
What’s in a Gene? The Outstanding Diversity of MAPT

 ,
,  , and
, and 
Abstract
:
{kind=link}
{kind=link}
{kind=link}
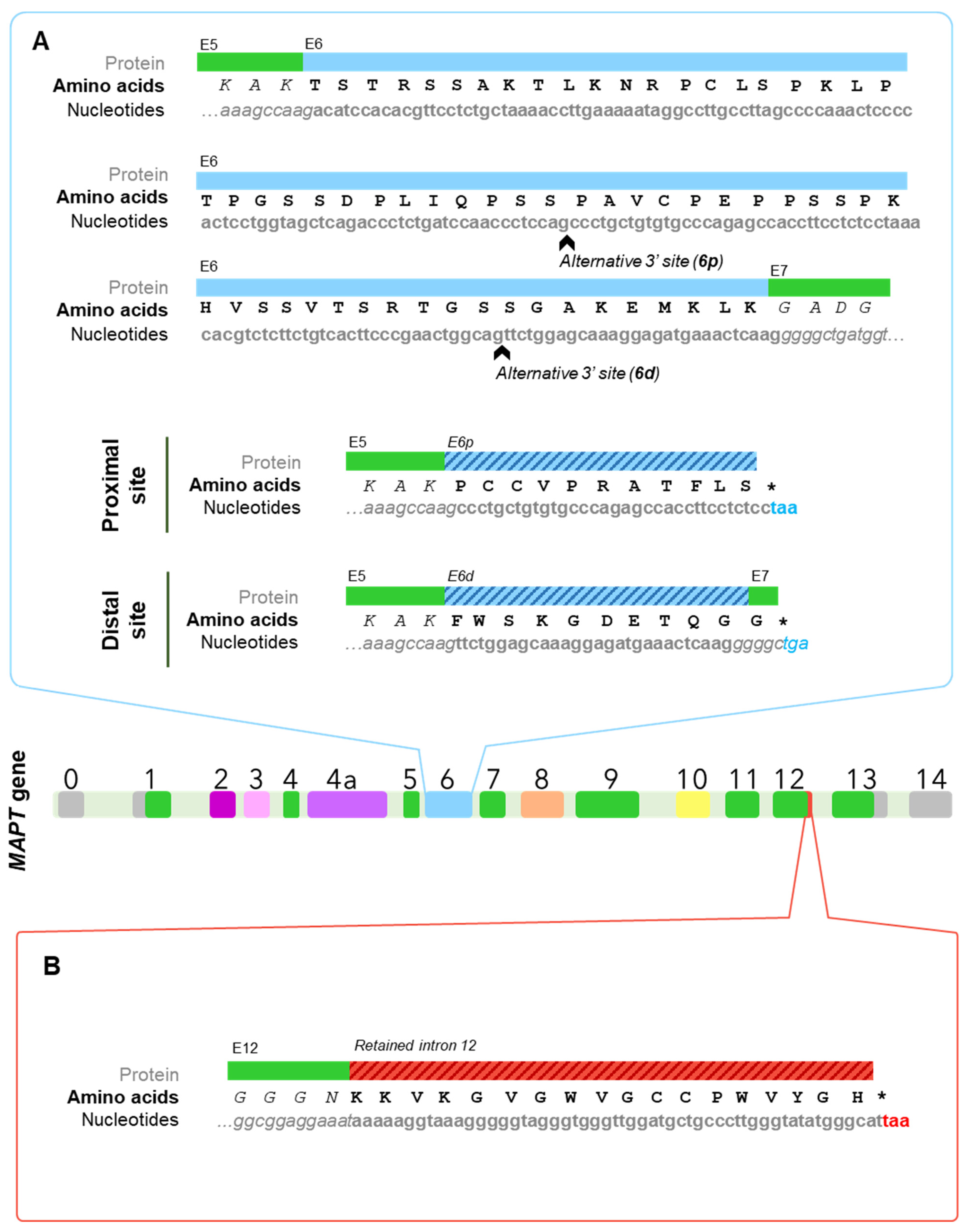{kind=link}
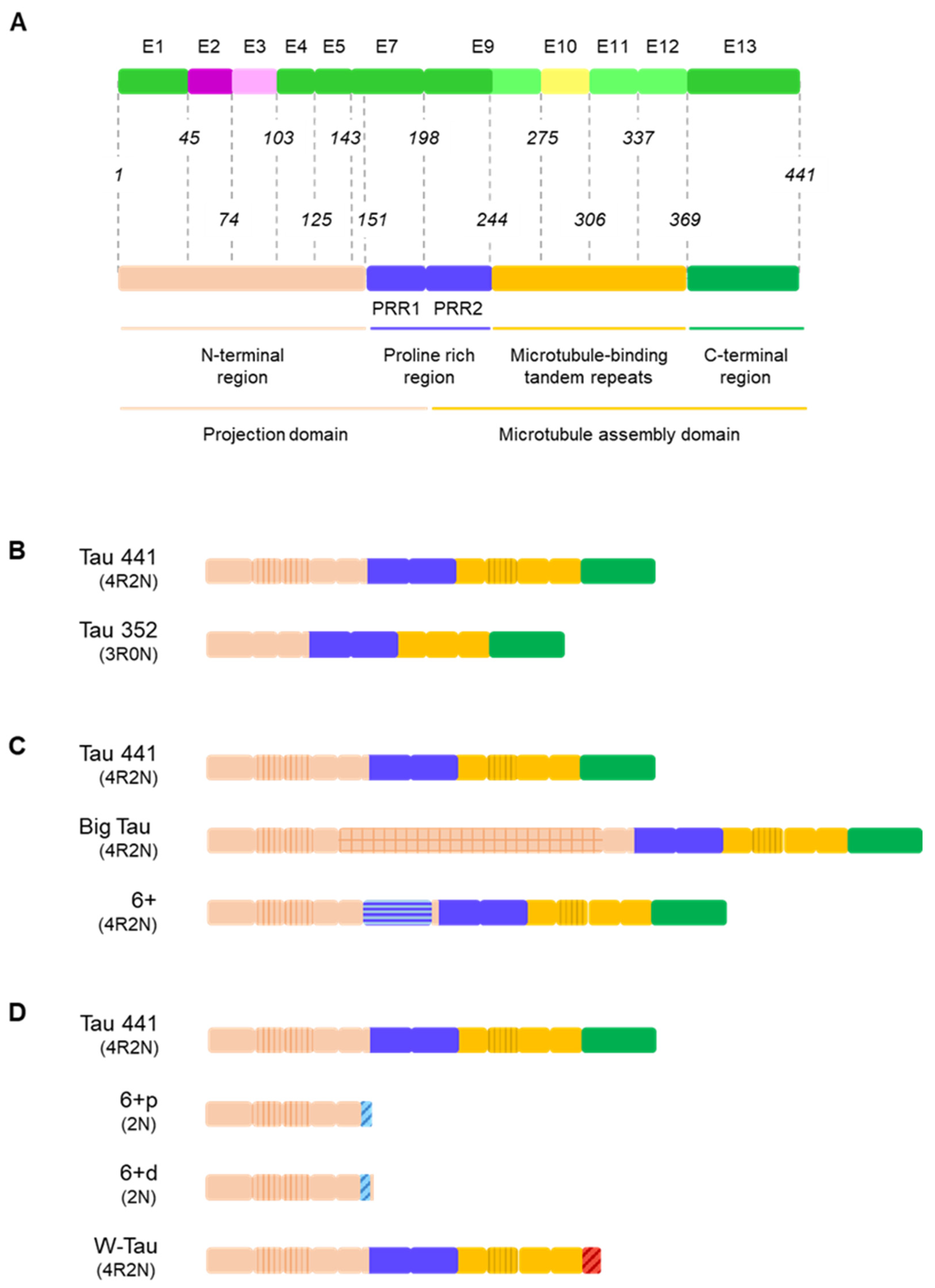{kind=link}
1. Introduction
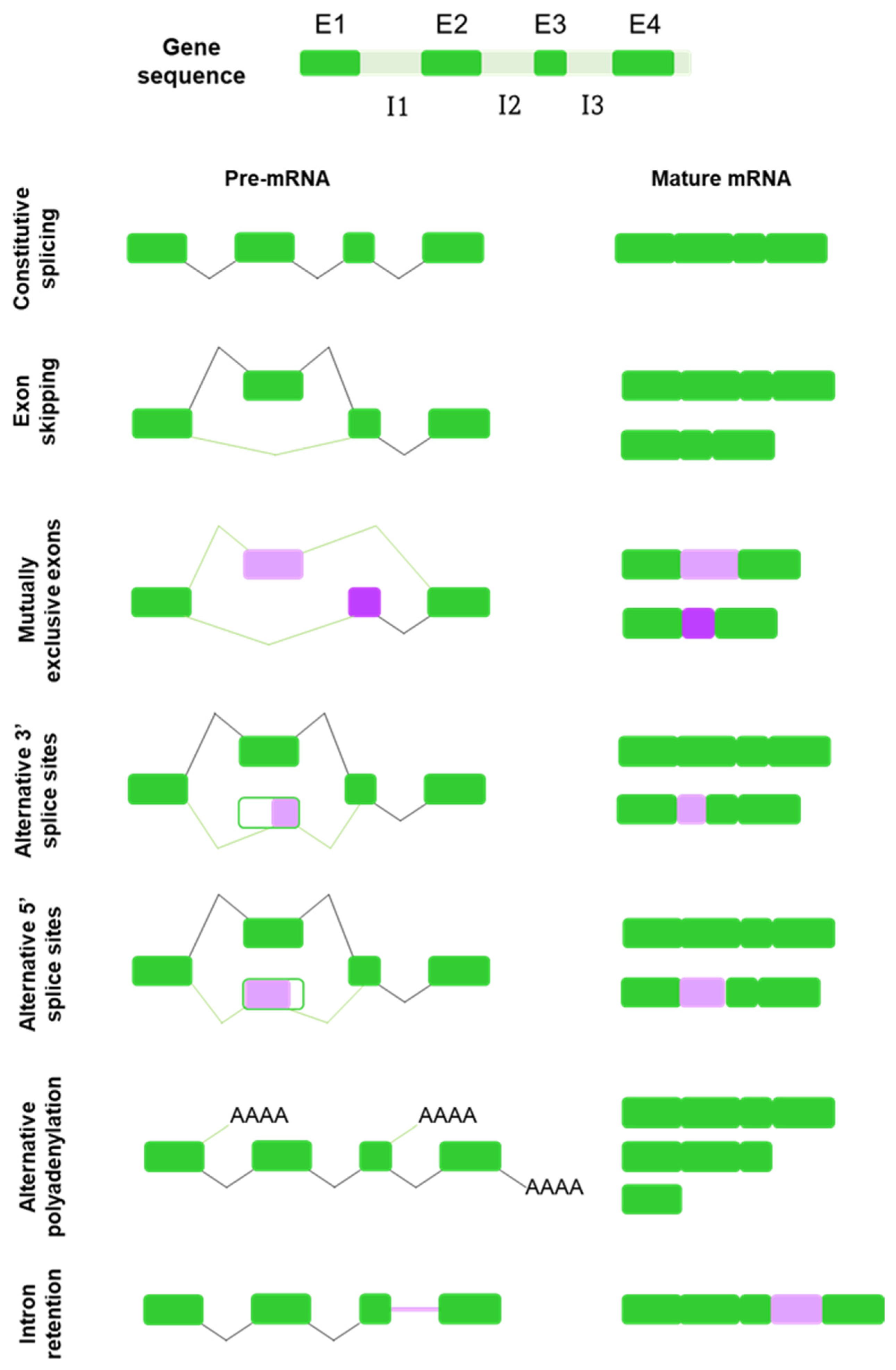
2. Alternative Splicing: A Force of Diversity
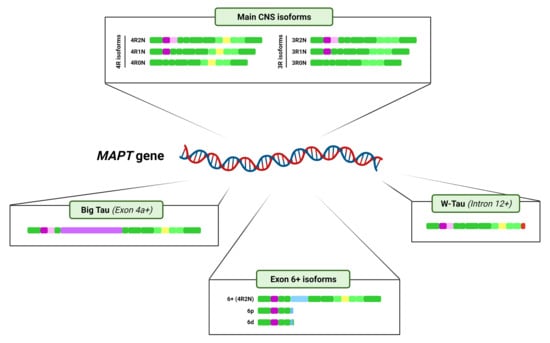
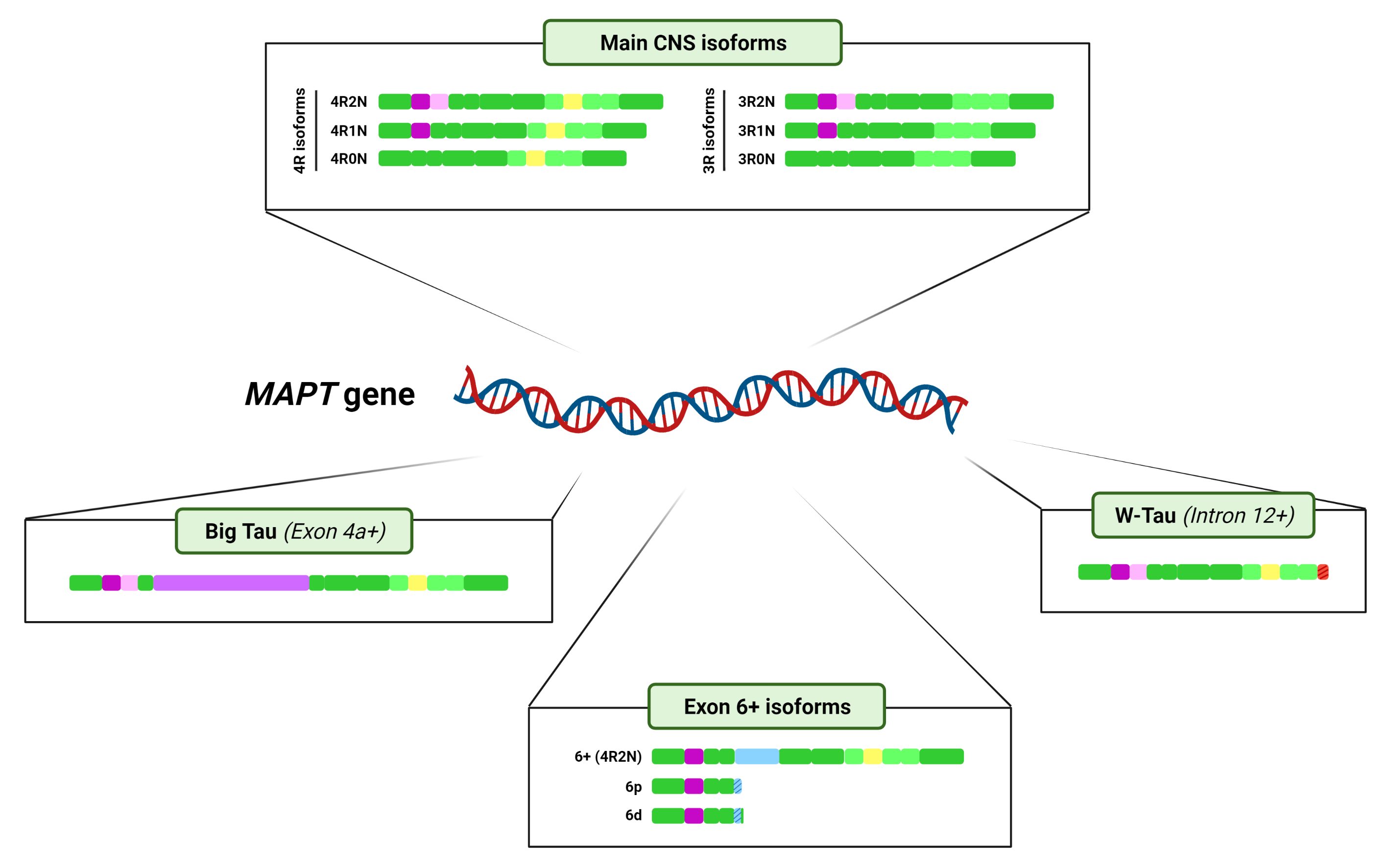
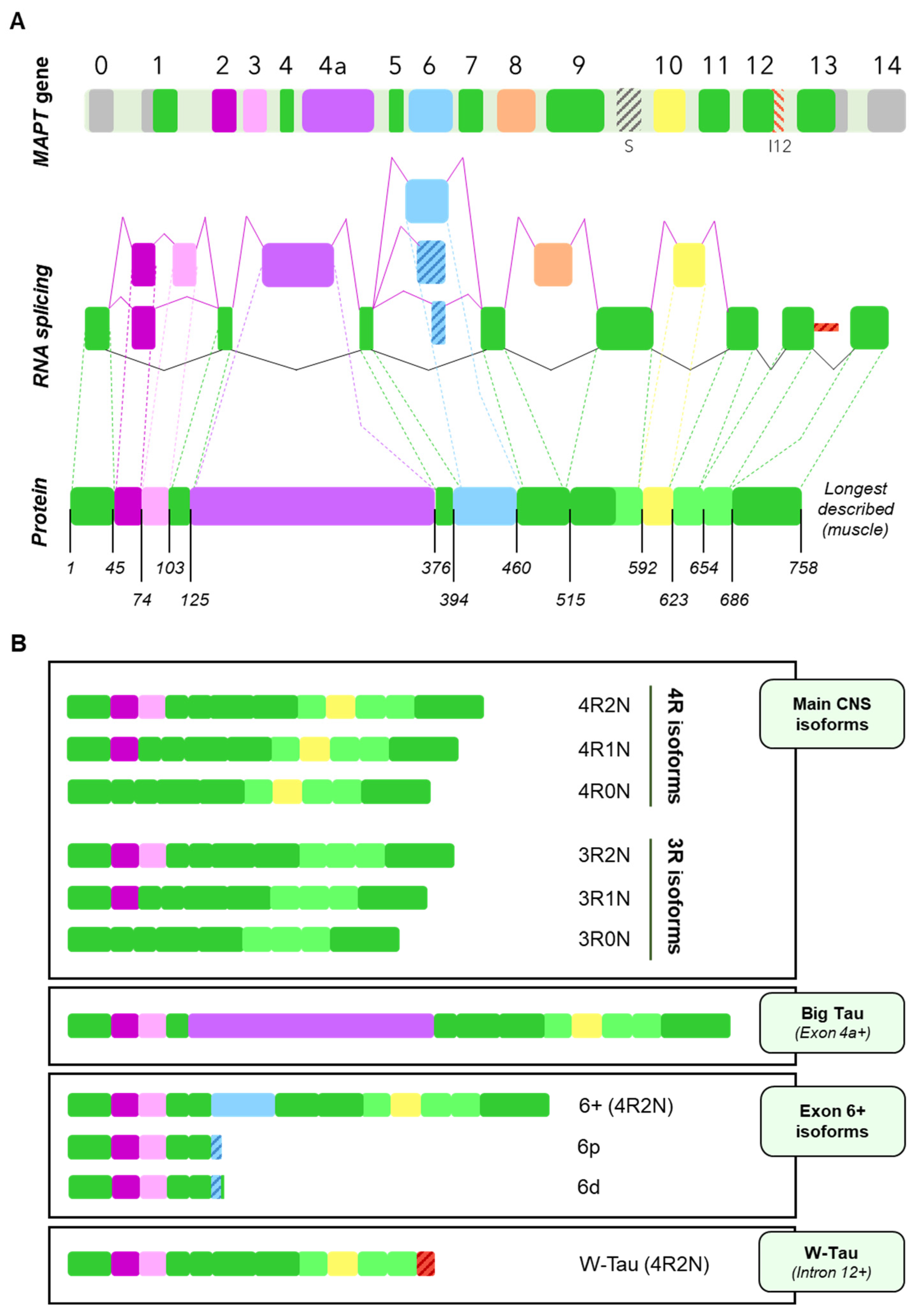
3. Tau Alternative Splicing: Diversity of Forms
3.1. Central Nervous System Isoforms: Meet the Classics
3.2. Big Tau: A Giant Outsider
3.3. Black Sheep: Isoforms including Exon 6
3.4. W-Tau: The Rara Avis
4. Tau Alternative Splicing: Diversity of Functions
4.1. The Projection Domain: Tau’s Versatile N-Terminal End and the Extension of Big Tau
4.2. Proline-Rich Region and Isoforms Including Exon 6
4.3. Microtubule-Binding Domain: A Repetitive Region
4.4. W-Tau: The Uncharted Territory of Novel Isoforms and New Mechanisms
4.5. The Interplay: Interaction between Functional Regions
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Buée, L.; Bussière, T.; Buée-Scherrer, V.; Delacourte, A.; Hof, P.R. Tau protein isoforms, phosphorylation and role in neurodegenerative disorders. Brain Res. Rev. 2000, 33, 95–130. [Google Scholar] [CrossRef]
- Motoi, Y.; Sahara, N.; Kambe, T.; Hattori, N. Tau and neurodegenerative disorders. Biomol. Concepts 2010, 1, 131–145. [Google Scholar] [CrossRef]
- Wang, Y.; Mandelkow, E. Tau in physiology and pathology. Nat. Rev. Neurosci. 2016, 17, 22–35. [Google Scholar] [CrossRef] [PubMed]
- Guo, T.; Noble, W.; Hanger, D.P. Roles of tau protein in health and disease. Acta Neuropathol. 2017, 133, 665–704. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kovacech, B.; Novak, M. Tau truncation is a productive posttranslational modification of neurofibrillary degeneration in Alzheimer’s disease. Curr. Alzheimer Res. 2010, 7, 708–716. [Google Scholar] [CrossRef] [PubMed]
- Gu, J.; Xu, W.; Jin, N.; Li, L.; Zhou, Y.; Chu, D.; Gong, C.-X.; Iqbal, K.; Liu, F. Truncation of Tau selectively facilitates its pathological activities. J. Biol. Chem. 2020, 295, 13812–13828. [Google Scholar] [CrossRef]
- Alquezar, C.; Arya, S.; Kao, A.W. Tau post-translational modifications: Dynamic transformers of tau function, degradation, and aggregation. Front. Neurol. 2021, 11, 1826. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Shi, J.; Chu, D.; Hu, W.; Guan, Z.; Gong, C.-X.; Iqbal, K.; Liu, F. Relevance of phosphorylation and truncation of tau to the etiopathogenesis of Alzheimer’s disease. Front. Aging Neurosci. 2018, 10, 27. [Google Scholar] [CrossRef]
- Andreadis, A. Tau gene alternative splicing: Expression patterns, regulation and modulation of function in normal brain and neurodegenerative diseases. Biochim. Biophys. Acta 2005, 1739, 91–103. [Google Scholar] [CrossRef] [Green Version]
- Qian, W.; Liu, F. Regulation of alternative splicing of tau exon 10. Neurosci. Bull. 2014, 30, 367–377. [Google Scholar] [CrossRef] [Green Version]
- Black, D.L. Mechanisms of alternative pre-messenger RNA splicing. Annu. Rev. Biochem. 2003, 72, 291–336. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blencowe, B.J. Alternative splicing: New insights from global analyses. Cell 2006, 126, 37–47. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lopez, A.J. Alternative splicing of pre-mRNA: Developmental consequences and mechanisms of regulation. Annu. Rev. Genet. 1998, 32, 279–305. [Google Scholar] [CrossRef] [PubMed]
- Tress, M.L.; Abascal, F.; Valencia, A. Alternative splicing may not be the key to proteome complexity. Trends Biochem. Sci. 2017, 42, 98–110. [Google Scholar] [CrossRef] [Green Version]
- Abascal, F.; Ezkurdia, I.; Rodriguez-Rivas, J.; Rodriguez, J.M.; del Pozo, A.; Vazquez, J.; Valencia, A.; Tress, M.L. Alternatively spliced homologous exons have ancient origins and are highly expressed at the protein level. PLoS Comput. Biol. 2015, 11, e1004325. [Google Scholar] [CrossRef] [Green Version]
