
Modeling Down Syndrome Myeloid Leukemia by Sequential Introduction of GATA1 and STAG2 Mutations in Induced Pluripotent Stem Cells with Trisomy 21

 , ,
, , {kind=link}
{kind=link}
{kind=link}
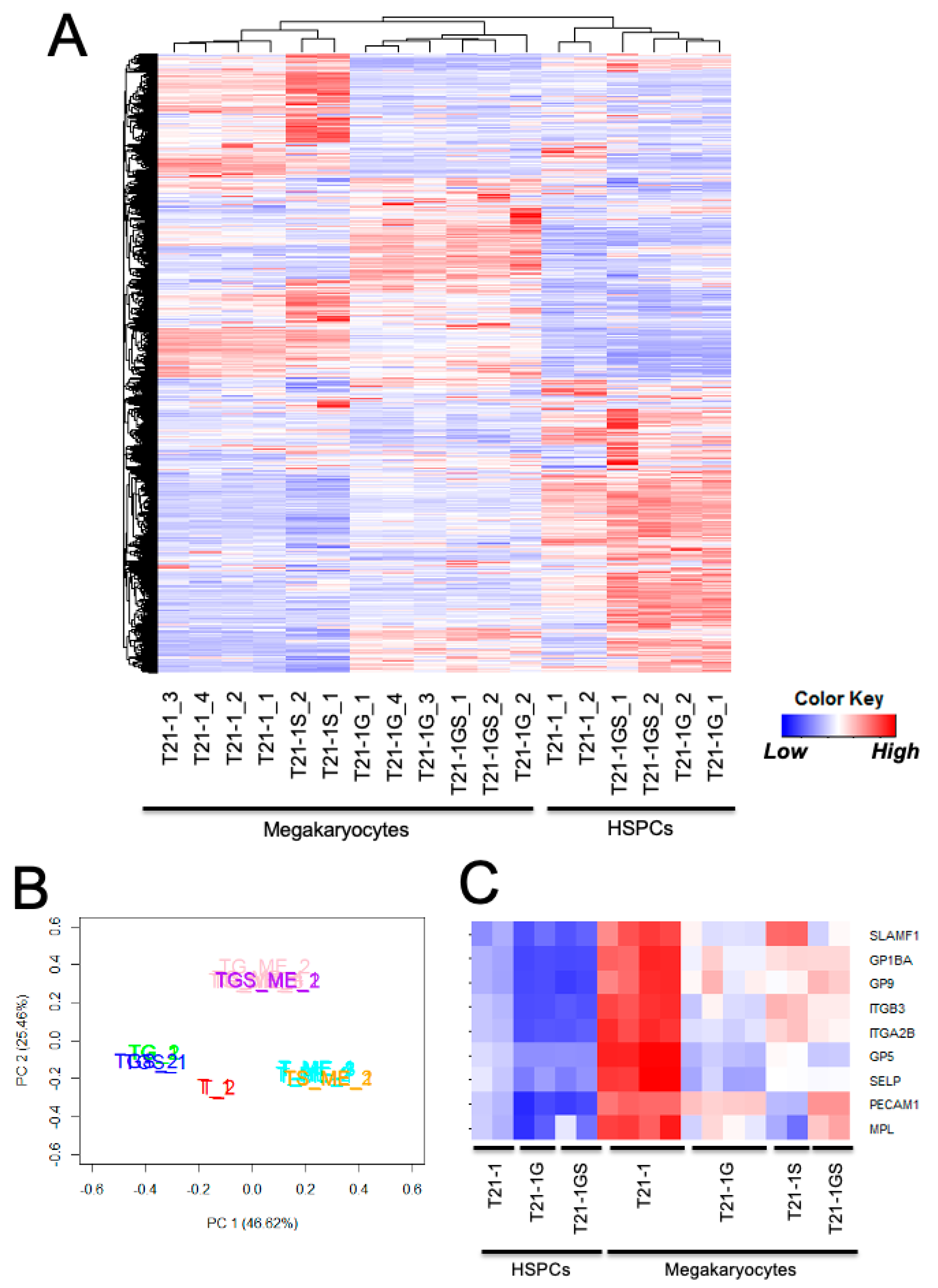{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. iPSC Lines and Culture
2.2. CRISPR Design and Cloning
2.3. Hematopoietic Differentiation and Lineage Expansion
2.4. Immunoblot Analysis
2.5. Flow Cytometry
2.6. Colony-Forming Unit Assay
2.7. RNA Sequencing and Data Analysis
2.8. Statistical Analysis
3. Results
3.1. GATA1 and STAG2 Knockout in Trisomic iPSCs by CRISPR/Cas9 Mutagenesis
3.2. Effect of STAG2 Knockout in the Presence or Absence of GATA1 Mutation on Erythroid Differentiation
3.3. Effect of STAG2 Knockout in the Presence or Absence of GATA1 Mutation on Megakaryoid Differentiation
3.4. Effect of STAG2 Knockout in the Presence or Absence of GATA1 Mutation on Myeloid Differentiation
3.5. Effect of GATA1 and STAG2 Mutation on Megakaryocyte Maturation
3.6. Effect of STAG2 and GATA1 Mutations on the Immunophenotype of Megakaryocyte Lineage Expanded Cell Population
3.7. GATA1 Mutation with or without STAG2 Knockout Altered Distinct Signaling Pathways
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Xavier, A.C.; Taub, J.W. Acute leukemia in children with Down syndrome. Haematologica 2010, 95, 1043–1045. [Google Scholar] [CrossRef] [PubMed]
- Saida, S. Evolution of myeloid leukemia in children with Down syndrome. Int. J. Hematol. 2016, 103, 365–372. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Vita, S.; Canzonetta, C.; Mulligan, C.; Delom, F.; Groet, J.; Baldo, C.; Vanes, L.; Dagna-Bricarelli, F.; Hoischen, A.; Veltman, J.; et al. Trisomic dose of several chromosome 21 genes perturbs haematopoietic stem and progenitor cell differentiation in Down’s syndrome. Oncogene 2010, 29, 6102–6114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Malinge, S.; Izraeli, S.; Crispino, J.D. Insights into the manifestations, outcomes, and mechanisms of leukemogenesis in Down syndrome. Blood 2009, 113, 2619–2628. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahmed, M.; Sternberg, A.; Hall, G.; Thomas, A.; Smith, O.; O’Marcaigh, A.; Wynn, R.; Stevens, R.; Addison, M.; King, D.; et al. Natural history of GATA1 mutations in Down syndrome. Blood 2004, 103, 2480–2489. [Google Scholar] [CrossRef]
- Crispino, J.D. GATA1 mutations in Down syndrome: Implications for biology and diagnosis of children with transient myeloproliferative disorder and acute megakaryoblastic leukemia. Pediatr. Blood Cancer 2005, 44, 40–44. [Google Scholar] [CrossRef]
