
miRNA-Dependent Regulation of AKT1 Phosphorylation

 , ,
, , 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Mammalian Cell Culture
2.2. Transfections
2.3. RT-qPCR
2.4. Protein Extraction
2.5. Western Blotting
2.6. Quantification and Statistical Analysis
3. Results
3.1. AKT1 Phosphorylation and let-7 Abundance in Epithelial Ovarian Cancer Cells
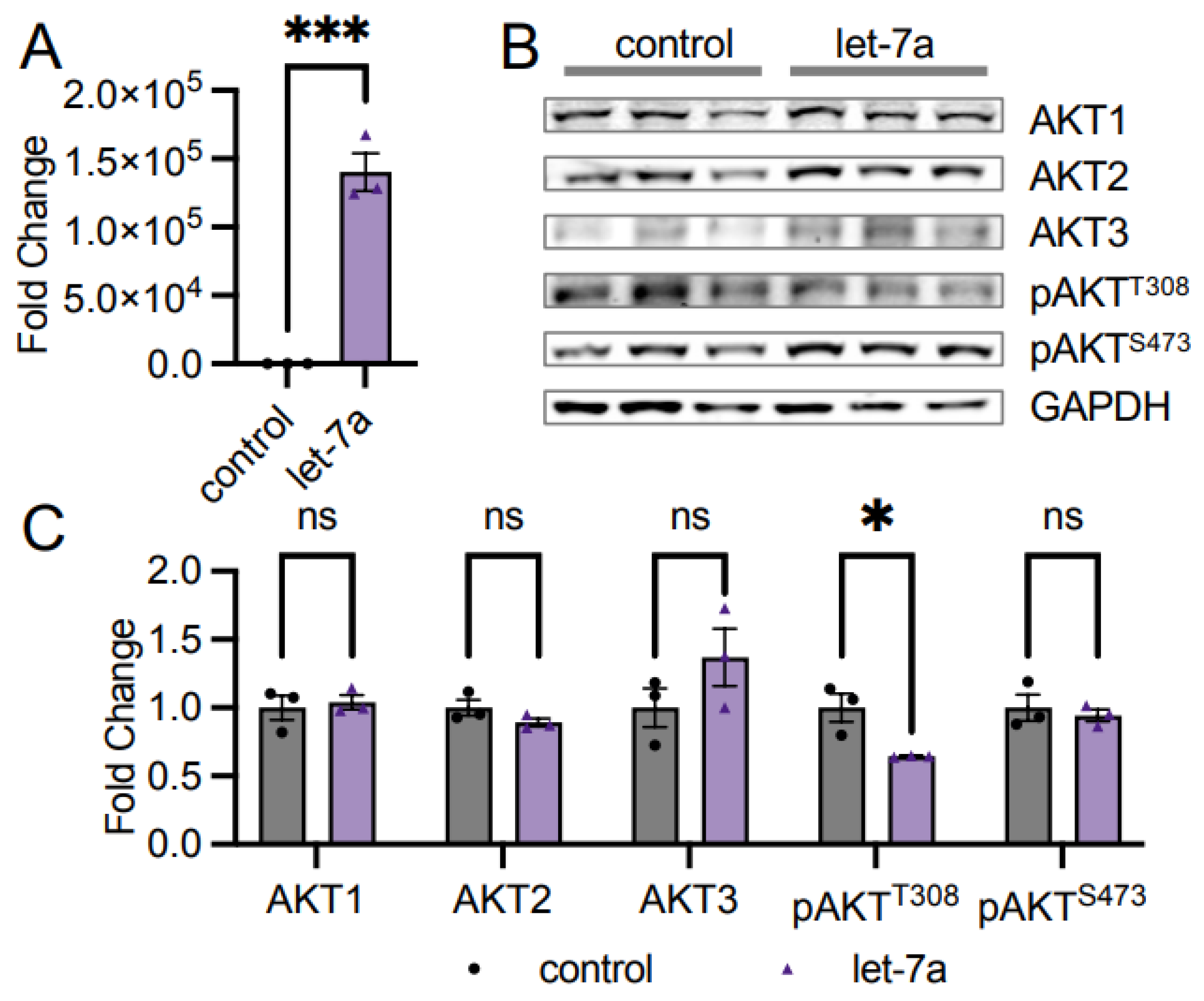
3.2. Let-7a miRNA Does Not Control AKT1 Abundance
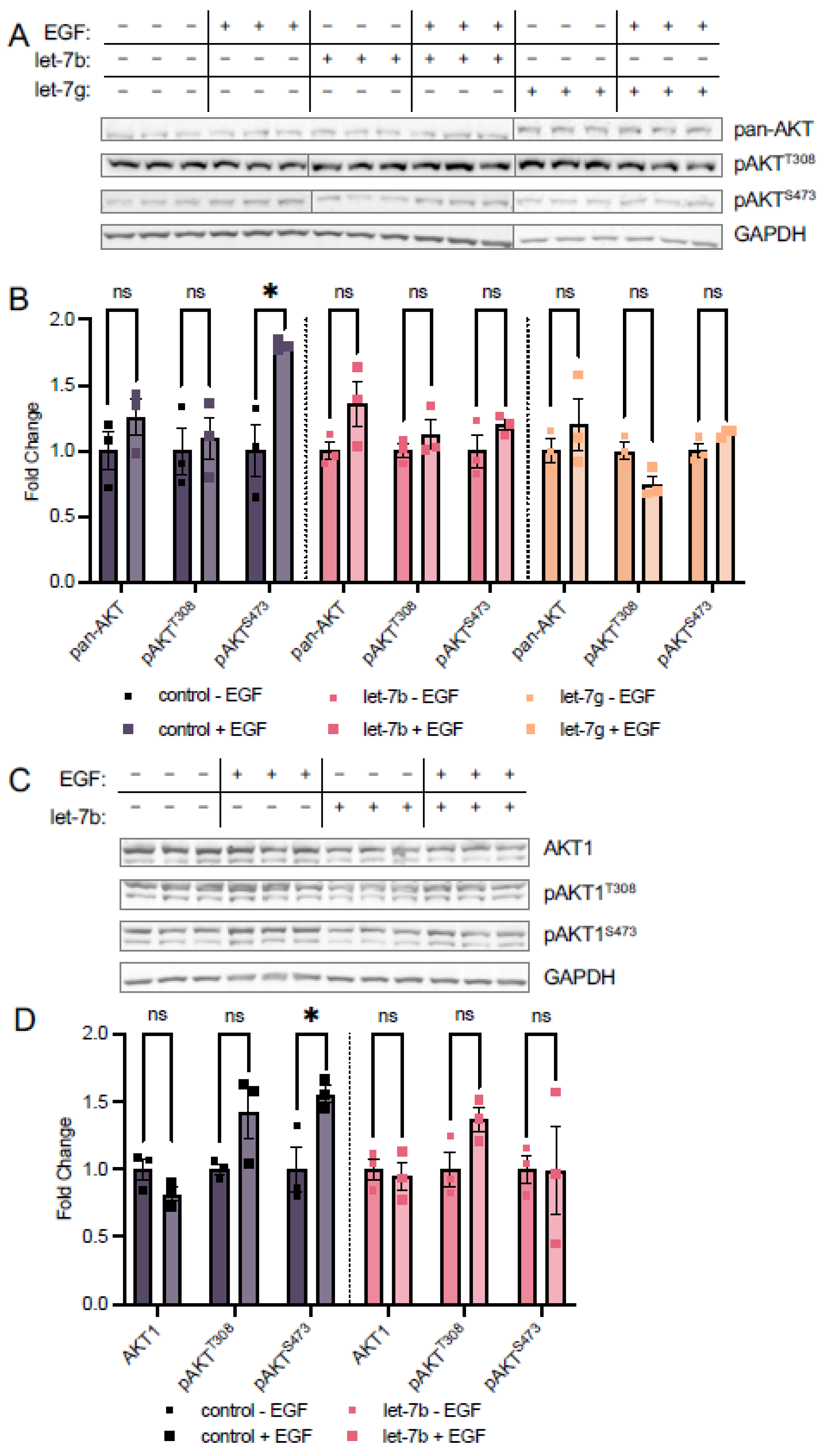
3.3. Let-7 miRNAs Regulate AKT Phosphorylation
3.3.1. Let-7a Increases AKT1 Phosphorylation at T308
3.3.2. Let-7a Regulation of T308 Causes Downstream Changes in AKT1 Signaling
3.4. Let-7b and Let-7g Also Counteract EGF Stimulation at S473
3.5. Regulation of AKT1 Phosphorylation
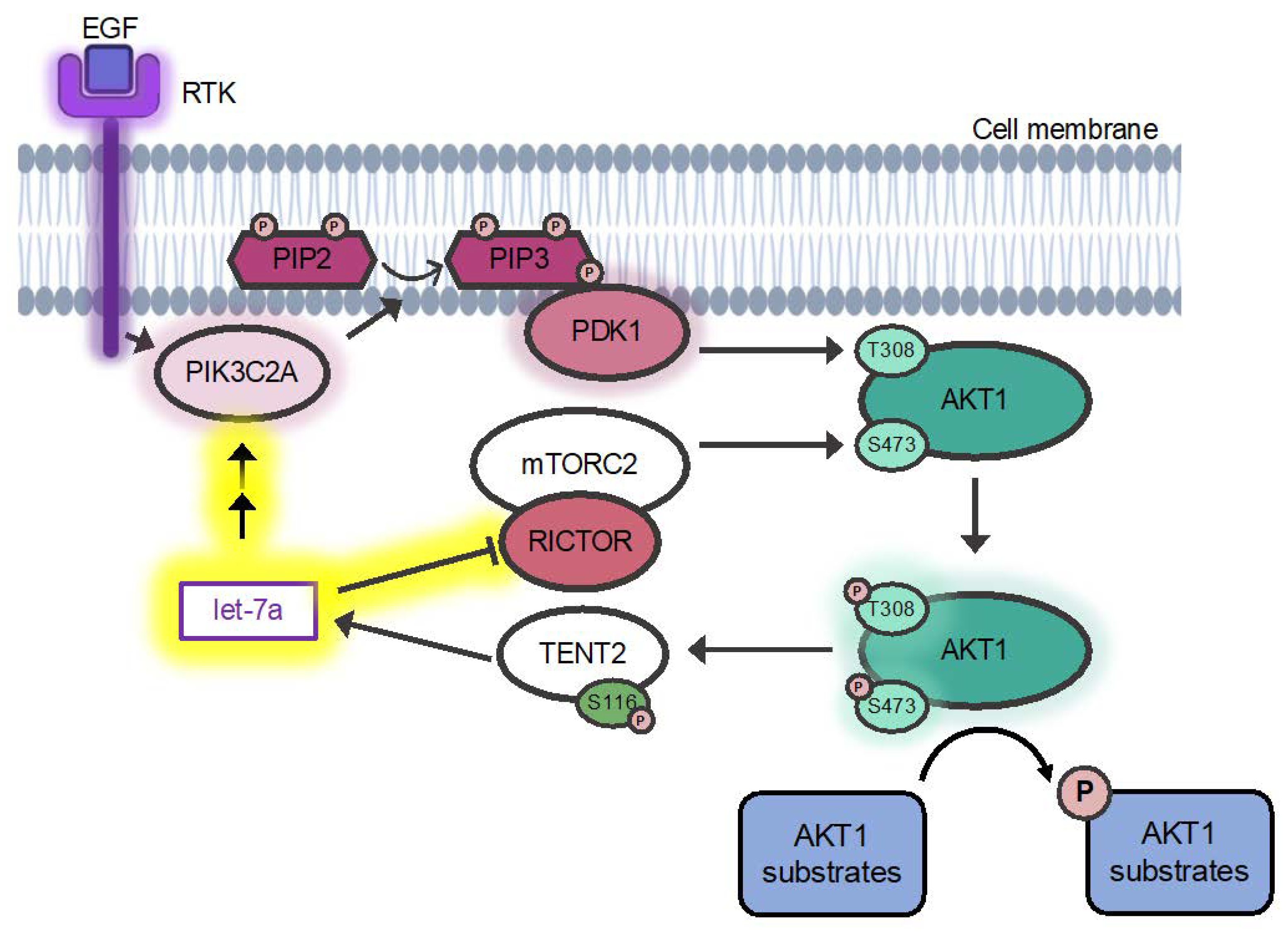
3.5.1. A Mechanism for Let-7a Regulation of AKT1 Phosphorylation at T308
3.5.2. Let-7 miRNAs Regulate AKT1 Phosphorylation at S473
4. Discussion
4.1. Let-7 miRNAs in Ovarian Cancer Cells
4.2. AKT Isoforms and Let-7a
4.3. Let-7 miRNAs Regulate AKT1 Ser473 Phosphorylation via RICTOR
4.4. Let-7a Regulates AKT1 T308 Phosphorylation via PDK1 and PIK3C2A
4.5. Expanding Connections between AKT1 and the Let-7 Family
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Karege, F.; Perroud, N.; Schurhoff, F.; Méary, A.; Marillier, G.; Burkhardt, S.; Ballmann, E.; Fernández, R.; Jamain, S.; Leboyer, M.; et al. Association ofAKT1gene variants and protein expression in both schizophrenia and bipolar disorder. Genes Brain Behav. 2010, 9, 503–511. [Google Scholar] [CrossRef] [PubMed]
- Correa, R.J.M.; Peart, T.; Valdes, Y.R.; DiMattia, G.E.; Shepherd, T.G. Modulation of AKT activity is associated with reversible dormancy in ascites-derived epithelial ovarian cancer spheroids. Carcinogenesis 2011, 33, 49–58. [Google Scholar] [CrossRef] [PubMed]
- Peart, T.M.; Correa, R.J.M.; Valdes, Y.R.; DiMattia, G.E.; Shepherd, T.G. BMP signalling controls the malignant potential of ascites-derived human epithelial ovarian cancer spheroids via AKT kinase activation. Clin. Exp. Metast. 2012, 29, 293–313. [Google Scholar] [CrossRef] [PubMed]
- Manning, B.D.; Toker, A. AKT/PKB Signaling: Navigating the Network. Cell 2017, 169, 381–405. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Linnerth-Petrik, N.M.; Santry, L.A.; Moorehead, R.; Jücker, M.; Wootton, S.K.; Petrik, J. Akt isoform specific effects in ovarian cancer progression. Oncotarget 2016, 7, 74820–74833. [Google Scholar] [CrossRef] [Green Version]