- Angarola, B.L.; Anczuków, O. Splicing alterations in healthy aging and disease. Wiley Interdiscip. Rev. RNA 2021, 12, e1643. [Google Scholar] [CrossRef]
- Bhadra, M.; Howell, P.; Dutta, S.; Heintz, C.; Mair, W.B. Alternative splicing in aging and longevity. Hum. Genet. 2020, 139, 357–369. [Google Scholar] [CrossRef]
- Li, H.; Wang, Z.; Ma, T.; Wei, G.; Ni, T. Alternative splicing in aging and age-related diseases. Transl. Med. Aging 2017, 1, 32–40. [Google Scholar] [CrossRef]
- Kar, A.; Kuo, D.; He, R.; Zhou, J.; Wu, J.Y. Tau alternative splicing and frontotemporal dementia. Alzheimer Dis. Assoc. Disord. 2005, 19, S29. [Google Scholar] [CrossRef]
- Ishunina, T.A. Alternative splicing in aging and Alzheimer’s disease: Highlighting the role of tau and estrogen receptor α isoforms in the hypothalamus. In Handbook of Clinical Neurology; Elsevier: Amsterdam, The Netherlands, 2021; Volume 182, pp. 177–189. [Google Scholar]
- Sakabe, N.J.; De Souza, S.J. Sequence features responsible for intron retention in human. BMC Genom. 2007, 8, 59. [Google Scholar] [CrossRef] [Green Version]
- Koren, E.; Lev-Maor, G.; Ast, G. The emergence of alternative 3′ and 5′ splice site exons from constitutive exons. PLoS Comput. Biol. 2007, 3, e95. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Breitbart, R.E.; Andreadis, A.; Nadal-Ginard, B. Alternative splicing: A ubiquitous mechanism for the generation of multiple protein isoforms from single genes. Annu. Rev. Biochem. 1987, 56, 467–495. [Google Scholar] [CrossRef] [PubMed]
- Kim, E.; Goren, A.; Ast, G. Alternative splicing: Current perspectives. Bioessays 2008, 30, 38–47. [Google Scholar] [CrossRef]
- Jin, Y.; Dong, H.; Shi, Y.; Bian, L. Mutually exclusive alternative splicing of pre-mRNAs. Wiley Interdiscip. Rev. RNA 2018, 9, e1468. [Google Scholar] [CrossRef]
- Lutz, C.S. Alternative polyadenylation: A twist on mRNA 3′ end formation. ACS Chem. Biol. 2008, 3, 609–617. [Google Scholar] [CrossRef] [PubMed]
- Ner-Gaon, H.; Halachmi, R.; Savaldi-Goldstein, S.; Rubin, E.; Ophir, R.; Fluhr, R. Intron retention is a major phenomenon in alternative splicing in Arabidopsis. Plant J. 2004, 39, 877–885. [Google Scholar] [CrossRef] [PubMed]
- Ule, J.; Blencowe, B.J. Alternative splicing regulatory networks: Functions, mechanisms, and evolution. Mol. Cell 2019, 76, 329–345. [Google Scholar] [CrossRef]
- House, A.E.; Lynch, K.W. Regulation of alternative splicing: More than just the ABCs. J. Biol. Chem. 2008, 283, 1217–1221. [Google Scholar] [CrossRef] [Green Version]
- Liu, F.; Gong, C.-X. Tau exon 10 alternative splicing and tauopathies. Mol. Neurodegener. 2008, 3, 8. [Google Scholar] [CrossRef] [Green Version]
- Latorre, E.; Harries, L.W. Splicing regulatory factors, ageing and age-related disease. Ageing Res. Rev. 2017, 36, 165–170. [Google Scholar] [CrossRef]
- Goedert, M. Tau filaments in neurodegenerative diseases. FEBS Lett. 2018, 592, 2383–2391. [Google Scholar] [CrossRef] [PubMed]
- Bachmann, S.; Bell, M.; Klimek, J.; Zempel, H. Differential effects of the six human TAU isoforms: Somatic retention of 2N-TAU and increased microtubule number induced by 4R-TAU. Front. Neurosci. 2021, 15. [Google Scholar] [CrossRef] [PubMed]
- Park, S.A.; Ahn, S.I.; Gallo, J.-M. Tau mis-splicing in the pathogenesis of neurodegenerative disorders. BMB Rep. 2016, 49, 405. [Google Scholar] [CrossRef] [Green Version]
- Arikan, M.C.; Memmott, J.; Broderick, J.A.; Lafyatis, R.; Screaton, G.; Stamm, S.; Andreadis, A. Modulation of the membrane-binding projection domain of tau protein: Splicing regulation of exon 3. Mol. Brain Res. 2002, 101, 109–121. [Google Scholar] [CrossRef]
- Nelson, P.T.; Stefansson, K.; Gulcher, J.; Saper, C.B. Molecular evolution of τ protein: Implications for Alzheimer’s disease. J. Neurochem. 1996, 67, 1622–1632. [Google Scholar] [CrossRef] [PubMed]
- García-Escudero, V.; Ruiz-Gabarre, D.; Gargini, R.; Pérez, M.; García, E.; Cuadros, R.; Hernández, I.H.; Cabrera, J.R.; García-Escudero, R.; Lucas, J.J. A new non-aggregative splicing isoform of human Tau is decreased in Alzheimer’s disease. Acta Neuropathol. 2021, 142, 159–177. [Google Scholar] [CrossRef]
