
Stabilization of PIM Kinases in Hypoxia Is Mediated by the Deubiquitinase USP28


Abstract
:1. Introduction
2. Materials and Methods
2.1. Tissue Culture
2.2. Plasmids and Reagents
2.3. Quantitative Polymerase Chain Reaction (qPCR)
2.4. Western Blotting
2.5. Immunoprecipitation and Protein Degradation Assays
2.6. In Vivo Experiments
2.7. Statistical Analysis
3. Results
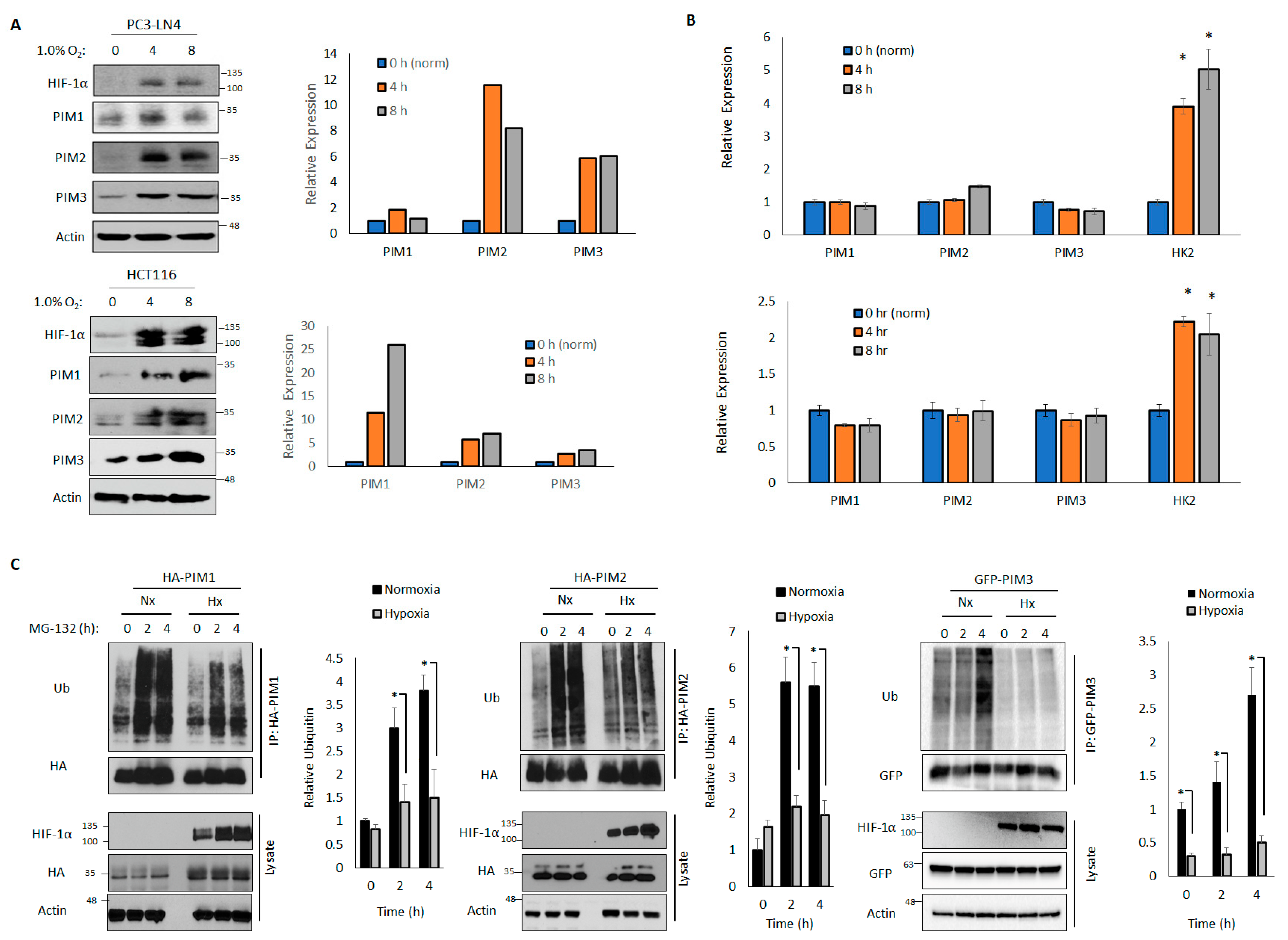
3.1. PIM Kinases Are Upregulated in Hypoxia at the Protein Level
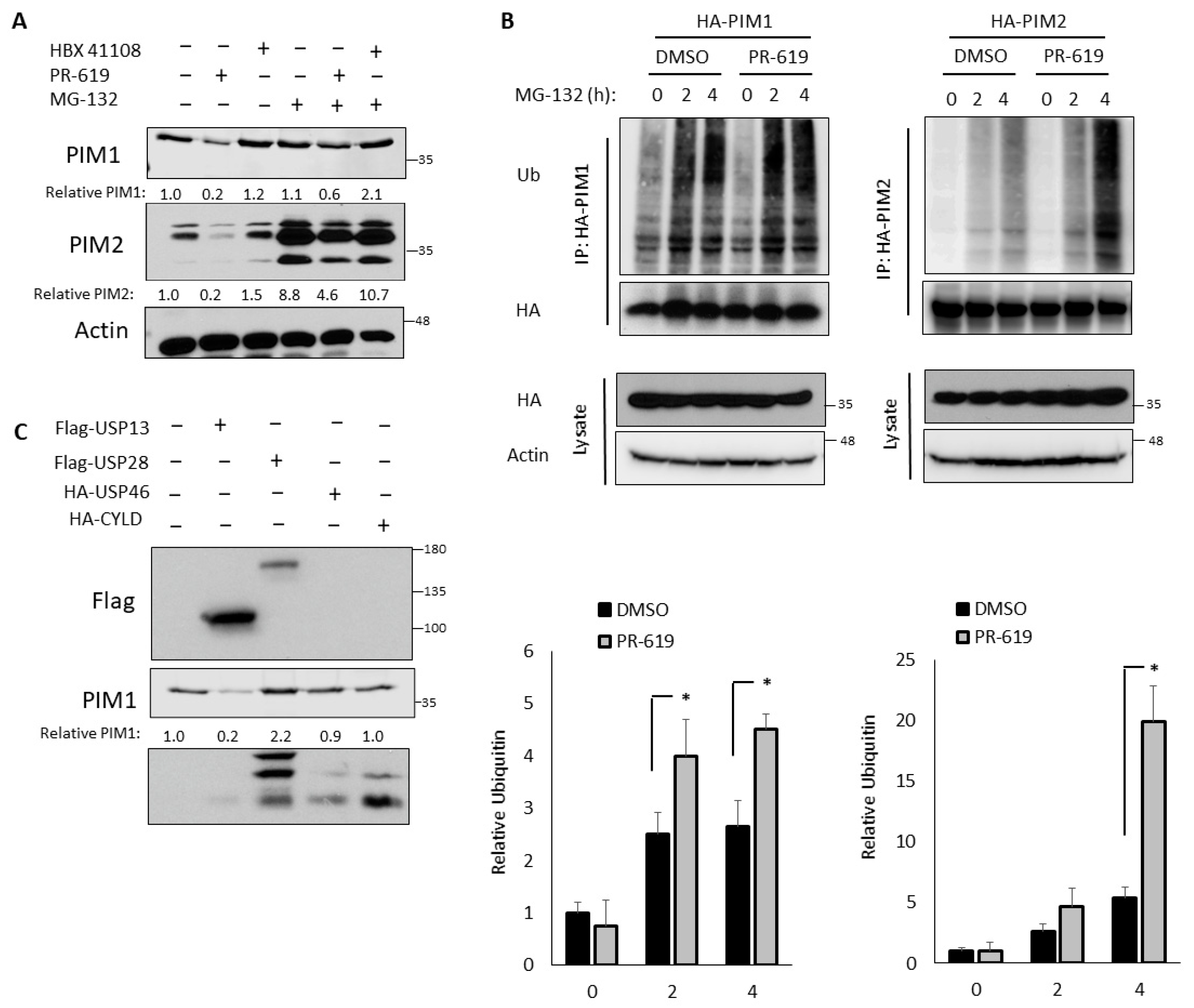
3.2. PIM Kinases Are Regulated by DUBs
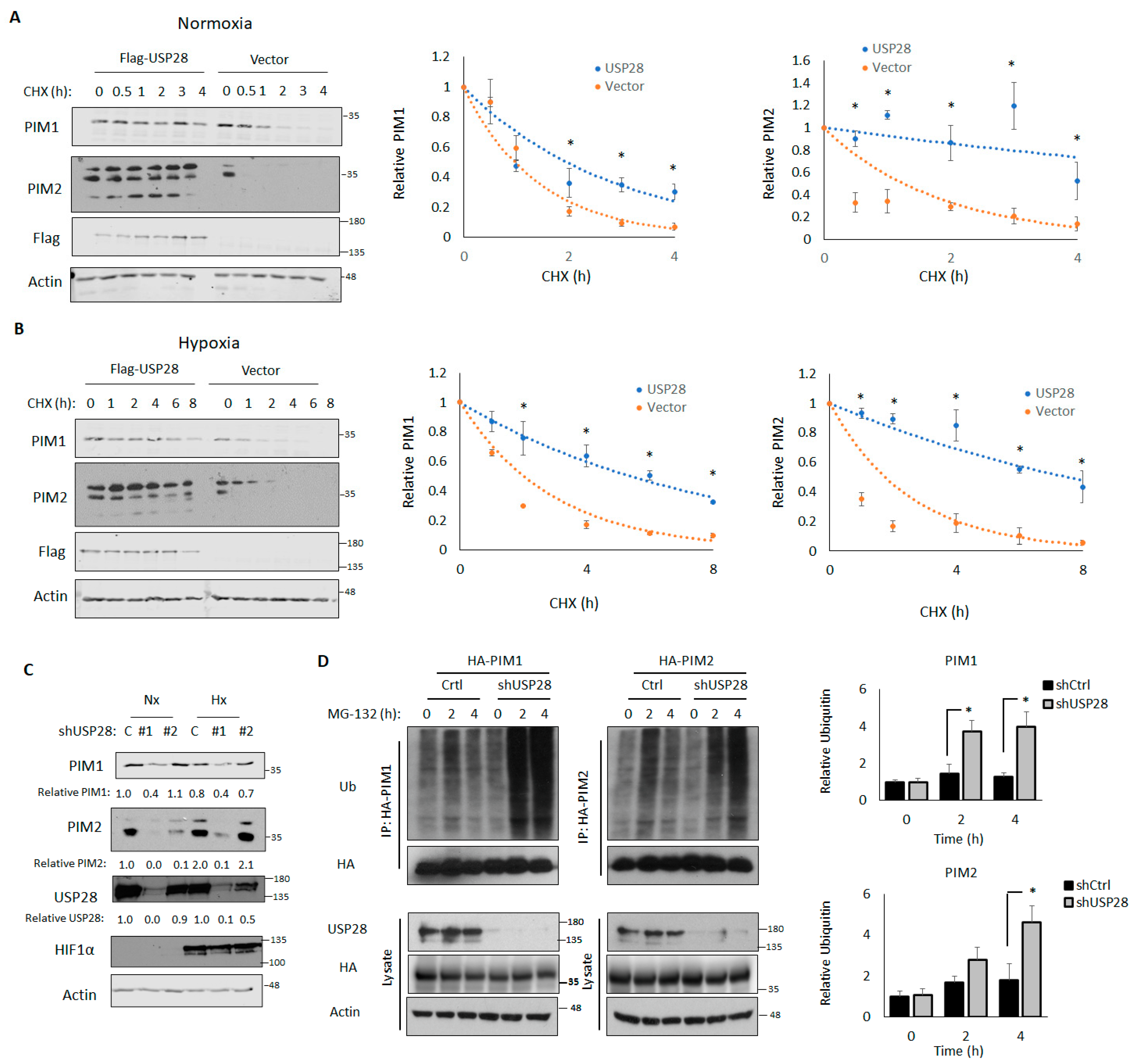
3.3. USP28 Increases PIM Stability
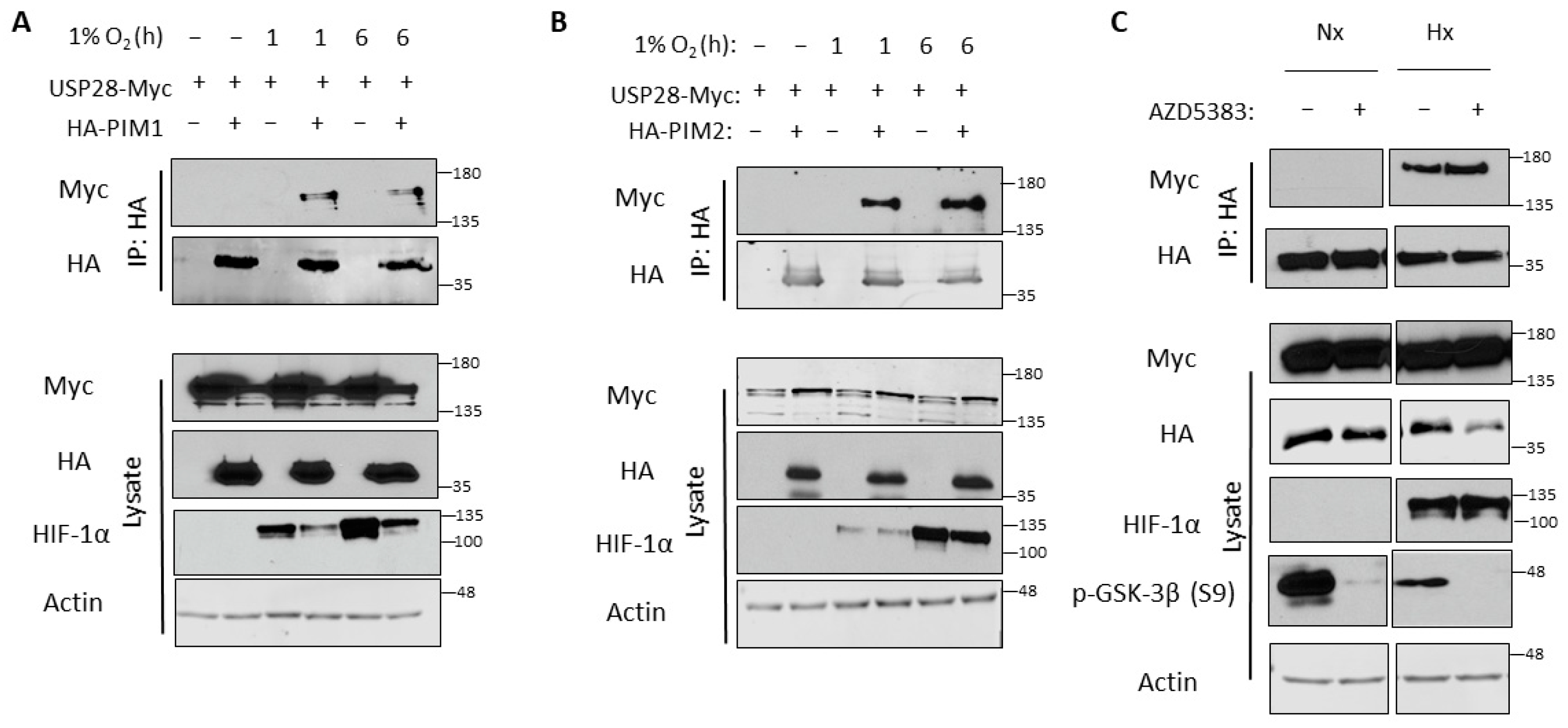
3.4. USP28 Interacts with PIM Kinases Preferentially in Hypoxia
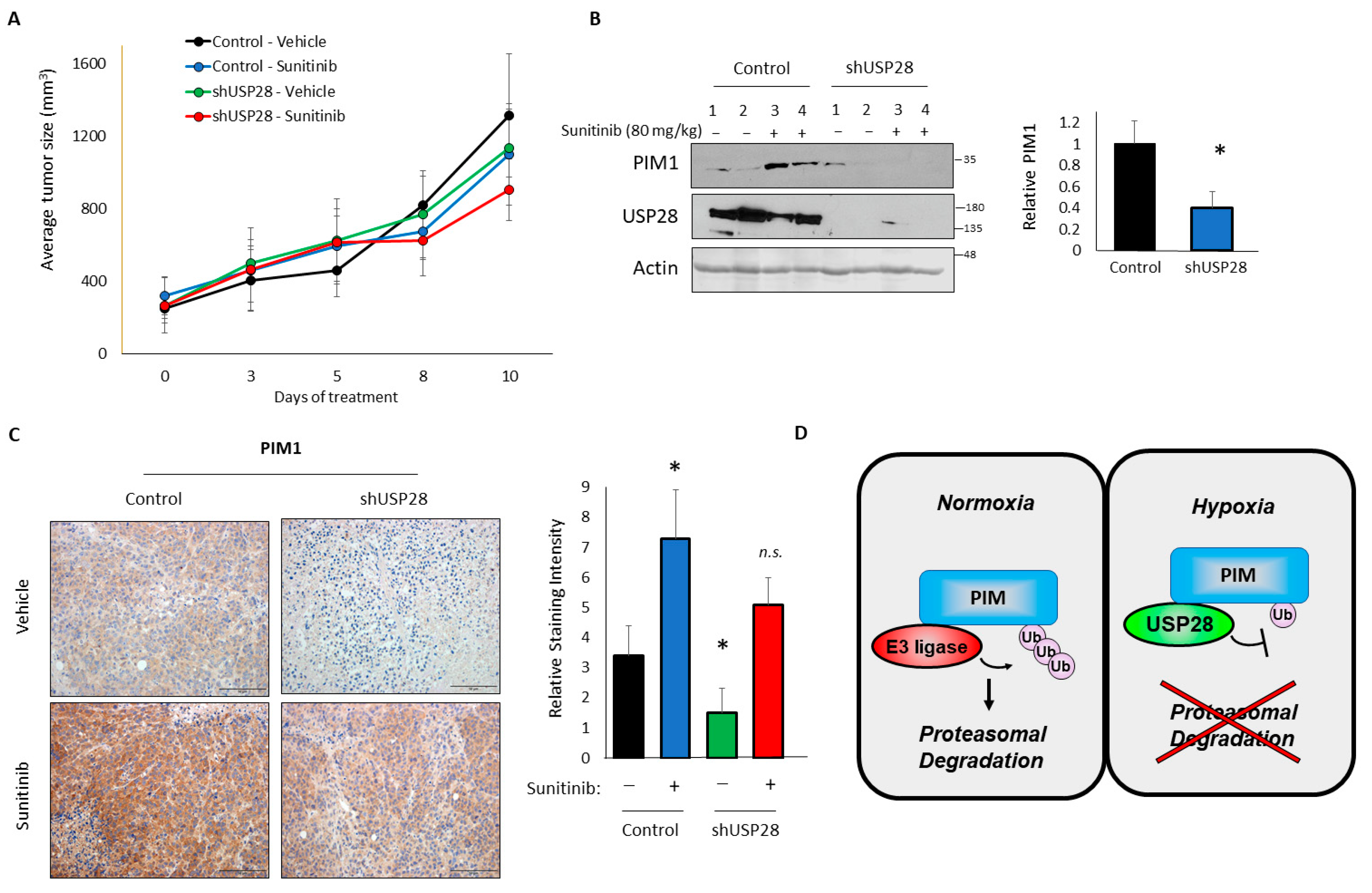
3.5. USP28 Regulates PIM Protein Levels In Vivo
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Semenza, G.L. Regulation of mammalian O2 homeostasis by hypoxia-inducible factor 1. Annu. Rev. Cell Dev. Biol. 1999, 15, 551–578. [Google Scholar] [CrossRef] [PubMed]
- Harris, A.L. Hypoxia—A key regulatory factor in tumour growth. Nat. Rev. Cancer 2002, 2, 38–47. [Google Scholar] [CrossRef] [PubMed]
- Bachmann, M.; Möröy, T. The serine/threonine kinase Pim-1. Int. J. Biochem. Cell Biol. 2005, 37, 726–730. [Google Scholar] [CrossRef] [PubMed]
- Cibull, T.L.; Jones, T.D.; Li, L.; Eble, J.N.; Baldridge, L.A.; Malott, S.R.; Luo, Y.; Cheng, L. Overexpression of Pim-1 during progression of prostatic adenocarcinoma. J. Clin. Pathol. 2006, 59, 285–288. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nawijn, M.C.; Alendar, A.; Berns, A. For better or for worse: The role of Pim oncogenes in tumorigenesis. Nat. Rev. Cancer 2011, 11, 23–34. [Google Scholar] [CrossRef] [PubMed]
- Peltola, K.; Hollmen, M.; Maula, S.M.; Rainio, E.; Ristamäki, R.; Luukkaa, M.; Sandholm, J.; Sundvall, M.; Elenius, K.; Koskinen, P.J.; et al. Pim-1 kinase expression predicts radiation response in squamocellular carcinoma of head and neck and is under the control of epidermal growth factor receptor. Neoplasia 2009, 11, 629–636. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xue, C.; He, Y.; Hu, Q.; Yu, Y.; Chen, X.; Chen, J.; Ren, F.; Li, J.; Ren, Z.; Cui, G.; et al. Downregulation of PIM1 regulates glycolysis and suppresses tumor progression in gallbladder cancer. Cancer Manag. Res. 2018, 10, 5101–5112. [Google Scholar] [CrossRef] [Green Version]