- Balasuriya, N.; Kunkel, M.T.; Liu, X.; Biggar, K.K.; Li, S.S.-C.; Newton, A.C.; O’Donoghue, P. Genetic code expansion and live cell imaging reveal that Thr-308 phosphorylation is irreplaceable and sufficient for Akt1 activity. J. Biol. Chem. 2018, 293, 10744–10756. [Google Scholar] [CrossRef] [Green Version]
- Balasuriya, N.; Davey, N.E.; Johnson, J.L.; Liu, H.; Biggar, K.K.; Cantley, L.C.; Li, S.S.-C.; O’Donoghue, P. Phosphorylation-dependent substrate selectivity of protein kinase B (AKT1). J. Biol. Chem. 2020, 295, 8120–8134. [Google Scholar] [CrossRef]
- Alessi, D.R.; James, S.R.; Downes, C.P.; Holmes, A.B.; Gaffney, P.R.; Reese, C.B.; Cohen, P. Characterization of a 3-phosphoinositide-dependent protein kinase which phosphorylates and activates protein kinase Balpha. Curr. Biol. 1997, 7, 261–269. [Google Scholar] [CrossRef] [Green Version]
- Sarbassov, D.D.; Guertin, D.A.; Ali, S.M.; Sabatini, D.M. Phosphorylation and regulation of Akt/PKB by the rictor-mTOR complex. Science 2005, 307, 1098–1101. [Google Scholar] [CrossRef] [Green Version]
- Gao, T.; Furnari, F.; Newton, A.C. PHLPP: A Phosphatase that Directly Dephosphorylates Akt, Promotes Apoptosis, and Suppresses Tumor Growth. Mol. Cell 2005, 18, 13–24. [Google Scholar] [CrossRef]
- Kuo, Y.-C.; Huang, K.-Y.; Yang, C.-H.; Yang, Y.-S.; Lee, W.-Y.; Chiang, C.-W. Regulation of Phosphorylation of Thr-308 of Akt, Cell Proliferation, and Survival by the B55α Regulatory Subunit Targeting of the Protein Phosphatase 2A Holoenzyme to Akt. J. Biol. Chem. 2008, 283, 1882–1892. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wei, W.-T.; Nian, X.-X.; Wang, S.-Y.; Jiao, H.-L.; Wang, Y.-X.; Xiao, Z.-Y.; Yang, R.-W.; Ding, Y.-Q.; Ye, Y.-P.; Liao, W.-T. miR-422a inhibits cell proliferation in colorectal cancer by targeting AKT1 and MAPK1. Cancer Cell Int. 2017, 17, 91. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Que, T.; Song, Y.; Liu, Z.; Zheng, S.; Long, H.; Li, Z.; Liu, Y.; Wang, G.; Zhou, J.; Zhang, X.; et al. Decreased miRNA-637 is an unfavorable prognosis marker and promotes glioma cell growth, migration and invasion via direct targeting Akt1. Oncogene 2015, 34, 4952–4963. [Google Scholar] [CrossRef] [PubMed]
- Fan, J.; Li, S.; Wang, D. MicroRNA-149 suppresses osteogenic differentiation of mesenchymal stem cells via inhibition of AKT1-dependent Twist1 phosphorylation. Cell Death Discov. 2022, 8, 2. [Google Scholar] [CrossRef]