- Conrad, C.; Vianna, C.; Freeman, M.; Davies, P. A polymorphic gene nested within an intron of the tau gene: Implications for Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 2002, 99, 7751–7756. [Google Scholar] [CrossRef] [Green Version]
- Andreadis, A. Tau splicing and the intricacies of dementia. J. Cell. Physiol. 2012, 227, 1220–1225. [Google Scholar] [CrossRef] [Green Version]
- Bowles, K.R.; Pugh, D.A.; Oja, L.-M.; Jadow, B.M.; Farrell, K.; Whitney, K.; Sharma, A.; Cherry, J.D.; Raj, T.; Pereira, A.C. Dysregulated coordination of MAPT exon 2 and exon 10 splicing underlies different tau pathologies in PSP and AD. Acta Neuropathol. 2022, 143, 225–243. [Google Scholar] [CrossRef]
- Xing, S.; Wang, J.; Wu, R.; Hefti, M.M.; Crary, J.F.; Lu, Y. Identification of HnRNPC as a novel Tau exon 10 splicing factor using RNA antisense purification mass spectrometry. RNA Biol. 2021, 19, 104–116. [Google Scholar] [CrossRef]
- Chen, J.L.; Moss, W.N.; Spencer, A.; Zhang, P.; Childs-Disney, J.L.; Disney, M.D. The RNA encoding the microtubule-associated protein tau has extensive structure that affects its biology. PLoS ONE 2019, 14, e0219210. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garuti, R.; Croce, M.; Tiozzo, R.; Dotti, M.; Federico, A.; Bertolini, S.; Calandra, S. Four novel mutations of sterol 27-hydroxylase gene in Italian patients with cerebrotendinous xanthomatosis. J. Lipid Res. 1997, 38, 2322–2334. [Google Scholar] [CrossRef]
- Shikama, Y.; Hu, H.; Ohno, M.; Matsuoka, I.; Shichishima, T.; Kimura, J. Transcripts expressed using a bicistronic vector pIREShyg2 are sensitized to nonsense-mediated mRNA decay. BMC Mol. Biol. 2010, 11, 42. [Google Scholar] [CrossRef] [Green Version]
- LaPointe, N.E.; Horowitz, P.M.; Guillozet-Bongaarts, A.L.; Silva, A.; Andreadis, A.; Binder, L.I. Tau 6D and 6P isoforms inhibit polymerization of full-length tau in vitro. Biochemistry 2009, 48, 12290–12297. [Google Scholar] [CrossRef] [Green Version]
- Holzer, M.; Craxton, M.; Jakes, R.; Arendt, T.; Goedert, M. Tau gene (MAPT) sequence variation among primates. Gene 2004, 341, 313–322. [Google Scholar] [CrossRef] [PubMed]
- Takuma, H.; Arawaka, S.; Mori, H. Isoforms changes of tau protein during development in various species. Dev. Brain Res. 2003, 142, 121–127. [Google Scholar] [CrossRef]
- Wegmann, S.; Biernat, J.; Mandelkow, E. A current view on Tau protein phosphorylation in Alzheimer’s disease. Curr. Opin. Neurobiol. 2021, 69, 131–138. [Google Scholar] [CrossRef]
- Biamonti, G.; Amato, A.; Belloni, E.; Di Matteo, A.; Infantino, L.; Pradella, D.; Ghigna, C. Alternative splicing in Alzheimer’s disease. Aging Clin. Exp. Res. 2021, 33, 747–758. [Google Scholar] [CrossRef]
- Niblock, M.; Gallo, J.-M. Tau alternative splicing in familial and sporadic tauopathies. Biochem. Soc. Trans. 2012, 40, 677–680. [Google Scholar] [CrossRef]
- Miguel, L.; Rovelet-Lecrux, A.; Feyeux, M.; Frebourg, T.; Nassoy, P.; Campion, D.; Lecourtois, M. Detection of all adult Tau isoforms in a 3D culture model of iPSC-derived neurons. Stem Cell Res. 2019, 40, 101541. [Google Scholar] [CrossRef]
- Hefti, M.M.; Farrell, K.; Kim, S.; Bowles, K.R.; Fowkes, M.E.; Raj, T.; Crary, J.F. High-resolution temporal and regional mapping of MAPT expression and splicing in human brain development. PLoS ONE 2018, 13, e0195771. [Google Scholar] [CrossRef] [Green Version]
- Sotiropoulos, I.; Galas, M.-C.; Silva, J.M.; Skoulakis, E.; Wegmann, S.; Maina, M.B.; Blum, D.; Sayas, C.L.; Mandelkow, E.-M.; Mandelkow, E. Atypical, non-standard functions of the microtubule associated Tau protein. Acta Neuropathol. Commun. 2017, 5, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Bukar Maina, M.; Al-Hilaly, Y.K.; Serpell, L.C. Nuclear tau and its potential role in Alzheimer’s disease. Biomolecules 2016, 6, 9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, C.; Götz, J. Profiling murine tau with 0N, 1N and 2N isoform-specific antibodies in brain and peripheral organs reveals distinct subcellular localization, with the 1N isoform being enriched in the nucleus. PLoS ONE 2013, 8, e84849. [Google Scholar] [CrossRef] [Green Version]