- Aho, T.L.T.; Sandholm, J.; Peltola, K.J.; Mankonen, H.P.; Lilly, M.; Koskinen, P.J. Pim-1 kinase promotes inactivation of the pro-apoptotic Bad protein by phosphorylating it on the Ser112 gatekeeper site. FEBS Lett. 2004, 571, 43–49. [Google Scholar] [CrossRef] [Green Version]
- Yan, B.; Zemskova, M.; Holder, S.; Chin, V.; Kraft, A.; Koskinen, P.J.; Lilly, M. The PIM-2 kinase phosphorylates BAD on serine 112 and reverses BAD-induced cell death. J. Biol. Chem. 2003, 278, 45358–45367. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Macdonald, A.; Campbell, D.G.; Toth, R.; McLauchlan, H.; Hastie, C.J.; Arthur, J.S.C. Pim kinases phosphorylate multiple sites in Bad and promote 14-3-3 binding and dissociation from Bcl-XL. BMC Cell Biol. 2006, 7, 1. [Google Scholar] [CrossRef] [Green Version]
- Warfel, N.A.; Sainz, A.G.; Song, J.H.; Kraft, A.S. PIM Kinase inhibitors kill hypoxic tumor cells by reducing Nrf2 signaling and increasing reactive oxygen species. Mol. Cancer Ther. 2016, 15, 1637–1647. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chauhan, S.S.; Toth, R.K.; Jensen, C.C.; Casillas, A.L.; Kashatus, D.F.; Warfel, N.A. PIM kinases alter mitochondrial dynamics and chemosensitivity in lung cancer. Oncogene 2020, 39, 2597–2611. [Google Scholar] [CrossRef] [PubMed]
- Beharry, Z.; Mahajan, S.; Zemskova, M.; Lin, Y.W.; Tholanikunnel, B.G.; Xia, Z.; Smith, C.D.; Kraft, A.S. The Pim protein kinases regulate energy metabolism and cell growth. Proc. Natl. Acad. Sci. USA 2011, 108, 528–533. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Casillas, A.L.; Toth, R.K.; Sainz, A.G.; Singh, N.; Desai, A.A.; Kraft, A.S.; Warfel, N.A. Hypoxia-inducible PIM kinase expression promotes resistance to antiangiogenic agents. Clin. Cancer Res. 2018, 24, 169–180. [Google Scholar] [CrossRef] [Green Version]