- Wang, X.; Cao, L.; Wang, Y.; Wang, X.; Liu, N.; You, Y. Regulation of let-7 and its target oncogenes (Review). Oncol. Lett. 2012, 3, 955–960. [Google Scholar] [CrossRef]
- Akao, Y.; Nakagawa, Y.; Naoe, T. let-7 MicroRNA Functions as a Potential Growth Suppressor in Human Colon Cancer Cells. Biol. Pharm. Bull. 2006, 29, 903–906. [Google Scholar] [CrossRef] [Green Version]
- Barh, D.; Malhotra, R.; Ravi, B.; Sindhurani, P. Microrna Let-7: An Emerging Next-Generation Cancer Therapeutic. Curr. Oncol. 2010, 17, 70–80. [Google Scholar] [CrossRef] [Green Version]
- Johnson, S.M.; Grosshans, H.; Shingara, J.; Byrom, M.; Jarvis, R.; Cheng, A.; Labourier, E.; Reinert, K.L.; Brown, D.; Slack, F.J. RAS Is Regulated by the let-7 MicroRNA Family. Cell 2005, 120, 635–647. [Google Scholar] [CrossRef] [Green Version]
- Molenaar, J.J.; Domingo-Fernández, R.; Ebus, M.E.; Lindner, S.; Koster, J.; Drabek, K.; Mestdagh, P.; Van Sluis, P.; Valentijn, L.J.; van Nes, J.; et al. LIN28B induces neuroblastoma and enhances MYCN levels via let-7 suppression. Nat. Genet. 2012, 44, 1199–1206. [Google Scholar] [CrossRef]
- Sampson, V.B.; Rong, N.H.; Han, J.; Yang, Q.; Aris, V.; Soteropoulos, P.; Petrelli, N.J.; Dunn, S.P.; Krueger, L.J. MicroRNA Let-7a Down-regulates MYC and Reverts MYC-Induced Growth in Burkitt Lymphoma Cells. Cancer Res. 2007, 67, 9762–9770. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.-H.; Wang, X.-J.; Li, G.-X.; Yang, E.; Yang, N.-M. Detection of let-7a microRNA by real-time PCR in gastric carcinoma. World J. Gastroenterol. 2007, 13, 2883–2888. [Google Scholar] [CrossRef] [PubMed]
- Turk, M.A.; Chung, C.; Manni, E.; Zukowski, S.A.; Engineer, A.; Badakhshi, Y.; Bi, Y.; Heinemann, I.U. MiRAR—miRNA Activity Reporter for Living Cells. Genes 2018, 9, 305. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chung, C.; Jaramillo, J.E.; Ellis, M.J.; Bour, D.Y.N.; Seidl, L.E.; Jo, D.H.S.; Turk, M.A.; Mann, M.R.; Bi, Y.; Haniford, D.B.; et al. RNA surveillance by uridylation-dependent RNA decay inSchizosaccharomyces pombe. Nucleic Acids Res. 2019, 47, 3045–3057. [Google Scholar] [CrossRef]
- Fan, H.; Jiang, M.; Li, B.; He, Y.; Huang, C.; Luo, D.; Xu, H.; Yang, L.; Zhou, J. MicroRNA-let-7a regulates cell autophagy by targeting Rictor in gastric cancer cell lines MGC-803 and SGC-7901. Oncol. Rep. 2018, 39, 1207–1214. [Google Scholar] [CrossRef] [Green Version]