- Fischer, I.; Baas, P.W. Resurrecting the mysteries of big tau. Trends Neurosci. 2020, 43, 493–504. [Google Scholar] [CrossRef] [PubMed]
- Majounie, E.; Cross, W.; Newsway, V.; Dillman, A.; Vandrovcova, J.; Morris, C.M.; Nalls, M.A.; Ferrucci, L.; Owen, M.J.; O’Donovan, M.C. Variation in tau isoform expression in different brain regions and disease states. Neurobiol. Aging 2013, 34, 1922.e7–1922.e1912. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wesseling, H.; Mair, W.; Kumar, M.; Schlaffner, C.N.; Tang, S.; Beerepoot, P.; Fatou, B.; Guise, A.J.; Cheng, L.; Takeda, S. Tau PTM profiles identify patient heterogeneity and stages of Alzheimer’s disease. Cell 2020, 183, 1699–1713.e13. [Google Scholar] [CrossRef] [PubMed]
- Twine, N.A.; Janitz, K.; Wilkins, M.R.; Janitz, M. Whole transcriptome sequencing reveals gene expression and splicing differences in brain regions affected by Alzheimer’s disease. PLoS ONE 2011, 6, e16266. [Google Scholar] [CrossRef]
- Drubin, D.; Kobayashi, S.; Kellogg, D.; Kirschner, M. Regulation of microtubule protein levels during cellular morphogenesis in nerve growth factor-treated PC12 cells. J. Cell Biol. 1988, 106, 1583–1591. [Google Scholar] [CrossRef] [Green Version]
- Oblinger, M.M.; Argasinski, A.; Wong, J.; Kosik, K.S. Tau gene expression in rat sensory neurons during development and regeneration. J. Neurosci. 1991, 11, 2453–2459. [Google Scholar] [CrossRef] [Green Version]
- Goedert, M.; Spillantini, M.; Crowther, R. Cloning of a big tau microtubule-associated protein characteristic of the peripheral nervous system. Proc. Natl. Acad. Sci. USA 1992, 89, 1983–1987. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Couchie, D.; Mavilia, C.; Georgieff, I.S.; Liem, R.; Shelanski, M.L.; Nunez, J. Primary structure of high molecular weight tau present in the peripheral nervous system. Proc. Natl. Acad. Sci. USA 1992, 89, 4378–4381. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wei, M.L.; Andreadis, A. Splicing of a regulated exon reveals additional complexity in the axonal microtubule-associated protein tau. J. Neurochem. 1998, 70, 1346–1356. [Google Scholar] [CrossRef]
- Luo, M.H.; Leski, M.L.; Andreadis, A. Tau isoforms which contain the domain encoded by exon 6 and their role in neurite elongation. J. Cell. Biochem. 2004, 91, 880–895. [Google Scholar] [CrossRef]
- Wei, M.-L.; Memmott, J.; Screaton, G.; Andreadis, A. The splicing determinants of a regulated exon in the axonal MAP tau reside within the exon and in its upstream intron. Mol. Brain Res. 2000, 80, 207–218. [Google Scholar] [CrossRef]
- Brandt, R.; Trushina, N.I.; Bakota, L. Much more than a cytoskeletal protein: Physiological and pathological functions of the non-microtubule binding region of tau. Front. Neurol. 2020, 11, 1269. [Google Scholar] [CrossRef] [PubMed]
- Evans, H.T.; Taylor, D.; Kneynsberg, A.; Bodea, L.-G.; Götz, J. Altered ribosomal function and protein synthesis caused by tau. Acta Neuropathol. Commun. 2021, 9, 110. [Google Scholar] [CrossRef] [PubMed]
- Pooler, A.M.; Hanger, D.P. Functional implications of the association of tau with the plasma membrane. Biochem. Soc. Trans. 2010, 38, 1012–1015. [Google Scholar] [CrossRef] [Green Version]
- Brandt, R. The tau proteins in neuronal growth and development. Front. Biosci. 1996, 1, 118–130. [Google Scholar] [CrossRef] [Green Version]
- Kobayashi, S.; Tanaka, T.; Soeda, Y.; Takashima, A. Enhanced tau protein translation by hyper-excitation. Front. Aging Neurosci. 2019, 11, 322. [Google Scholar] [CrossRef] [Green Version]
- Trabzuni, D.; Wray, S.; Vandrovcova, J.; Ramasamy, A.; Walker, R.; Smith, C.; Luk, C.; Gibbs, J.R.; Dillman, A.; Hernandez, D.G. MAPT expression and splicing is differentially regulated by brain region: Relation to genotype and implication for tauopathies. Hum. Mol. Genet. 2012, 21, 4094–4103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jacob, A.G.; Smith, C.W. Intron retention as a component of regulated gene expression programs. Hum. Genet. 2017, 136, 1043–1057. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Braunschweig, U.; Gueroussov, S.; Plocik, A.M.; Graveley, B.R.; Blencowe, B.J. Dynamic integration of splicing within gene regulatory pathways. Cell 2013, 152, 1252–1269. [Google Scholar] [CrossRef] [Green Version]
