe, J.L.; Wang, Z.; et al. Rare copy number variation discovery and cross-disorder comparisons identify risk genes for ADHD. Sci. Transl. Med. 2011, 3, 95ra75. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, J.W.; Zou, M.M.; Li, Y.F.; Chen, W.J.; Liu, J.C.; Chen, H.; Fang, L.P.; Zhang, Y.; Wang, Z.T.; Chen, J.B.; et al. Absence of TRIM32 Leads to Reduced GABAergic Interneuron Generation and Autism-like Behaviors in Mice via Suppressing mTOR Signaling. Cereb. Cortex 2020, 30, 3240–3258. [Google Scholar] [CrossRef] [PubMed]
- Nicklas, S.; Otto, A.; Wu, X.L.; Miller, P.; Stelzer, S.; Wen, Y.F.; Kuang, S.H.; Wrogemann, K.; Patel, K.; Ding, H.; et al. TRIM32 Regulates Skeletal Muscle Stem Cell Differentiation and Is Necessary for Normal Adult Muscle Regeneration. PLoS ONE 2012, 7, e30445. [Google Scholar] [CrossRef] [Green Version]
- Mitsuhashi, T.; Takahashi, T. Genetic regulation of proliferation/differentiation characteristics of neural progenitor cells in the developing neocortex. Brain Dev. 2009, 31, 553–557. [Google Scholar] [CrossRef]
- Tarabykin, V.; Stoykova, A.; Usman, N.; Gruss, P. Cortical upper layer neurons derive from the subventricular zone as indicated by Svet1 gene expression. Development 2001, 128, 1983–1993. [Google Scholar] [CrossRef]
- Cubelos, B.; Sebastian-Serrano, A.; Kim, S.; Moreno-Ortiz, C.; Redondo, J.M.; Walsh, C.A.; Nieto, M. Cux-2 controls the proliferation of neuronal intermediate precursors of the cortical subventricular zone. Cereb. Cortex 2008, 18, 1758–1770. [Google Scholar] [CrossRef] [Green Version]
- McConnell, S.K.; Kaznowski, C.E. Cell cycle dependence of laminar determination in developing neocortex. Science 1991, 254, 282–285. [Google Scholar] [CrossRef]
- Price, J.; Thurlow, L. Cell lineage in the rat cerebral cortex: A study using retroviral-mediated gene transfer. Development 1988, 104, 473–482. [Google Scholar] [CrossRef]
- Reid, C.B.; Liang, I.; Walsh, C. Systematic widespread clonal organization in cerebral cortex. Neuron 1995, 15, 299–310. [Google Scholar] [CrossRef] [Green Version]
- Buss, R.R.; Sun, W.; Oppenheim, R.W. Adaptive roles of programmed cell death during nervous system development. Annu. Rev. Neurosci. 2006, 29, 1–35. [Google Scholar] [CrossRef] [PubMed]
- Fang, W.Q.; Chen, W.W.; Jiang, L.; Liu, K.; Yung, W.H.; Fu AK, Y.; Ip, N.Y. Overproduction of upper-layer neurons in the neocortex leads to autism-like features in mice. Cell Rep. 2014, 9, 1635–1643. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lipton, J.O.; Sahin, M. The neurology of mTOR. Neuron 2014, 84, 275–291. [Google Scholar] [CrossRef] [Green Version]
- Crino, P.B. The mTOR signalling cascade: Paving new roads to cure neurological disease. Nat. Rev. Neurol. 2016, 12, 379–392. [Google Scholar] [CrossRef]
- Costa-Mattioli, M.; Monteggia, L.M. mTOR complexes in neurodevelopmental and neuropsychiatric disorders. Nat. Neurosci. 2013, 16, 1537–1543. [Google Scholar] [CrossRef]
- Imayoshi, I.; Sakamoto, M.; Yamaguchi, M.; Mori, K.; Kageyama, R. Essential roles of Notch signaling in maintenance of neural stem cells in developing and adult brains. J. Neurosci. 2010, 30, 3489–3498. [Google Scholar] [CrossRef] [Green Version]
