
Retrospective Analysis of Autologous Chondrocyte-Based Cytotherapy Production for Clinical Use: GMP Process-Based Manufacturing Optimization in a Swiss University Hospital

 ,
,  ,
,  and
and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials Available for Retrospective Analysis and Ethical Compliance of the Study
2.1.1. Clinical Trial Orthopedic Patient Files and Related GMP Manufacturing Records Analyses
2.1.2. Ethical, Regulatory, and Clinical Protocols of the ACI CHUV Clinical Trial
- Patient age > 15 years and <50 years of age.
- Presence of symptomatic focal chondral and osteochondral defects of traumatic origin, grades III and IV of the defects according to ICRS classifications, and defect size < 15 cm2.
- The lesion may result from a failure of autologous osteochondral transplantation (i.e., mosaicplasty) or of microfractures.
- Presence of an adequate biomechanical environment (i.e., ligamentary stability, preserved or restored meniscus, neutral axial mechanical axis).
- Patient in good overall health, documented by an ASA score ≤ 2.
- Patient assessed as compliant and as capable of participating in pre/post-operative follow-up and reeducation.
- Patient consent for participation in the study.
- Procedure covered by basic health insurance or by accident insurance.
- Patient non-responsive to conservative treatment (>6 months).
- All degenerative inflammatory pathologies and synovial pathologies (e.g., arthritis).
- Diffuse chondral lesions, of traumatic or non-traumatic nature (e.g., gonarthrotic).
- Non-favorable biomechanical environment (e.g., subtotal or total meniscectomy in the same compartment, ligamentary instability, deviation of the mechanical axis leading to an overload of the treated compartment).