- Mazin, P.; Xiong, J.; Liu, X.; Yan, Z.; Zhang, X.; Li, M.; He, L.; Somel, M.; Yuan, Y.; Phoebe Chen, Y.P. Widespread splicing changes in human brain development and aging. Mol. Syst. Biol. 2013, 9, 633. [Google Scholar] [CrossRef]
- Drummond, E.; Wisniewski, T. Alzheimer’s disease: Experimental models and reality. Acta Neuropathol. 2017, 133, 155–175. [Google Scholar] [CrossRef] [PubMed]
- Moon, H.; Cho, S.; Loh, T.J.; Jang, H.N.; Liu, Y.; Choi, N.; Oh, J.; Ha, J.; Zhou, J.; Cho, S. SRSF2 directly inhibits intron splicing to suppresses cassette exon inclusion. BMB Rep. 2017, 50, 423. [Google Scholar] [CrossRef] [Green Version]
- Hernandez, F.; Perez, M.; Lucas, J.J.; Mata, A.M.; Bhat, R.; Avila, J. Glycogen synthase kinase-3 plays a crucial role in tau exon 10 splicing and intranuclear distribution of SC35. Implications for Alzheimer’s disease. J. Biol. Chem. 2004, 279, 3801–3806. [Google Scholar] [CrossRef] [Green Version]
- Chen, K.-L.; Yuan, R.-Y.; Hu, C.-J.; Hsu, C.Y. Amyloid-β peptide alteration of tau exon-10 splicing via the GSK3β-SC35 pathway. Neurobiol. Dis. 2010, 40, 378–385. [Google Scholar] [CrossRef]
- Zempel, H.; Dennissen, F.J.; Kumar, Y.; Luedtke, J.; Biernat, J.; Mandelkow, E.-M.; Mandelkow, E. Axodendritic sorting and pathological missorting of Tau are isoform-specific and determined by axon initial segment architecture. J. Biol. Chem. 2017, 292, 12192–12207. [Google Scholar] [CrossRef] [Green Version]
- Kolarova, M.; García-Sierra, F.; Bartos, A.; Ricny, J.; Ripova, D. Structure and pathology of tau protein in Alzheimer disease. Int. J. Alzheimer’s Dis. 2012, 2012, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Brandt, R.; Léger, J.; Lee, G. Interaction of tau with the neural plasma membrane mediated by tau’s amino-terminal projection domain. J. Cell Biol. 1995, 131, 1327–1340. [Google Scholar] [CrossRef] [PubMed]
- Amadoro, G.; Latina, V.; Corsetti, V.; Calissano, P. N-terminal tau truncation in the pathogenesis of Alzheimer’s disease (AD): Developing a novel diagnostic and therapeutic approach. Biochim. Et Biophys. Acta (BBA)-Mol. Basis Dis. 2020, 1866, 165584. [Google Scholar] [CrossRef] [PubMed]
- Decker, J.M.; Mandelkow, E.-M. Presynaptic Pathophysiology Encoded in Different Domains of Tau–Hyper-Versus Hypoexcitability? Tau Biol. 2019, 97–103. [Google Scholar] [CrossRef]
- Pooler, A.M.; Usardi, A.; Evans, C.J.; Philpott, K.L.; Noble, W.; Hanger, D.P. Dynamic association of tau with neuronal membranes is regulated by phosphorylation. Neurobiol. Aging 2012, 33, 431.e427–431.e438. [Google Scholar] [CrossRef]
- Magnani, E.; Fan, J.; Gasparini, L.; Golding, M.; Williams, M.; Schiavo, G.; Goedert, M.; Amos, L.A.; Spillantini, M.G. Interaction of tau protein with the dynactin complex. EMBO J. 2007, 26, 4546–4554. [Google Scholar] [CrossRef] [Green Version]
- Ritter, M.L.; Avila, J.; García-Escudero, V.; Hernández, F.; Pérez, M. Frontotemporal dementia-associated N279K tau mutation localizes at the nuclear compartment. Front. Cell. Neurosci. 2018, 12, 202. [Google Scholar] [CrossRef] [Green Version]
- Corces, V.G.; Salas, J.; Salas, M.L.; Ávila, J. Binding of microtubule proteins to DNA: Specificity of the interaction. Eur. J. Biochem. 1978, 86, 473–479. [Google Scholar] [CrossRef]
- Villasante, A.; Corces, V.G.; Manso-Martinez, R.; Avila, J. Binding of microtubule protein to DNA and chromatin: Possibility of simultaneous linkage of microtubule to nucleic acid and assembly of the microtubule structure. Nucleic Acids Res. 1981, 9, 895–908. [Google Scholar] [CrossRef]