- Greene, M.E.; Mundschau, G.; Wechsler, J.; McDevitt, M.; Gamis, A.; Karp, J.; Gurbuxani, S.; Arceci, R.; Crispino, J.D. Mutations in GATA1 in both transient myeloproliferative disorder and acute megakaryoblastic leukemia of Down syndrome. Blood Cells Mol. Dis. 2003, 31, 351–356. [Google Scholar] [CrossRef]
- Wechsler, J.; Greene, M.; McDevitt, M.A.; Anastasi, J.; Karp, J.E.; Le Beau, M.M.; Crispino, J.D. Acquired mutations in GATA1 in the megakaryoblastic leukemia of Down syndrome. Nat. Genet. 2002, 32, 148–152. [Google Scholar] [CrossRef]
- Kolb, E.A.; Meshinchi, S. Acute myeloid leukemia in children and adolescents: Identification of new molecular targets brings promise of new therapies. Hematol. Am. Soc. Hematol. Educ. Program 2015, 2015, 507–513. [Google Scholar] [CrossRef] [Green Version]
- Gruber, T.A.; Downing, J.R. The biology of pediatric acute megakaryoblastic leukemia. Blood 2015, 126, 943–949. [Google Scholar] [CrossRef] [Green Version]
- Nikolaev, S.I.; Santoni, F.; Vannier, A.; Falconnet, E.; Giarin, E.; Basso, G.; Hoischen, A.; Veltman, J.A.; Groet, J.; Nizetic, D.; et al. Exome sequencing identifies putative drivers of progression of transient myeloproliferative disorder to AMKL in infants with Down syndrome. Blood 2013, 122, 554–561. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoshida, K.; Toki, T.; Okuno, Y.; Kanezaki, R.; Shiraishi, Y.; Sato-Otsubo, A.; Sanada, M.; Park, M.J.; Terui, K.; Suzuki, H.; et al. The landscape of somatic mutations in Down syndrome-related myeloid disorders. Nat. Genet. 2013, 45, 1293–1299. [Google Scholar] [CrossRef] [PubMed]
- Solomon, D.A.; Kim, T.; Diaz-Martinez, L.A.; Fair, J.; Elkahloun, A.G.; Harris, B.T.; Toretsky, J.A.; Rosenberg, S.A.; Shukla, N.; Ladanyi, M.; et al. Mutational inactivation of STAG2 causes aneuploidy in human cancer. Science 2011, 333, 1039–1043. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Viny, A.D.; Levine, R.L. Cohesin mutations in myeloid malignancies made simple. Curr. Opin. Hematol. 2018, 25, 61–66. [Google Scholar] [CrossRef] [PubMed]
- Barwe, S.P.; Sidhu, I.; Kolb, E.A.; Gopalakrishnapillai, A. Modeling Transient Abnormal Myelopoiesis Using Induced Pluripotent Stem Cells and CRISPR/Cas9 Technology. Mol. Ther. Methods Clin. Dev. 2020, 19, 201–209. [Google Scholar] [CrossRef]
- Chen, C.; Jiang, P.; Xue, H.; Peterson, S.E.; Tran, H.T.; McCann, A.E.; Parast, M.M.; Li, S.; Pleasure, D.E.; Laurent, L.C.; et al. Role of astroglia in Down’s syndrome revealed by patient-derived human-induced pluripotent stem cells. Nat. Commun. 2014, 5, 4430. [Google Scholar] [CrossRef] [Green Version]
- Park, I.H.; Arora, N.; Huo, H.; Maherali, N.; Ahfeldt, T.; Shimamura, A.; Lensch, M.W.; Cowan, C.; Hochedlinger, K.; Daley, G.Q. Disease-specific induced pluripotent stem cells. Cell 2008, 134, 877–886. [Google Scholar] [CrossRef] [Green Version]