- Szydłowski, M.; Prochorec-Sobieszek, M.; Szumera-Ciećkiewicz, A.; Derezińska, E.; Hoser, G.; Wasilewska, D.; Szymańska-Giemza, O.; Jabłońska, E.; Białopiotrowicz, E.; Sewastianik, T.; et al. Expression of PIM kinases in Reed-Sternberg cells fosters immune privilege and tumor cell survival in Hodgkin lymphoma. Blood 2017, 130, 1418–1429. [Google Scholar] [CrossRef] [Green Version]
- Santio, N.M.; Vahakoski, R.L.; Rainio, E.M.; Sandholm, J.A.; Virtanen, S.S.; Prudhomme, M.; Anizon, F.; Moreau, P.; Koskinen, P.J. Pim-selective inhibitor DHPCC-9 reveals Pim kinases as potent stimulators of cancer cell migration and invasion. Mol. Cancer 2010, 9, 279. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qian, K.C.; Wang, L.; Hickey, E.R.; Studts, J.; Barringer, K.; Peng, C.; Kronkaitis, A.; Li, J.; White, A.; Mische, S.; et al. Structural basis of constitutive activity and a unique nucleotide binding mode of human Pim-1 kinase. J. Biol. Chem. 2005, 280, 6130–6137. [Google Scholar] [CrossRef] [Green Version]
- Kim, O.; Jiang, T.; Xie, Y.; Guo, Z.; Chen, H.; Qiu, Y. Synergism of cytoplasmic kinases in IL6-induced ligand-independent activation of androgen receptor in prostate cancer cells. Oncogene 2004, 23, 1838–1844. [Google Scholar] [CrossRef] [Green Version]
- Bullock, A.N.; Debreczeni, J.; Amos, A.L.; Knapp, S.; Turk, B.E. Structure and substrate specificity of the Pim-1 kinase. J. Biol. Chem. 2005, 280, 41675–41682. [Google Scholar] [CrossRef] [Green Version]
- Wernig, G.; Gonneville, J.R.; Crowley, B.J.; Rodrigues, M.S.; Reddy, M.M.; Hudon, H.E.; Walz, C.; Reiter, A.; Podar, K.; Royer, Y.; et al. The Jak2V617F oncogene associated with myeloproliferative diseases requires a functional FERM domain for transformation and for expression of the Myc and Pim proto-oncogenes. Blood 2008, 111, 3751–3759. [Google Scholar] [CrossRef] [Green Version]
- Zhu, N.; Ramirez, L.M.; Lee, R.L.; Magnuson, N.S.; Bishop, G.A.; Gold, M.R. CD40 Signaling in B Cells Regulates the Expression of the Pim-1 Kinase Via the NF-κB Pathway. J. Immunol. 2002, 168, 744–754. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saris, C.J.M.; Domen, J.; Berns, A. The pim-1 oncogene encodes two related protein-serine/threonine kinases by alternative initiation at AUG and CUG. EMBO J. 1991, 10, 655–664. [Google Scholar] [CrossRef] [PubMed]