- Mansuroglu, Z.; Benhelli-Mokrani, H.; Marcato, V.; Sultan, A.; Violet, M.; Chauderlier, A.; Delattre, L.; Loyens, A.; Talahari, S.; Bégard, S. Loss of Tau protein affects the structure, transcription and repair of neuronal pericentromeric heterochromatin. Sci. Rep. 2016, 6, 33047. [Google Scholar] [CrossRef] [Green Version]
- Lester, E.; Ooi, F.K.; Bakkar, N.; Ayers, J.; Woerman, A.L.; Wheeler, J.; Bowser, R.; Carlson, G.A.; Prusiner, S.B.; Parker, R. Tau aggregates are RNA-protein assemblies that mislocalize multiple nuclear speckle components. Neuron 2021, 109, 1675–1691.e1679. [Google Scholar] [CrossRef]
- Sultan, A.; Nesslany, F.; Violet, M.; Bégard, S.; Loyens, A.; Talahari, S.; Mansuroglu, Z.; Marzin, D.; Sergeant, N.; Humez, S. Nuclear tau, a key player in neuronal DNA protection. J. Biol. Chem. 2011, 286, 4566–4575. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eftekharzadeh, B.; Daigle, J.G.; Kapinos, L.E.; Coyne, A.; Schiantarelli, J.; Carlomagno, Y.; Cook, C.; Miller, S.J.; Dujardin, S.; Amaral, A.S. Tau protein disrupts nucleocytoplasmic transport in Alzheimer’s disease. Neuron 2018, 99, 925–940.e927. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Diez, L.; Wegmann, S. Nuclear transport deficits in tau-related neurodegenerative diseases. Front. Neurol. 2020, 11, 1056. [Google Scholar] [CrossRef]
- Koren, S.A.; Hamm, M.J.; Meier, S.E.; Weiss, B.E.; Nation, G.K.; Chishti, E.A.; Arango, J.P.; Chen, J.; Zhu, H.; Blalock, E.M. Tau drives translational selectivity by interacting with ribosomal proteins. Acta Neuropathol. 2019, 137, 571–583. [Google Scholar] [CrossRef] [Green Version]
- Maina, M.B.; Bailey, L.J.; Wagih, S.; Biasetti, L.; Pollack, S.J.; Quinn, J.P.; Thorpe, J.R.; Doherty, A.J.; Serpell, L.C. The involvement of tau in nucleolar transcription and the stress response. Acta Neuropathol. Commun. 2018, 6, 70. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Portillo, M.; Eremenko, E.; Kaluski, S.; Garcia-Venzor, A.; Onn, L.; Stein, D.; Slobodnik, Z.; Zaretsky, A.; Ueberham, U.; Einav, M. SIRT6-CBP-dependent nuclear Tau accumulation and its role in protein synthesis. Cell Rep. 2021, 35, 109035. [Google Scholar] [CrossRef] [PubMed]
- Gil, L.; Federico, C.; Pinedo, F.; Bruno, F.; Rebolledo, A.B.; Montoya, J.J.; Olazabal, I.M.; Ferrer, I.; Saccone, S. Aging dependent effect of nuclear tau. Brain Res. 2017, 1677, 129–137. [Google Scholar] [CrossRef] [Green Version]
- Georgieff, I.S.; Liem, R.; Couchie, D.; Mavilia, C.; Nunez, J.; Shelanski, M.L. Expression of high molecular weight tau in the central and peripheral nervous systems. J. Cell Sci. 1993, 105, 729–737. [Google Scholar] [CrossRef]
- Liu, Y.; Wang, C.; Destin, G.; Szaro, B.G. Microtubule-associated protein tau promotes neuronal class II β-tubulin microtubule formation and axon elongation in embryonic X enopus laevis. Eur. J. Neurosci. 2015, 41, 1263–1275. [Google Scholar] [CrossRef]
- Sibille, N.; Huvent, I.; Fauquant, C.; Verdegem, D.; Amniai, L.; Leroy, A.; Wieruszeski, J.M.; Lippens, G.; Landrieu, I. Structural characterization by nuclear magnetic resonance of the impact of phosphorylation in the proline-rich region of the disordered Tau protein. Proteins Struct. Funct. Bioinform. 2012, 80, 454–462. [Google Scholar] [CrossRef]
- Goode, B.L.; Denis, P.E.; Panda, D.; Radeke, M.J.; Miller, H.P.; Wilson, L.; Feinstein, S. Functional interactions between the proline-rich and repeat regions of tau enhance microtubule binding and assembly. Mol. Biol. Cell 1997, 8, 353–365. [Google Scholar] [CrossRef] [PubMed]
- He, H.J.; Wang, X.S.; Pan, R.; Wang, D.L.; Liu, M.N.; He, R.Q. The proline-rich domain of tau plays a role in interactions with actin. BMC Cell Biol. 2009, 10, 81. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McKibben, K.M.; Rhoades, E. Independent tubulin binding and polymerization by the proline-rich region of Tau is regulated by Tau’s N-terminal domain. J. Biol. Chem. 2019, 294, 19381–19394. [Google Scholar] [CrossRef] [PubMed]
- Pérez, M.; Cuadros, R.; Hernández, F.; Avila, J. Secretion of full-length tau or tau fragments in a cell culture model. Neurosci. Lett. 2016, 634, 63–69. [Google Scholar] [CrossRef]