- Qualified obesity of grade ≥ 2, with a body-mass index value > 35 kg/m2.
- Active tobacco product consumption habit.
- Consumption of hard recreational drugs.
- Bad compliance of the patient.
- Current participation in an alternative clinical trial.
- Compromised overall patient health, documented by an ASA score ≥ 3.
- Vulnerable populations.
- Active or planned pregnancy.
- Qualified allergy to porcine collagen, penicillin, or to gentamycin.
- Qualified seropositivity for HIV, HBV, HCV, or for Treponema pallidum (i.e., assessed by serological testing before biopsy harvest).
- Presence of growth cartilage (i.e., presence of an open epiphyseal growth plate) in adolescents 15–18 years of age.
2.2. Original Data on Primary HAC Sourcing, Manufacturing, and Formulation
2.2.1. Biological Starting Material Procurement and Processing for In Vitro Cell Isolation
2.2.2. Initial Cellular API Manufacturing Process Optimization and Validation Steps
2.2.3. Cellular API GMP Manufacturing and Controls for the CHUV ACI Clinical Trial
2.2.4. Cellular Finished Product Manufacturing and Controls
2.3. Establishment of Optimized and Parametric Technical Workflows for HAC-Based API and Finished Product GMP Manufacture
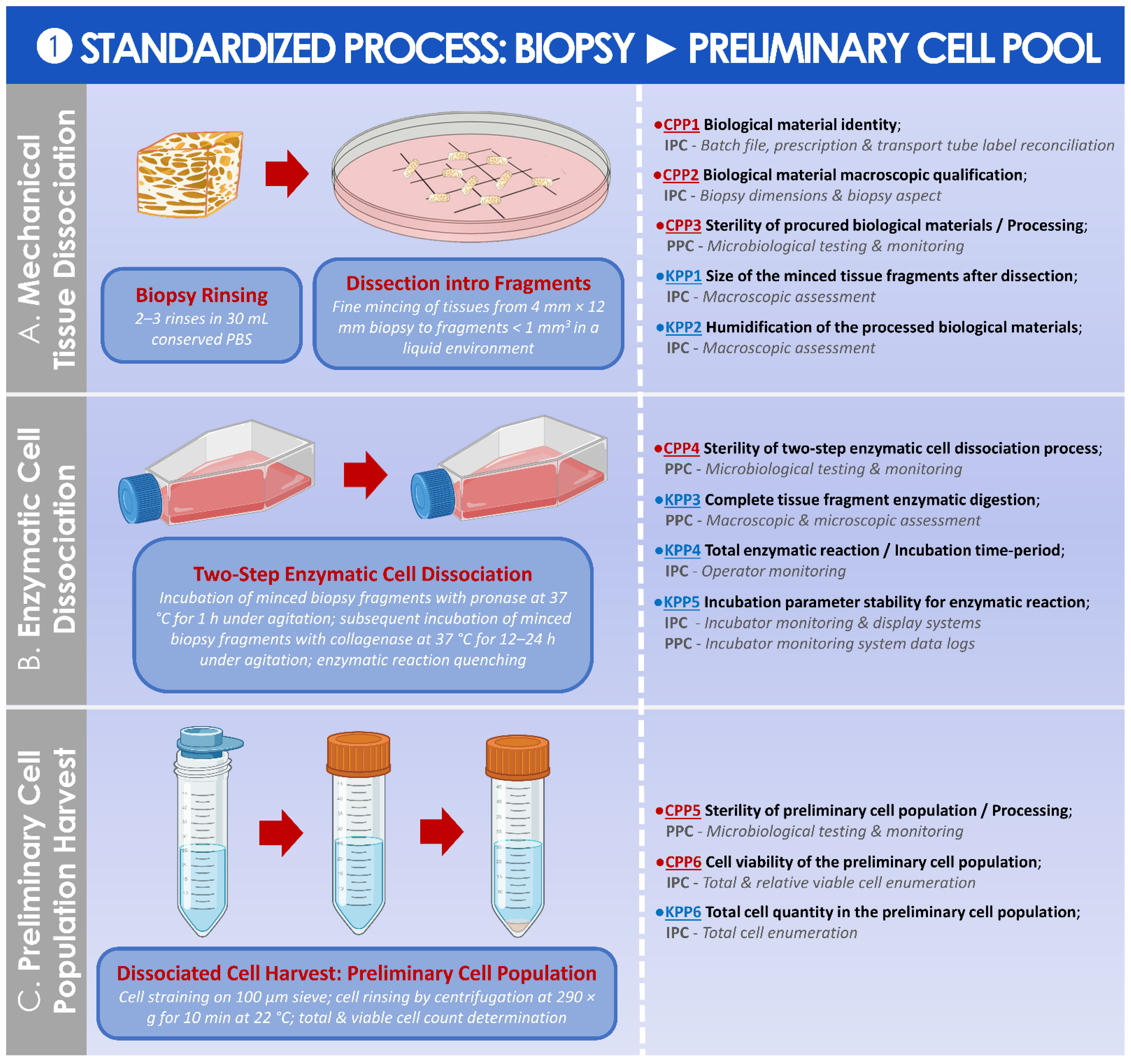
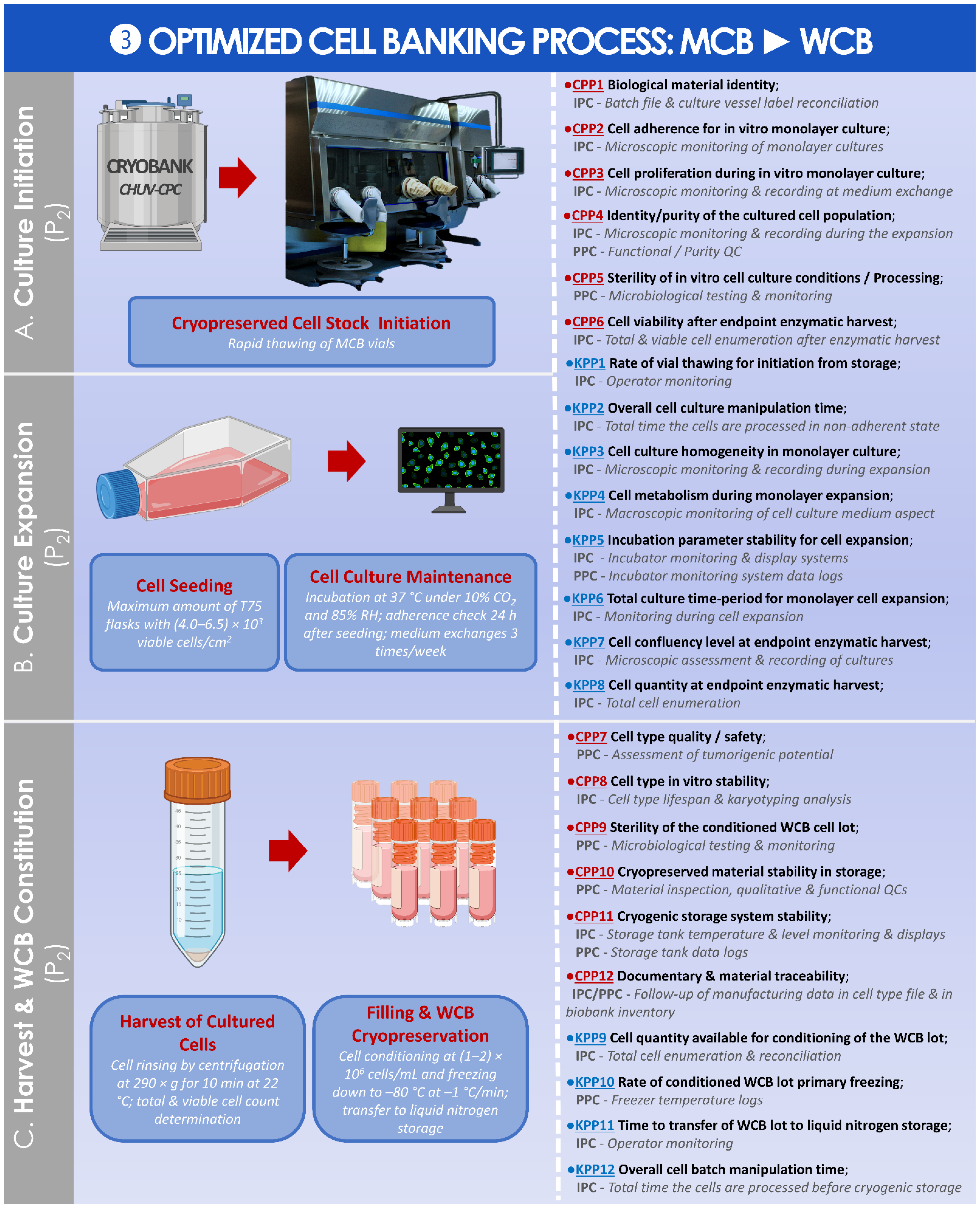
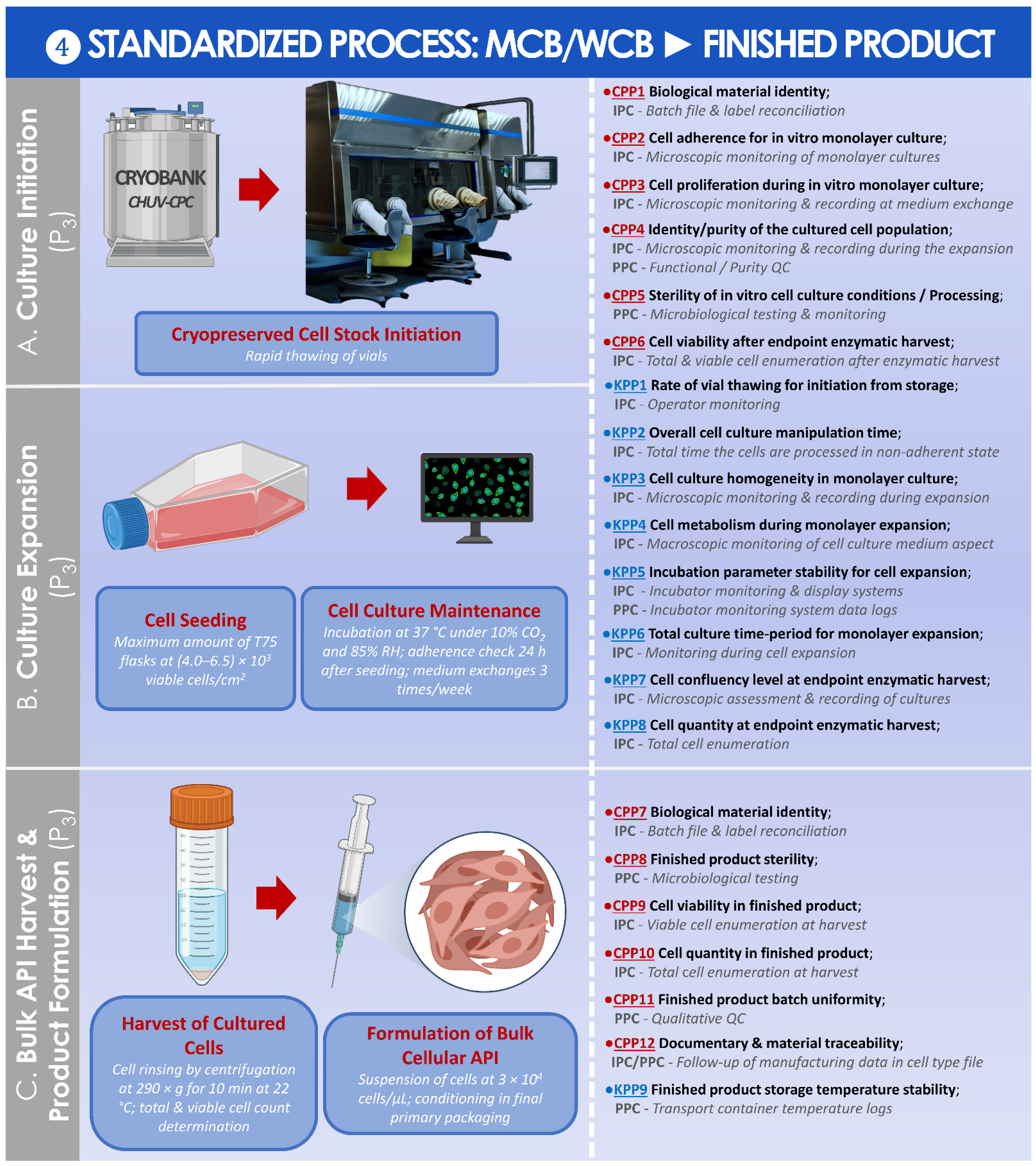
2.3.1. Risk Analysis-Based Process Approach for Parametric Definition of the Process, including Controls and Criteria
2.3.2. Synthetic Establishment of the Optimized Parametric Process Workflows
2.4. Numerical Data Processing, Statistical Analysis of Data, and Data Presentation
3. Results
3.1. Results of Compilation and Data Analysis for Clinical Workflows, Patient Files, and GMP Manufacturing Records