- Weirauch, U.; Beckmann, N.; Thomas, M.; Grünweller, A.; Huber, K.; Bracher, F.; Hartmann, R.K.; Aigner, A. Functional role and therapeutic potential of the Pim-1 kinase in colon carcinoma. Neoplasia (U. S.) 2013, 15, 783–794. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thomas, M.; Lange-Grünweller, K.; Weirauch, U.; Gutsch, D.; Aigner, A.; Grünweller, A.; Hartmann, R.K. The proto-oncogene Pim-1 is a target of miR-33a. Oncogene 2012, 31, 918–928. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liang, C.; Yu, X.J.; Guo, X.Z.; Sun, M.H.; Wang, Z.; Song, Y.; Ni, Q.X.; Li, H.Y.; Mukaida, N.; Li, Y.Y. MicroRNA-33a-mediated downregulation of Pim-3 kinase expression renders human pancreatic cancer cells sensitivity to gemcitabine. Oncotarget 2015, 6, 14440–14455. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mizuno, K.; Shirogane, T.; Shinohara, A.; Iwamatsu, A.; Hibi, M.; Hirano, T. Regulation of Pim-1 by Hsp90. Biochem. Biophys. Res. Commun. 2001, 281, 663–669. [Google Scholar] [CrossRef] [PubMed]
- Losman, J.A.; Chen, X.P.; Vuong, B.Q.; Fay, S.; Rothman, P.B. Protein phosphatase 2A regulates the stability of Pim protein kinases. J. Biol. Chem. 2003, 278, 4800–4805. [Google Scholar] [CrossRef] [Green Version]
- Shay, K.P.; Wang, Z.; Xing, P.X.; McKenzie, I.F.C.; Magnuson, N.S. Pim-1 kinase stability is regulated by heat shock proteins and the ubiquitin-proteasome pathway. Mol. Cancer Res. 2005, 3, 170–181. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.; Kobayashi, M.; Darmanin, S.; Qiao, Y.; Gully, C.; Zhao, R.; Yeung, S.C.; Lee, M.H. Pim-1 plays a pivotal role in hypoxia-induced chemoresistance. Oncogene 2009, 28, 2581–2592. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, J.; Kobayashi, M.; Darmanin, S.; Qiao, Y.; Gully, C.; Zhao, R.; Kondo, S.; Wang, H.; Wang, H.; Yeung, S.C.J.; et al. Hypoxia-mediated up-regulation of pim-1 contributes to solid tumor formation. Am. J. Pathol. 2009, 175, 400–411. [Google Scholar] [CrossRef] [Green Version]