- Prikas, E.; Ahel, H.; Stefanoska, K.; Asih, P.R.; Volkerling, A.; Ittner, L.M.; Ittner, A. Interaction between the guanylate kinase domain of PSD-95 and the proline-rich region and microtubule binding repeats 2 and 3 of tau. Biochem. Cell Biol. 2021, 99, 606–616. [Google Scholar] [CrossRef]
- Teravskis, P.J.; Oxnard, B.R.; Miller, E.C.; Kemper, L.; Ashe, K.H.; Liao, D. Phosphorylation in two discrete tau domains regulates a stepwise process leading to postsynaptic dysfunction. J. Physiol. 2021, 599, 2483–2498. [Google Scholar] [CrossRef]
- Horowitz, P.M.; LaPointe, N.; Guillozet-Bongaarts, A.L.; Berry, R.W.; Binder, L.I. N-terminal fragments of tau inhibit full-length tau polymerization in vitro. Biochemistry 2006, 45, 12859–12866. [Google Scholar] [CrossRef]
- Janning, D.; Igaev, M.; Sündermann, F.; Brühmann, J.; Beutel, O.; Heinisch, J.J.; Bakota, L.; Piehler, J.; Junge, W.; Brandt, R. Single-molecule tracking of tau reveals fast kiss-and-hop interaction with microtubules in living neurons. Mol. Biol. Cell 2014, 25, 3541–3551. [Google Scholar] [CrossRef]
- Niewidok, B.; Igaev, M.; Sündermann, F.; Janning, D.; Bakota, L.; Brandt, R. Presence of a carboxy-terminal pseudorepeat and disease-like pseudohyperphosphorylation critically influence tau’s interaction with microtubules in axon-like processes. Mol. Biol. Cell 2016, 27, 3537–3549. [Google Scholar] [CrossRef]
- Kellogg, E.H.; Hejab, N.M.; Poepsel, S.; Downing, K.H.; DiMaio, F.; Nogales, E. Near-atomic model of microtubule-tau interactions. Science 2018, 360, 1242–1246. [Google Scholar] [CrossRef] [Green Version]
- Cohen, T.J.; Friedmann, D.; Hwang, A.W.; Marmorstein, R.; Lee, V.M. The microtubule-associated tau protein has intrinsic acetyltransferase activity. Nat. Struct. Mol. Biol. 2013, 20, 756–762. [Google Scholar] [CrossRef] [Green Version]
- Trushina, N.I.; Bakota, L.; Mulkidjanian, A.Y.; Brandt, R. The evolution of tau phosphorylation and interactions. Front. Aging Neurosci. 2019, 11, 256. [Google Scholar] [CrossRef] [PubMed]
- Pérez, M.; Valpuesta, J.M.; Medina, M.; Montejo de Garcini, E.; Avila, J. Polymerization of τ into filaments in the presence of heparin: The minimal sequence required for τ-τ interaction. J. Neurochem. 1996, 67, 1183–1190. [Google Scholar] [CrossRef] [PubMed]
- Goedert, M.; Spillantini, M.; Jakes, R.; Rutherford, D.; Crowther, R. Multiple isoforms of human microtubule-associated protein tau: Sequences and localization in neurofibrillary tangles of Alzheimer’s disease. Neuron 1989, 3, 519–526. [Google Scholar] [CrossRef]
- Ingelsson, M.; Ramasamy, K.; Cantuti-Castelvetri, I.; Skoglund, L.; Matsui, T.; Orne, J.; Kowa, H.; Raju, S.; Vanderburg, C.R.; Augustinack, J.C. No alteration in tau exon 10 alternative splicing in tangle-bearing neurons of the Alzheimer’s disease brain. Acta Neuropathol. 2006, 112, 439–449. [Google Scholar] [CrossRef]
- Fitzpatrick, A.W.; Falcon, B.; He, S.; Murzin, A.G.; Murshudov, G.; Garringer, H.J.; Crowther, R.A.; Ghetti, B.; Goedert, M.; Scheres, S.H. Cryo-EM structures of tau filaments from Alzheimer’s disease. Nature 2017, 547, 185–190. [Google Scholar] [CrossRef] [Green Version]
- Zhang, W.; Tarutani, A.; Newell, K.L.; Murzin, A.G.; Matsubara, T.; Falcon, B.; Vidal, R.; Garringer, H.J.; Shi, Y.; Ikeuchi, T. Novel tau filament fold in corticobasal degeneration. Nature 2020, 580, 283–287. [Google Scholar] [CrossRef]
- Zhang, Z.; Song, M.; Liu, X.; Kang, S.S.; Kwon, I.-S.; Duong, D.M.; Seyfried, N.T.; Hu, W.T.; Liu, Z.; Wang, J.-Z. Cleavage of tau by asparagine endopeptidase mediates the neurofibrillary pathology in Alzheimer’s disease. Nat. Med. 2014, 20, 1254–1262. [Google Scholar] [CrossRef] [Green Version]
- Fasulo, L.; Ugolini, G.; Visintin, M.; Bradbury, A.; Brancolini, C.; Verzillo, V.; Novak, M.; Cattaneo, A. The neuronal microtubule-associated protein tau is a substrate for caspase-3 and an effector of apoptosis. J. Neurochem. 2000, 75, 624–633. [Google Scholar] [CrossRef] [Green Version]