- Zhou, B.; Shan, H.; Su, Y.; Xia, K.; Zou, R.; Shao, Q. Let-7a inhibits migration, invasion and tumor growth by targeting AKT2 in papillary thyroid carcinoma. Oncotarget 2017, 8, 69746–69755. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Y.; Wang, X. miRDB: An online database for prediction of functional microRNA targets. Nucleic Acids Res. 2020, 48, D127–D131. [Google Scholar] [CrossRef] [Green Version]
- Liu, W.; Wang, X. Prediction of functional microRNA targets by integrative modeling of microRNA binding and target expression data. Genome Biol. 2019, 20, 18. [Google Scholar] [CrossRef]
- Kiyatkin, A.; Aksamitiene, E.; Markevich, N.I.; Borisov, N.M.; Hoek, J.B.; Kholodenko, B.N. Scaffolding protein Grb2-associated binder 1 sustains epidermal growth factor-induced mitogenic and survival signaling by multiple positive feedback loops. J. Biol. Chem. 2006, 281, 19925–19938. [Google Scholar] [CrossRef] [Green Version]
- McKenna, M.; Balasuriya, N.; Zhong, S.; Li, S.S.-C.; O’Donoghue, P. Phospho-Form Specific Substrates of Protein Kinase B (AKT1). Front. Bioeng. Biotechnol. 2021, 8, 619252. [Google Scholar] [CrossRef]
- Hart, J.R.; Vogt, P.K. Phosphorylation of AKT: A Mutational Analysis. Oncotarget 2011, 2, 467–476. [Google Scholar] [CrossRef] [Green Version]
- Altomare, D.A.; Testa, J.R. Perturbations of the AKT signaling pathway in human cancer. Oncogene 2005, 24, 7455–7464. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dai, D.L.; Martinka, M.; Li, G. Prognostic Significance of Activated Akt Expression in Melanoma: A Clinicopathologic Study of 292 Cases. J. Clin. Oncol. 2005, 23, 1473–1482. [Google Scholar] [CrossRef] [PubMed]
- Freudlsperger, C.; Horn, D.; Weißfuß, S.; Weichert, W.; Weber, K.J.; Saure, D.; Sharma, S.; Dyckhoff, G.; Grabe, N.; Plinkert, P. Phosphorylation of AKT (S er473) serves as an independent prognostic marker for radiosensitivity in advanced head and neck squamous cell carcinoma. Int. J. Cancer 2015, 136, 2775–2785. [Google Scholar] [CrossRef] [PubMed]
- Tsao, A.S.; McDonnell, T.; Lam, S.; Putnam, J.B.; Bekele, N.; Hong, W.K.; Kurie, J.M. Increased phospho-AKT (Ser473) expression in bronchial dysplasia: Implications for lung cancer prevention studies. Cancer Epidemiol. Biomark. Prev. 2003, 12, 660–664. [Google Scholar]
- Kearney, A.L.; Cooke, K.C.; Norris, D.M.; Zadoorian, A.; Krycer, J.R.; Fazakerley, D.J.; Burchfield, J.G.; James, D.E. Serine 474 phosphorylation is essential for maximal Akt2 kinase activity in adipocytes. J. Biol. Chem. 2019, 294, 16729–16739. [Google Scholar] [CrossRef]