- Nijman, S.M.B.; Luna-Vargas, M.P.A.; Velds, A.; Brummelkamp, T.R.; Dirac, A.M.G.; Sixma, T.K.; Bernards, R. A Genomic and Functional Inventory of Deubiquitinating Enzymes. Cell 2005, 123, 773–786. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mennerich, D.; Kubaichuk, K.; Kietzmann, T. DUBs, Hypoxia, and Cancer. Trends Cancer 2019, 5, 632–653. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Flügel, D.; Görlach, A.; Kietzmann, T. GSK-3β regulates cell growth, migration, and angiogenesis via Fbw7 and USP28-dependent degradation of HIF-1α. Blood 2012, 119, 1292–1301. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.; Liu, Z.; Zhang, L.; Yang, Z.; Chen, X.; Luo, J.; Zhou, Z.; Mei, X.; Yu, X.; Shao, Z.; et al. Targeting deubiquitinase USP28 for cancer therapy. Cell Death Dis. 2018, 9, 1–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, D.; Zaugg, K.; Mak, T.W.; Elledge, S.J. A Role for the Deubiquitinating Enzyme USP28 in Control of the DNA-Damage Response. Cell 2006, 126, 529–542. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lambrus, B.G.; Daggubati, V.; Uetake, Y.; Scott, P.M.; Clutario, K.M.; Sluder, G.; Holland, A.J. A USP28-53BP1-p53-p21 signaling axis arrests growth after centrosome loss or prolonged mitosis. J. Cell Biol. 2016, 214, 143–153. [Google Scholar] [CrossRef]
- Fong, C.S.; Mazo, G.; Das, T.; Goodman, J.; Kim, M.; O’Rourke, B.P.; Izquierdo, D.; Tsou, M.F.B. 53BP1 and USP28 mediate p53- dependent cell cycle arrest in response to centrosome loss and prolonged mitosis. Elife 2016, 5, e16270. [Google Scholar] [CrossRef]
- Popov, N.; Wanzel, M.; Madiredjo, M.; Zhang, D.; Beijersbergen, R.; Bernards, R.; Moll, R.; Elledge, S.J.; Eilers, M. The ubiquitin-specific protease USP28 is required for MYC stability. Nat. Cell Biol. 2007, 9, 765–774. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Roh, M.; Abdulkadir, S.A. Pim1 promotes human prostate cancer cell tumorigenicity and c-MYC transcriptional activity. BMC Cancer 2010, 10, 248. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Kim, J.; Roh, M.; Franco, O.E.; Hayward, S.W.; Wills, M.L.; Abdulkadir, S.A. Pim1 kinase synergizes with c-MYC to induce advanced prostate carcinoma. Oncogene 2010, 29, 2477–2487. [Google Scholar] [CrossRef] [Green Version]