- Griner, S.L.; Seidler, P.; Bowler, J.; Murray, K.A.; Yang, T.P.; Sahay, S.; Sawaya, M.R.; Cascio, D.; Rodriguez, J.A.; Philipp, S. Structure-based inhibitors of amyloid beta core suggest a common interface with tau. Elife 2019, 8, e46924. [Google Scholar] [CrossRef]
- Park, C.; Raines, R.T. Adjacent cysteine residues as a redox switch. Protein Eng. 2001, 14, 939–942. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mattaj, I.W.; Englmeier, L. Nucleocytoplasmic transport: The soluble phase. Annu. Rev. Biochem. 1998, 67, 265–306. [Google Scholar] [CrossRef] [PubMed]
- Dingwall, C.; Robbins, J.; Dilworth, S.M.; Roberts, B.; Richardson, W.D. The nucleoplasmin nuclear location sequence is larger and more complex than that of SV-40 large T antigen. J. Cell Biol. 1988, 107, 841–849. [Google Scholar] [CrossRef] [PubMed]
- Hinrich, A.J.; Jodelka, F.M.; Chang, J.L.; Brutman, D.; Bruno, A.M.; Briggs, C.A.; James, B.D.; Stutzmann, G.E.; Bennett, D.A.; Miller, S.A. Therapeutic correction of ApoER2 splicing in Alzheimer’s disease mice using antisense oligonucleotides. EMBO Mol. Med. 2016, 8, 328–345. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, R.; Tikoo, S.; Jain, R.; Pinello, N.; Au, A.Y.; Nagarajah, R.; Porse, B.; Rasko, J.E.; Wong, J.J.-L. Dynamic intron retention modulates gene expression in the monocytic differentiation pathway. Immunology 2021, 165, 274–286. [Google Scholar] [CrossRef]
- Novak, P.; Cehlar, O.; Skrabana, R.; Novak, M. Tau conformation as a target for disease-modifying therapy: The role of truncation. J. Alzheimer’s Dis. 2018, 64, S535–S546. [Google Scholar] [CrossRef]
- Jeganathan, S.; Chinnathambi, S.; Mandelkow, E.-M.; Mandelkow, E. Conformations of microtubule-associated protein Tau mapped by fluorescence resonance energy transfer. In Amyloid Proteins; Springer: Berlin/Heidelberg, Germany, 2012; pp. 85–99. [Google Scholar]
- Zhou, X.-W.; Li, X.; Bjorkdahl, C.; Sjogren, M.J.; Alafuzoff, I.; Soininen, H.; Grundke-Iqbal, I.; Iqbal, K.; Winblad, B.; Pei, J.-J. Assessments of the accumulation severities of amyloid β-protein and hyperphosphorylated tau in the medial temporal cortex of control and Alzheimer’s brains. Neurobiol. Dis. 2006, 22, 657–668. [Google Scholar] [CrossRef]
- Goedert, M.; Jakes, R.; Crowther, R.; Six, J.; Lübke, U.; Vandermeeren, M.; Cras, P.; Trojanowski, J.; Lee, V. The abnormal phosphorylation of tau protein at Ser-202 in Alzheimer disease recapitulates phosphorylation during development. Proc. Natl. Acad. Sci. USA 1993, 90, 5066–5070. [Google Scholar] [CrossRef] [Green Version]
- Goedert, M.; Jakes, R.; Vanmechelen, E. Monoclonal antibody AT8 recognises tau protein phosphorylated at both serine 202 and threonine 205. Neurosci. Lett. 1995, 189, 167–170. [Google Scholar] [CrossRef]
- Braak, H.; Thal, D.R.; Ghebremedhin, E.; Del Tredici, K. Stages of the pathologic process in Alzheimer disease: Age categories from 1 to 100 years. J. Neuropathol. Exp. Neurol. 2011, 70, 960–969. [Google Scholar] [CrossRef]
- Nelson, P.T.; Alafuzoff, I.; Bigio, E.H.; Bouras, C.; Braak, H.; Cairns, N.J.; Castellani, R.J.; Crain, B.J.; Davies, P.; Tredici, K.D. Correlation of Alzheimer disease neuropathologic changes with cognitive status: A review of the literature. J. Neuropathol. Exp. Neurol. 2012, 71, 362–381. [Google Scholar] [CrossRef] [PubMed]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ruiz-Gabarre, D.; Carnero-Espejo, A.; Ávila, J.; García-Escudero, V. What’s in a Gene? The Outstanding Diversity of MAPT. Cells 2022, 11, 840. https://doi.org/10.3390/cells11050840
Ruiz-Gabarre D, Carnero-Espejo A, Ávila J, García-Escudero V. What’s in a Gene? The Outstanding Diversity of MAPT. Cells. 2022; 11(5):840. https://doi.org/10.3390/cells11050840
Chicago/Turabian StyleRuiz-Gabarre, Daniel, Almudena Carnero-Espejo, Jesús Ávila, and Vega García-Escudero. 2022. "What’s in a Gene? The Outstanding Diversity of MAPT" Cells 11, no. 5: 840. https://doi.org/10.3390/cells11050840
APA StyleRuiz-Gabarre, D., Carnero-Espejo, A., Ávila, J., & García-Escudero, V. (2022). What’s in a Gene? The Outstanding Diversity of MAPT. Cells, 11(5), 840. https://doi.org/10.3390/cells11050840