- Concordet, J.P.; Haeussler, M. CRISPOR: Intuitive guide selection for CRISPR/Cas9 genome editing experiments and screens. Nucleic Acids Res. 2018, 46, W242–W245. [Google Scholar] [CrossRef]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef]
- Liao, Y.; Smyth, G.K.; Shi, W. featureCounts: An efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 2014, 30, 923–930. [Google Scholar] [CrossRef] [Green Version]
- Risso, D.; Ngai, J.; Speed, T.P.; Dudoit, S. Normalization of RNA-seq data using factor analysis of control genes or samples. Nat. Biotechnol. 2014, 32, 896–902. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuleshov, M.V.; Jones, M.R.; Rouillard, A.D.; Fernandez, N.F.; Duan, Q.; Wang, Z.; Koplev, S.; Jenkins, S.L.; Jagodnik, K.M.; Lachmann, A.; et al. Enrichr: A comprehensive gene set enrichment analysis web server 2016 update. Nucleic Acids Res. 2016, 44, W90–W97. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.; Vasaikar, S.; Shi, Z.; Greer, M.; Zhang, B. WebGestalt 2017: A more comprehensive, powerful, flexible and interactive gene set enrichment analysis toolkit. Nucleic Acids Res. 2017, 45, W130–W137. [Google Scholar] [CrossRef] [PubMed]
- Banno, K.; Omori, S.; Hirata, K.; Nawa, N.; Nakagawa, N.; Nishimura, K.; Ohtaka, M.; Nakanishi, M.; Sakuma, T.; Yamamoto, T.; et al. Systematic Cellular Disease Models Reveal Synergistic Interaction of Trisomy 21 and GATA1 Mutations in Hematopoietic Abnormalities. Cell Rep. 2016, 15, 1228–1241. [Google Scholar] [CrossRef] [Green Version]
- Viny, A.D.; Bowman, R.L.; Liu, Y.; Lavallee, V.P.; Eisman, S.E.; Xiao, W.; Durham, B.H.; Navitski, A.; Park, J.; Braunstein, S.; et al. Cohesin Members Stag1 and Stag2 Display Distinct Roles in Chromatin Accessibility and Topological Control of HSC Self-Renewal and Differentiation. Cell Stem Cell 2019, 25, 682–696.e688. [Google Scholar] [CrossRef]
- Chou, S.T.; Opalinska, J.B.; Yao, Y.; Fernandes, M.A.; Kalota, A.; Brooks, J.S.; Choi, J.K.; Gewirtz, A.M.; Danet-Desnoyers, G.A.; Nemiroff, R.L.; et al. Trisomy 21 enhances human fetal erythro-megakaryocytic development. Blood 2008, 112, 4503–4506. [Google Scholar] [CrossRef]
- Katsumura, K.R.; Bresnick, E.H.; Group, G.F.M. The GATA factor revolution in hematology. Blood 2017, 129, 2092–2102. [Google Scholar] [CrossRef] [Green Version]
- Karandikar, N.J.; Aquino, D.B.; McKenna, R.W.; Kroft, S.H. Transient myeloproliferative disorder and acute myeloid leukemia in Down syndrome. An immunophenotypic analysis. Am. J. Clin. Pathol. 2001, 116, 204–210. [Google Scholar] [CrossRef] [Green Version]