- Pettaway, C.A.; Pathak, S.; Greene, G.; Ramirez, E.; Wilson, M.R.; Killion, J.J.; Fidler, I.J. Selection of highly metastatic variants of different human prostatic carcinomas using orthotopic implantation in nude mice. Clin. Cancer Res. 1996, 2, 1627–1636. [Google Scholar] [PubMed]
- Chen, Q.; Wang, Y.; Shi, S.; Li, K.; Zhang, L.; Gao, J. Insights into the Interaction Mechanisms of the Proviral Integration Site of Moloney Murine Leukemia Virus (Pim) Kinases with Pan-Pim Inhibitors PIM447 and AZD1208: A Molecular Dynamics Simulation and MM/GBSA Calculation Study. Int. J. Mol. Sci. 2019, 20, 5410. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saurabh, K.; Scherzer, M.T.; Shah, P.P.; Mims, A.S.; Lockwood, W.W.; Kraft, A.S.; Beverly, L.J. The PIM family of oncoproteins: Small kinases with huge implications in myeloid leukemogenesis and as therapeutic targets. Oncotarget 2014, 5, 8503. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ritorto, M.S.; Ewan, R.; Perez-Oliva, A.B.; Knebel, A.; Buhrlage, S.J.; Wightman, M.; Kelly, S.M.; Wood, N.T.; Virdee, S.; Gray, N.S.; et al. Screening of DUB activity and specificity by MALDI-TOF mass spectrometry. Nat. Commun. 2014, 5, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Welcker, M.; Clurman, B.E. FBW7 ubiquitin ligase: A tumour suppressor at the crossroads of cell division, growth and differentiation. Nat. Rev. Cancer 2008, 8, 83–93. [Google Scholar] [CrossRef]
- Sampson, N.; Untergasser, G.; Plas, E.; Berger, P. The ageing male reproductive tract. J. Pathol. 2007, 211, 206–218. [Google Scholar] [CrossRef]
- Guzy, R.D.; Schumacker, P.T. Oxygen sensing by mitochondria at complex III: The paradox of increased reactive oxygen species during hypoxia. Exp. Physiol. 2006, 91, 807–819. [Google Scholar] [CrossRef]
- Belanger, A.J.; Lu, H.; Date, T.; Liu, L.X.; Vincent, K.A.; Akita, G.Y.; Cheng, S.H.; Gregory, R.J.; Jiang, C. Hypoxia Up-regulates Expression of Peroxisome Proliferator-activated Receptor γ Angiopoietin-related Gene (PGAR) in Cardiomyocytes: Role of Hypoxia Inducible Factor 1α. J. Mol. Cell. Cardiol. 2002, 34, 765–774. [Google Scholar] [CrossRef]