- Jacinto, E.; Facchinetti, V.; Liu, D.; Soto, N.; Wei, S.; Jung, S.Y.; Huang, Q.; Qin, J.; Su, B. SIN1/MIP1 Maintains rictor-mTOR Complex Integrity and Regulates Akt Phosphorylation and Substrate Specificity. Cell 2006, 127, 125–137. [Google Scholar] [CrossRef] [Green Version]
- Yang, J.; Cron, P.; Thompson, V.; Good, V.M.; Hess, D.; Hemmings, B.A.; Barford, D. Molecular Mechanism for the Regulation of Protein Kinase B/Akt by Hydrophobic Motif Phosphorylation. Mol. Cell 2002, 9, 1227–1240. [Google Scholar] [CrossRef]
- Correa, R.J.; Valdes, Y.R.; Peart, T.M.; Fazio, E.N.; Bertrand, M.; McGee, J.; Préfontaine, M.; Sugimoto, A.; DiMattia, G.E.; Shepherd, T.G. Combination of AKT inhibition with autophagy blockade effectively reduces ascites-derived ovarian cancer cell viability. Carcinogenesis 2014, 35, 1951–1961. [Google Scholar] [CrossRef] [Green Version]
- Correa, R.J.M.; Valdes, Y.R.; Shepherd, T.G.; DiMattia, G.E. Beclin-1 expression is retained in high-grade serous ovarian cancer yet is not essential for autophagy induction in vitro. J. Ovarian Res. 2015, 8, 52. [Google Scholar] [CrossRef] [Green Version]
- Kolendowski, B.; Valdes, Y.; Hirte, H.; Itamochi, H.; Lee, W.; Carey, M.; Shepherd, T.; DiMattia, G. Characterization of Mutational Status, Spheroid Formation, and Drug Response of a New Genomically-Stable Human Ovarian Clear Cell Carcinoma Cell Line, 105C. Cells 2020, 9, 2408. [Google Scholar] [CrossRef]
- Chirshev, E.; Oberg, K.; Ioffe, Y.J.; Unternaehrer, J.J. Let-7as biomarker, prognostic indicator, and therapy for precision medicine in cancer. Clin. Transl. Med. 2019, 8, 24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiao, M.; Cai, J.; Cai, L.; Jia, J.; Xie, L.; Zhu, Y.; Huang, B.; Jin, D.; Wang, Z. Let-7e sensitizes epithelial ovarian cancer to cisplatin through repressing DNA double strand break repair. J. Ovarian Res. 2017, 10, 24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Biamonte, F.; Santamaria, G.; Sacco, A.; Perrone, F.M.; Di Cello, A.; Battaglia, A.M.; Salatino, A.; Di Vito, A.; Aversa, I.; Venturella, R.; et al. MicroRNA let-7g acts as tumor suppressor and predictive biomarker for chemoresistance in human epithelial ovarian cancer. Sci. Rep. 2019, 9, 5668. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dai, X.; Jiang, Y.; Tan, C. Let-7 Sensitizes KRAS Mutant Tumor Cells to Chemotherapy. PLoS ONE 2015, 10, e0126653. [Google Scholar] [CrossRef] [Green Version]