- Ma, J.; Meng, Y.; Kwiatkowski, D.J.; Chen, X.; Peng, H.; Sun, Q.; Zha, X.; Wang, F.; Wang, Y.; Jing, Y.; et al. Mammalian target of rapamycin regulates murine and human cell differentiation through STAT3/p63/Jagged/Notch cascade. J. Clin. Investig. 2010, 120, 103–114. [Google Scholar] [CrossRef] [Green Version]
- Maamar, H.; Cabili, M.N.; Rinn, J.; Raj, A. linc-HOXA1 is a noncoding RNA that represses Hoxa1 transcription in cis. Gene Dev. 2013, 27, 1260–1271. [Google Scholar] [CrossRef] [Green Version]
- De Kumar, B.; Parker, H.J.; Paulson, A.; Parrish, M.E.; Zeitlinger, J.; Krumlauf, R. Hoxa1 targets signaling pathways during neural differentiation of ES cells and mouse embryogenesis. Dev. Biol. 2017, 432, 151–164. [Google Scholar] [CrossRef]
- Zhang, L.; Liu, X.L.; Yuan, Z.; Cui, J.; Zhang, H. MiR-99a suppressed cell proliferation and invasion by directly targeting HOXA1 through regulation of the AKT/mTOR signaling pathway and EMT in ovarian cancer. Eur. Rev. Med. Pharmacol. 2019, 23, 4663–4672. [Google Scholar]
- Wu, Q.X.; Lu, S.T.; Zhang, L.; Zhao, L.J. LncRNA HOXA-AS2 Activates the Notch Pathway to Promote Cervical Cancer Cell Proliferation and Migration. Reprod. Sci. 2021, 28, 3000–3009. [Google Scholar] [CrossRef] [PubMed]
- Kitagawa, M. Notch signalling in the nucleus: Roles of Mastermind-like (MAML) transcriptional coactivators. J. Biochem. 2016, 159, 287–294. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heynen, G.J.J.E.; Nevedomskaya, E.; Palit, S.; Basheer, N.J.; Lieftink, C.; Schlicker, A.; Zwart, W.; Bernards, R.; Bajpe, P.K. Mastermind-Like 3 Controls Proliferation and Differentiation in Neuroblastoma. Mol. Cancer Res. 2016, 14, 411–422. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Endo, Y.; Osumi, N.; Wakamatsu, Y. Deltex/Dtx mediates NOTCH signaling in regulation of Bmp4 expression in cranial neural crest formation during avian development. Dev. Growth Differ. 2003, 45, 241–248. [Google Scholar] [CrossRef]
- Gamez, B.; Rodriguez-Carballo, E.; Ventura, F. BMP signaling in telencephalic neural cell specification and maturation. Front. Cell. Neurosci. 2013, 7, 87. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ntim, M.; Li, Q.F.; Zhang, Y.; Liu, X.D.; Li, N.; Sun, H.L.; Zhang, X.; Khan, B.; Wang, B.; Wu, Q.; et al. TRIM32 Deficiency Impairs Synaptic Plasticity by Excitatory-Inhibitory Imbalance via Notch Pathway. Cereb. Cortex 2020, 30, 4617–4632. [Google Scholar] [CrossRef] [PubMed]
- Miyoshi, G.; Butt, S.J.B.; Takebayashi, H.; Fishell, G. Physiologically distinct temporal cohorts of cortical interneurons arise from telencephalic olig2-expressing precursors. J. Neurosci. 2007, 27, 7786–7798. [Google Scholar] [CrossRef] [Green Version]
- Sohal, V.S.; Rubenstein JL, R. Excitation-inhibition balance as a framework for investigating mechanisms in neuropsychiatric disorders. Mol. Psychiatry 2019, 24, 1248–1257. [Google Scholar] [CrossRef]