- Forsythe, J.A.; Jiang, B.H.; Iyer, N.V.; Agani, F.; Leung, S.W.; Koos, R.D.; Semenza, G.L. Activation of vascular endothelial growth factor gene transcription by hypoxia-inducible factor 1. Mol. Cell. Biol. 1996, 16, 4604–4613. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.; Gao, P.; Liu, Y.-C.; Semenza, G.L.; Dang, C.V. Hypoxia-inducible factor 1 and dysregulated c-Myc cooperatively induce vascular endothelial growth factor and metabolic switches hexokinase 2 and pyruvate dehydrogenase kinase 1. Mol. Cell. Biol. 2007, 27, 7381–7393. [Google Scholar] [CrossRef] [Green Version]
- Komatsu, D.E.; Hadjiargyrou, M. Activation of the transcription factor HIF-1 and its target genes, VEGF, HO-1, iNOS, during fracture repair. Bone 2004, 34, 680–688. [Google Scholar] [CrossRef] [PubMed]
- Du, S.C.; Zhu, L.; Wang, Y.X.; Liu, J.; Zhang, D.; Chen, Y.L.; Peng, Q.; Liu, W.; Liu, B. SENP1-mediated deSUMOylation of USP28 regulated HIF-1α accumulation and activation during hypoxia response 06 Biological Sciences 0601 Biochemistry and Cell Biology. Cancer Cell Int. 2019, 19, 1–8. [Google Scholar] [CrossRef]
- Fresno Vara, J.Á.; Casado, E.; de Castro, J.; Cejas, P.; Belda-Iniesta, C.; González-Barón, M. PI3K/Akt signalling pathway and cancer. Cancer Treat. Rev. 2004, 30, 193–204. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Huang, H.; Feng, X.; Song, H.; Zhang, Z.; Shen, A.; Qiu, X. Deubiquitinase USP28 inhibits ubiquitin ligase KLHL2-mediated uridine-cytidine kinase 1 degradation and confers sensitivity to 5′-azacytidine-resistant human leukemia cells. Theranostics 2020, 10, 1046–1059. [Google Scholar] [CrossRef]
- Bohgaki, M.; Hakem, A.; Halaby, M.J.; Bohgaki, T.; Li, Q.; Bissey, P.A.; Shloush, J.; Kislinger, T.; Sanchez, O.; Sheng, Y.; et al. The E3 ligase PIRH2 polyubiquitylates CHK2 and regulates its turnover. Cell Death Differ. 2013, 20, 812–822. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schumacher, F.R.; Sorrell, F.J.; Alessi, D.R.; Bullock, A.N.; Kurz, T. Structural and biochemical characterization of the KLHL3-WNK kinase interaction important in blood pressure regulation. Biochem. J. 2014, 460, 237–246. [Google Scholar] [CrossRef] [Green Version]