- Schulman, B.R.M.; Liang, X.; Stahlhut, C.; Delconte, C.; Stefani, G.; Slack, F.J. The let-7 microRNA target gene, Mlin41/Trim71 is required for mouse embryonic survival and neural tube closure. Cell Cycle 2008, 7, 3935–3942. [Google Scholar] [CrossRef]
- Hagan, J.; Piskounova, E.; Gregory, R.I. Lin28 recruits the TUTase Zcchc11 to inhibit let-7 maturation in mouse embryonic stem cells. Nat. Struct. Mol. Biol. 2009, 16, 1021–1025. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lippa, B.; Pan, G.; Corbett, M.; Li, C.; Kauffman, G.S.; Pandit, J.; Robinson, S.; Wei, L.; Kozina, E.; Marr, E.S.; et al. Synthesis and structure based optimization of novel Akt inhibitors. Bioorganic Med. Chem. Lett. 2008, 18, 3359–3363. [Google Scholar] [CrossRef]
- Alessi, D.R.; Andjelkovic, M.; Caudwell, B.; Cron, P.; Morrice, N.; Cohen, P.; Hemmings, B.A. Mechanism of activation of protein kinase B by insulin and IGF-1. EMBO J. 1996, 15, 6541–6551. [Google Scholar] [CrossRef] [Green Version]
- Zinzalla, V.; Stracka, D.; Oppliger, W.; Hall, M.N. Activation of mTORC2 by Association with the Ribosome. Cell 2011, 144, 757–768. [Google Scholar] [CrossRef] [Green Version]
- Betz, C.; Stracka, D.; Prescianotto-Baschong, C.; Frieden, M.; Demaurex, N.; Hall, M.N. mTOR complex 2-Akt signaling at mitochondria-associated endoplasmic reticulum membranes (MAM) regulates mitochondrial physiology. Proc. Natl. Acad. Sci. USA 2013, 110, 12526–12534. [Google Scholar] [CrossRef] [Green Version]
- Fu, W.; Hall, M.N. Regulation of mTORC2 Signaling. Genes 2020, 11, 1045. [Google Scholar] [CrossRef] [PubMed]
- Margaria, J.P.; Ratto, E.; Gozzelino, L.; Li, H.; Hirsch, E. Class II PI3Ks at the Intersection between Signal Transduction and Membrane Trafficking. Biomolecules 2019, 9, 104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hamatani, T.; Daikoku, T.; Wang, H.; Matsumoto, H.; Carter, M.G.; Ko, M.S.H.; Dey, S.K. Global gene expression analysis identifies molecular pathways distinguishing blastocyst dormancy and activation. Proc. Natl. Acad. Sci. USA 2004, 101, 10326–10331. [Google Scholar] [CrossRef] [Green Version]
- Chao, L.-M.; Sun, W.; Chen, H.; Liu, B.-Y.; Li, P.-F.; Zhao, D.-W. MicroRNA-31 inhibits osteosarcoma cell proliferation, migration and invasion by targeting PIK3C2A. Eur. Rev. Med. Pharmacol. Sci. 2018, 22, 7205–7213. [Google Scholar] [PubMed]