- Yumura-Yagi, K.; Hara, J.; Kurahashi, H.; Nishiura, T.; Kaneyama, Y.; Osugi, Y.; Sakata, N.; Inoue, M.; Tawa, A.; Okada, S.; et al. Mixed phenotype of blasts in acute megakaryocytic leukaemia and transient abnormal myelopoiesis in Down’s syndrome. Br. J. Haematol. 1992, 81, 520–525. [Google Scholar] [CrossRef]
- Gadgeel, M.; AlQanber, B.; Buck, S.; Taub, J.W.; Ravindranath, Y.; Savasan, S. Aberrant myelomonocytic CD56 expression in Down syndrome is frequent and not associated with leukemogenesis. Ann. Hematol. 2021, 100, 1695–1700. [Google Scholar] [CrossRef] [PubMed]
- Langebrake, C.; Klusmann, J.H.; Wortmann, K.; Kolar, M.; Puhlmann, U.; Reinhardt, D. Concomitant aberrant overexpression of RUNX1 and NCAM in regenerating bone marrow of myeloid leukemia of Down’s syndrome. Haematologica 2006, 91, 1473–1480. [Google Scholar] [PubMed]
- Mullenders, J.; Aranda-Orgilles, B.; Lhoumaud, P.; Keller, M.; Pae, J.; Wang, K.; Kayembe, C.; Rocha, P.P.; Raviram, R.; Gong, Y.; et al. Cohesin loss alters adult hematopoietic stem cell homeostasis, leading to myeloproliferative neoplasms. J. Exp. Med. 2015, 212, 1833–1850. [Google Scholar] [CrossRef] [PubMed]
- Wagenblast, E.; Araujo, J.; Gan, O.I.; Cutting, S.K.; Murison, A.; Krivdova, G.; Azkanaz, M.; McLeod, J.L.; Smith, S.A.; Gratton, B.A.; et al. Mapping the cellular origin and early evolution of leukemia in Down syndrome. Science 2021, 373, eabf6202. [Google Scholar] [CrossRef] [PubMed]
- Greig, K.T.; Carotta, S.; Nutt, S.L. Critical roles for c-Myb in hematopoietic progenitor cells. Semin. Immunol. 2008, 20, 247–256. [Google Scholar] [CrossRef] [PubMed]
- Rehwinkel, J.; Gack, M.U. RIG-I-like receptors: Their regulation and roles in RNA sensing. Nat. Rev. Immunol. 2020, 20, 537–551. [Google Scholar] [CrossRef]
- Wan, Q.; Yang, C.; Rao, Y.; Liao, Z.; Su, J. MDA5 Induces a Stronger Interferon Response than RIG-I to GCRV Infection through a Mechanism Involving the Phosphorylation and Dimerization of IRF3 and IRF7 in CIK Cells. Front. Immunol. 2017, 8, 189. [Google Scholar] [CrossRef] [Green Version]
- Monlish, D.A.; Bhatt, S.T.; Schuettpelz, L.G. The Role of Toll-Like Receptors in Hematopoietic Malignancies. Front. Immunol. 2016, 7, 390. [Google Scholar] [CrossRef] [Green Version]
- Brenner, A.K.; Bruserud, Ø. Functional Toll-Like Receptors (TLRs) Are Expressed by a Majority of Primary Human Acute Myeloid Leukemia Cells and Inducibility of the TLR Signaling Pathway Is Associated with a More Favorable Phenotype. Cancers 2019, 11, 973. [Google Scholar] [CrossRef] [Green Version]
- Cuartero, S.; Innes, A.J.; Merkenschlager, M. Towards a Better Understanding of Cohesin Mutations in AML. Front. Oncol. 2019, 9, 867. [Google Scholar] [CrossRef] [Green Version]