- Renard, S.; Paulin, R.; Breuils-Bonnet, S.; Simard, S.; Pibarot, P.; Bonnet, S.; Provencher, S. Pim-1: A new biomarker in pulmonary arterial hypertension. Pulm. Circ. 2013, 3, 74. [Google Scholar] [CrossRef] [Green Version]
- Lin, Y.; Lu, Z.; Kokontis, J.; Xiang, J. Androgen receptor primes prostate cancer cells to apoptosis through down-regulation of basal p21 expression. Biochem. Biophys. Res. Commun. 2013, 430, 289–293. [Google Scholar] [CrossRef] [Green Version]
- Leng, R.P.; Lin, Y.; Ma, W.; Wu, H.; Lemmers, B.; Chung, S.; Parant, J.M.; Lozano, G.; Hakem, R.; Benchimol, S. Pirh2, a p53-Induced Ubiquitin-Protein Ligase, Promotes p53 Degradation. Cell 2003, 112, 779–791. [Google Scholar] [CrossRef] [Green Version]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Target | Primer Sequence |
|---|---|
| PIM1 | CGACATCAAGGACGAAAACATC ACTCTGGAGGGCTATACACTC |
| PIM2 | GAACATCCTGATAGACCTACGC CATGGTACTGGTGTCGAGAG |
| PIM3 | GACATCCCCTTCGAGCAG ATGGGCCGCAATCTGATC |
| HK2 | Purchased from Qiagen (catalog # QT00013209) |
| ACTB | TGACGTGGACATCCGCAAAG CTGGAAGGTGGACAGCGAGG |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Toth, R.K.; Solomon, R.; Warfel, N.A. Stabilization of PIM Kinases in Hypoxia Is Mediated by the Deubiquitinase USP28. Cells 2022, 11, 1006. https://doi.org/10.3390/cells11061006
Toth RK, Solomon R, Warfel NA. Stabilization of PIM Kinases in Hypoxia Is Mediated by the Deubiquitinase USP28. Cells. 2022; 11(6):1006. https://doi.org/10.3390/cells11061006
Chicago/Turabian StyleToth, Rachel K., Regina Solomon, and Noel A. Warfel. 2022. "Stabilization of PIM Kinases in Hypoxia Is Mediated by the Deubiquitinase USP28" Cells 11, no. 6: 1006. https://doi.org/10.3390/cells11061006
APA StyleToth, R. K., Solomon, R., & Warfel, N. A. (2022). Stabilization of PIM Kinases in Hypoxia Is Mediated by the Deubiquitinase USP28. Cells, 11(6), 1006. https://doi.org/10.3390/cells11061006