- Shi, Y.; Gao, X.; Hu, Q.; Li, X.; Xu, J.; Lu, S.; Liu, Y.; Xu, C.; Jiang, D.; Lin, J.; et al. PIK3C2A is a gene-specific target of microRNA-518a-5p in imatinib mesylate-resistant gastrointestinal stromal tumor. Lab. Investig. 2016, 96, 652–660. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Emmanouilidi, A.; Falasca, M. Targeting PDK1 for Chemosensitization of Cancer Cells. Cancers 2017, 9, 140. [Google Scholar] [CrossRef]
- Zabkiewicz, J.; Pearn, L.; Hills, R.; Morgan, R.; Tonks, A.; Burnett, A.K.; Darley, R.L. The PDK1 master kinase is over-expressed in acute myeloid leukemia and promotes PKC-mediated survival of leukemic blasts. Haematologica 2013, 99, 858–864. [Google Scholar] [CrossRef] [Green Version]
- Zheng, N.; Ding, X.; Sun, A.; Jahan, R. PDK1 Activity Regulates Proliferation, Invasion and Growth of Hemangiomas. Cell. Physiol. Biochem. 2015, 36, 1903–1910. [Google Scholar] [CrossRef]
- Lin, J.-X.; Xie, X.-S.; Weng, X.-F.; Qiu, S.-L.; Yoon, C.; Lian, N.-Z.; Xie, J.-W.; Wang, J.-B.; Lu, J.; Chen, Q.-Y.; et al. UFM1 suppresses invasive activities of gastric cancer cells by attenuating the expression of PDK1 through PI3K/AKT signaling. J. Exp. Clin. Cancer Res. 2019, 38, 410. [Google Scholar] [CrossRef]
- Chung, C.; Balasuriya, N.; Manni, E.; Liu, X.; Li, S.S.-C.; O’Donoghue, P.; Heinemann, I.U. Gld2 activity is regulated by phosphorylation in the N-terminal domain. RNA Biol. 2019, 16, 1022–1033. [Google Scholar] [CrossRef]
- Chung, C.Z.; Balasuriya, N.; Siddika, T.; Frederick, M.I.; Heinemann, I.U. Gld2 activity and RNA specificity is dynamically regulated by phosphorylation and interaction with QKI-7. RNA Biol. 2021, 18, 397–408. [Google Scholar] [CrossRef] [PubMed]
- Frederick, M.I.; Heinemann, I.U. Regulation of RNA stability at the 3′ end. Biol. Chem. 2021, 402, 425–431. [Google Scholar] [CrossRef] [PubMed]
- Chung, C.Z.; Jo, D.H.S.; Heinemann, I.U. Nucleotide specificity of the human terminal nucleotidyltransferase Gld2 (TUT2). RNA 2016, 22, 1239–1249. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, H.; Shyh-Chang, N.; Segrè, A.V.; Shinoda, G.; Shah, S.P.; Einhorn, W.S.; Takeuchi, A.; Engreitz, J.M.; Hagan, J.; Kharas, M.G.; et al. The Lin28/let-7 Axis Regulates Glucose Metabolism. Cell 2011, 147, 81–94. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roush, S.; Slack, F.J. The let-7 family of microRNAs. Trends Cell Biol. 2008, 18, 505–516. [Google Scholar] [CrossRef] [PubMed]