- Orkin, S.H.; Shivdasani, R.A.; Fujiwara, Y.; McDevitt, M.A. Transcription factor GATA-1 in megakaryocyte development. Stem Cells 1998, 16 (Suppl. 2), 79–83. [Google Scholar] [CrossRef] [PubMed]
- Labuhn, M.; Perkins, K.; Matzk, S.; Varghese, L.; Garnett, C.; Papaemmanuil, E.; Metzner, M.; Kennedy, A.; Amstislavskiy, V.; Risch, T.; et al. Mechanisms of Progression of Myeloid Preleukemia to Transformed Myeloid Leukemia in Children with Down Syndrome. Cancer Cell 2019, 36, 123–138.e110. [Google Scholar] [CrossRef] [PubMed]
- Woo, A.J.; Wieland, K.; Huang, H.; Akie, T.E.; Piers, T.; Kim, J.; Cantor, A.B. Developmental differences in IFN signaling affect GATA1s-induced megakaryocyte hyperproliferation. J. Clin. Investig. 2013, 123, 3292–3304. [Google Scholar] [CrossRef] [PubMed]
- Mateos, M.K.; Barbaric, D.; Byatt, S.A.; Sutton, R.; Marshall, G.M. Down syndrome and leukemia: Insights into leukemogenesis and translational targets. Transl. Pediatr. 2015, 4, 76–92. [Google Scholar] [CrossRef]
- Sidhu, I.; Barwe, S.P.; Gopalakrishnapillai, A. The extracellular matrix: A key player in the pathogenesis of hematologic malignancies. Blood Rev. 2021, 48, 100787. [Google Scholar] [CrossRef]
- Sasca, D.; Szybinski, J.; Schüler, A.; Shah, V.; Heidelberger, J.; Haehnel, P.S.; Dolnik, A.; Kriege, O.; Fehr, E.M.; Gebhardt, W.H.; et al. NCAM1 (CD56) promotes leukemogenesis and confers drug resistance in AML. Blood 2019, 133, 2305–2319. [Google Scholar] [CrossRef]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Barwe, S.P.; Sebastian, A.; Sidhu, I.; Kolb, E.A.; Gopalakrishnapillai, A. Modeling Down Syndrome Myeloid Leukemia by Sequential Introduction of GATA1 and STAG2 Mutations in Induced Pluripotent Stem Cells with Trisomy 21. Cells 2022, 11, 628. https://doi.org/10.3390/cells11040628
Barwe SP, Sebastian A, Sidhu I, Kolb EA, Gopalakrishnapillai A. Modeling Down Syndrome Myeloid Leukemia by Sequential Introduction of GATA1 and STAG2 Mutations in Induced Pluripotent Stem Cells with Trisomy 21. Cells. 2022; 11(4):628. https://doi.org/10.3390/cells11040628
Chicago/Turabian StyleBarwe, Sonali P., Aimy Sebastian, Ishnoor Sidhu, Edward Anders Kolb, and Anilkumar Gopalakrishnapillai. 2022. "Modeling Down Syndrome Myeloid Leukemia by Sequential Introduction of GATA1 and STAG2 Mutations in Induced Pluripotent Stem Cells with Trisomy 21" Cells 11, no. 4: 628. https://doi.org/10.3390/cells11040628
APA StyleBarwe, S. P., Sebastian, A., Sidhu, I., Kolb, E. A., & Gopalakrishnapillai, A. (2022). Modeling Down Syndrome Myeloid Leukemia by Sequential Introduction of GATA1 and STAG2 Mutations in Induced Pluripotent Stem Cells with Trisomy 21. Cells, 11(4), 628. https://doi.org/10.3390/cells11040628