3.2. Results of the Standardized, Risk Analysis-Based, and Parametric Process Definition
4. Discussion
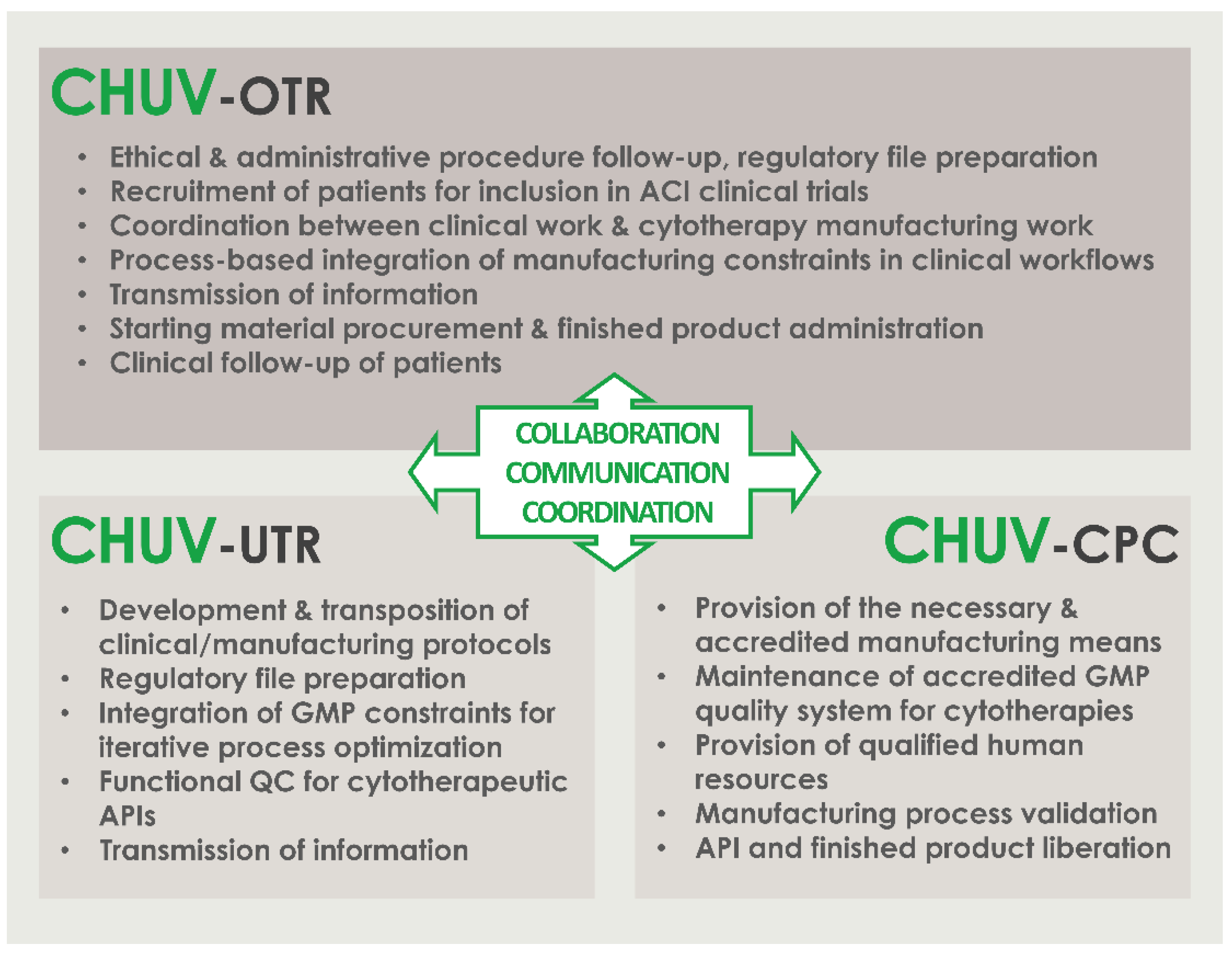
4.1. Critical Importance of Interdisciplinary Collaboration, Communication, and Coordination of Professional Stakeholders for the Successful Clinical Implementation of HAC-Based ATMPs/ATIMPs
- Communicate; establish clear, precise, and traceable transmission of information and data between all units for the appropriate meeting of general and specific clinical needs; establish regular exchanges for iterative assessments and optimization of process quality.
- Compliance; regularly assess the continued availability of accredited manufacturing means, the continued compliance of all activities with applicable institutional/legal frameworks and clinical trial authorizations, and the continued monitoring of ethical compliance with defined protocols.