- Kumar, M.; Ahmad, T.; Sharma, A.; Mabalirajan, U.; Kulshreshtha, A.; Agrawal, A.; Ghosh, B. Let-7 microRNA–mediated regulation of IL-13 and allergic airway inflammation. J. Allergy Clin. Immunol. 2011, 128, 1077–1085.e10. [Google Scholar] [CrossRef] [PubMed]
- Kumar, M.; Sahu, S.K.; Kumar, R.; Subuddhi, A.; Maji, R.K.; Jana, K.; Gupta, P.; Raffetseder, J.; Lerm, M.; Ghosh, Z.; et al. MicroRNA let-7 Modulates the Immune Response to Mycobacterium tuberculosis Infection via Control of A20, an Inhibitor of the NF-κB Pathway. Cell Host Microbe 2015, 17, 345–356. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Naderi, M.; Abdul Tehrani, H.; Soleimani, M.; Shabani, I.; Hashemi, S.M. A home-brew real-time PCR assay for reliable detection and quantification of mature miR-122. Appl. Immunohistochem. Mol. Morphol. 2015, 23, 601–606. [Google Scholar] [CrossRef]
- Boukhalfa, A.; Nascimbeni, A.C.; Ramel, D.; Dupont, N.; Hirsch, E.; Gayral, S.; Laffargue, M.; Codogno, P.; Morel, E. PI3KC2alpha-dependent and VPS34-independent generation of PI3P controls primary cilium-mediated autophagy in response to shear stress. Nat. Commun. 2020, 11, 1–9. [Google Scholar] [CrossRef]
- Barber, R.D.; Harmer, D.W.; Coleman, R.A.; Clark, B.J. GAPDH as a housekeeping gene: Analysis of GAPDH mRNA expression in a panel of 72 human tissues. Physiol. Genom. 2005, 21, 389–395. [Google Scholar] [CrossRef] [Green Version]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Frederick, M.I.; Siddika, T.; Zhang, P.; Balasuriya, N.; Turk, M.A.; O’Donoghue, P.; Heinemann, I.U. miRNA-Dependent Regulation of AKT1 Phosphorylation. Cells 2022, 11, 821. https://doi.org/10.3390/cells11050821
Frederick MI, Siddika T, Zhang P, Balasuriya N, Turk MA, O’Donoghue P, Heinemann IU. miRNA-Dependent Regulation of AKT1 Phosphorylation. Cells. 2022; 11(5):821. https://doi.org/10.3390/cells11050821
Chicago/Turabian StyleFrederick, Mallory I., Tarana Siddika, Pengcheng Zhang, Nileeka Balasuriya, Matthew A. Turk, Patrick O’Donoghue, and Ilka U. Heinemann. 2022. "miRNA-Dependent Regulation of AKT1 Phosphorylation" Cells 11, no. 5: 821. https://doi.org/10.3390/cells11050821
APA StyleFrederick, M. I., Siddika, T., Zhang, P., Balasuriya, N., Turk, M. A., O’Donoghue, P., & Heinemann, I. U. (2022). miRNA-Dependent Regulation of AKT1 Phosphorylation. Cells, 11(5), 821. https://doi.org/10.3390/cells11050821