- Fombonne, E.; Roge, B.; Claverie, J.; Courty, S.; Fremolle, J. Microcephaly and macrocephaly in autism. J. Autism Dev. Disord. 1999, 29, 113–119. [Google Scholar] [CrossRef]
- Wu, X.; Fleming, A.; Ricketts, T.; Pavel, M.; Virgin, H.; Menzies, F.M.; Rubinsztein, D.C. Autophagy regulates Notch degradation and modulates stem cell development and neurogenesis. Nat. Commun. 2016, 7, 10533. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Conciatori, M.; Stodgell, C.J.; Hyman, S.L.; O’Bara, M.; Militerni, R.; Bravaccio, C.; Trillo, S.; Montecchi, F.; Schneider, C.; Melmed, R.; et al. Association between the HOXA1 A218G polymorphism and increased head circumference in patients with autism. Biol. Psychiatry 2004, 55, 413–419. [Google Scholar] [CrossRef] [PubMed]
- Raznahan, A.; Lee, Y.; Vaituzis, C.; Tran, L.; Mackie, S.; Tiemeier, H.; Clasen, L.; Lalonde, F.; Greenstein, D.; Pierson, R.; et al. Allelic Variation Within the Putative Autism Spectrum Disorder Risk Gene Homeobox A1 and Cerebellar Maturation in Typically Developing Children and Adolescents. Autism Res. 2012, 5, 93–100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muscarella, L.A.; Guarnieri, V.; Sacco, R.; Curatolo, P.; Manzi, B.; Alessandrelli, R.; Giana, G.; Militerni, R.; Bravaccio, C.; Lenti, C.; et al. Candidate gene study of HOXB1 in autism spectrum disorder. Mol. Autism 2010, 1, 9. [Google Scholar] [CrossRef] [Green Version]
- Mukhopadhyay, A.; Nieves, E.; Che, F.Y.; Wang, J.; Jin, L.J.; Murray, J.W.; Gordon, K.; Angeletti, R.H.; Wolkoff, A.W. Proteomic analysis of endocytic vesicles: Rab1a regulates motility of early endocytic vesicles. J. Cell Sci. 2011, 124, 765–775. [Google Scholar] [CrossRef] [Green Version]
- Hutagalung, A.H.; Novick, P.J. Role of Rab GTPases in Membrane Traffic and Cell Physiology. Physiol. Rev. 2011, 91, 119–149. [Google Scholar] [CrossRef] [Green Version]








Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sun, Y.-Y.; Chen, W.-J.; Huang, Z.-P.; Yang, G.; Wu, M.-L.; Xu, D.-E.; Yang, W.-L.; Luo, Y.-C.; Xiao, Z.-C.; Xu, R.-X.; et al. TRIM32 Deficiency Impairs the Generation of Pyramidal Neurons in Developing Cerebral Cortex. Cells 2022, 11, 449. https://doi.org/10.3390/cells11030449
Sun Y-Y, Chen W-J, Huang Z-P, Yang G, Wu M-L, Xu D-E, Yang W-L, Luo Y-C, Xiao Z-C, Xu R-X, et al. TRIM32 Deficiency Impairs the Generation of Pyramidal Neurons in Developing Cerebral Cortex. Cells. 2022; 11(3):449. https://doi.org/10.3390/cells11030449
Chicago/Turabian StyleSun, Yan-Yun, Wen-Jin Chen, Ze-Ping Huang, Gang Yang, Ming-Lei Wu, De-En Xu, Wu-Lin Yang, Yong-Chun Luo, Zhi-Cheng Xiao, Ru-Xiang Xu, and et al. 2022. "TRIM32 Deficiency Impairs the Generation of Pyramidal Neurons in Developing Cerebral Cortex" Cells 11, no. 3: 449. https://doi.org/10.3390/cells11030449
APA StyleSun, Y. -Y., Chen, W. -J., Huang, Z. -P., Yang, G., Wu, M. -L., Xu, D. -E., Yang, W. -L., Luo, Y. -C., Xiao, Z. -C., Xu, R. -X., & Ma, Q. -H. (2022). TRIM32 Deficiency Impairs the Generation of Pyramidal Neurons in Developing Cerebral Cortex. Cells, 11(3), 449. https://doi.org/10.3390/cells11030449