- Clarify; establish clear roles and responsibilities of the involved personnel and units; identify individual responsibilities at each step of the considered processes.
- Collaborate; mutualize resources for an enhanced detection of risks and provision of efficient solutions; collaboration of research and clinical units for understanding of clinical needs and provision through development of adequate solutions; collaboration of research and GMP manufacturing units for transposition of the developed processes in response to clinical needs; collaboration of GMP manufacturing and clinical units for meeting of individual patient needs and clinician requirements.
- Coordinate; coordinate activities between GMP manufacturing and clinical units for meeting of clinician expectations and patient needs; continually seek to identify potential process gaps to be corrected by complementary responsibility attribution.
- Control; iterative and step-wise verification of information comprehension following communication between the stakeholders; verification of resulting action performance.
- Check; validation and revalidation of the processes after technical specification updates or material changes; regular reassessment of the entire process for verification of the adequation between objectives and available data/records/results.
4.2. High Inter-Patient Variability: Standardized Manufacturing Processes for Patient-Specific Cytotherapies
4.3. Confirmation That the Use of hPL Is Appropriate for Primary HAC Culture in Clinical ACI Applications
4.4. Possibility to Optimize HAC Cell Banking Strategies for Enhanced Material Sustainability: Multi-Tiered Primary Cell Banking
4.5. Advantages of Risk Analysis-Based and Quality-Oriented GMP Manufacturing Approaches for HAC-Based Therapeutic Products in a Public Hospital Setting
4.6. Current GMP Manufacturing Process Limitations: The Open Question of HAC Cell Population Purity
4.7. Technical and Clinical Future Directions: Cell Dose Considerations and Use of a Cell Scaffold for HAC-Based Therapeutic ACI in the CHUV
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| ACI | autologous chondrocyte implantation |
| aHS API | autologous human serum active pharmaceutical ingredient |
| ASA | American Society of Anesthesiologists |
| ATIMP | advanced therapy investigational medicinal product |
| ATMP | advanced therapy medicinal product |
| cATMP | combined advanced therapy medicinal product |
| cGMP | current good manufacturing practices |
| CH | Helvetic Confederation |
| CHUV | centre hospitalier universitaire vaudois |
| CPC | cell production center |
| CPP | critical process parameter |
| CPR | Plastic and Reconstructive Surgery Service |
| CQA | critical quality attribute |
| DMSO | dimethyl sulfoxide |
| DNA | deoxyribonucleic acid |
| EC | European Commission |
| EDTA | ethylenediaminetetraacetic acid |
| EMA | European Medicines Agency |
| EU | European Union |
| FBS | fetal bovine serum |
| FDA | US Food and Drug Administration |
| GMP | good manufacturing practices |
| HAC | human articular chondrocytes |
| HCV | hepatitis C virus |
| HIV | human immunodeficiency virus |
| ICRS | International Cartilage Regeneration and Joint Preservation Society |
| IMPD | investigational medicinal product dossier |
| IPC | in-process control |
| KPP | key process parameter |
| KQA | key quality attribute |
| MCB | master cell bank |
| OTR | Orthopedics and Traumatology Service |
| Ph. Eur. | European pharmacopoeia |
| PPC | post-process control |
| PRP | platelet-rich plasma |
| QA | quality assurance |
| QC | quality control |
| RAM | risk analysis matrix |
| RH | relative humidity |
| RT-PCR | real-time polymerase chain reaction |
| TrSt | standardized transplant product |
| USA | United States of America |
| UTR | Regenerative Therapy Unit |
| WCB | working cell bank |
References
- Lechanteur, C.; Briquet, A.; Bettonville, V.; Baudoux, E.; Beguin, Y. MSC manufacturing for academic clinical trials: From a clinical-grade to a full GMP-compliant process. Cells 2021, 10, 1320. [Google Scholar] [CrossRef] [PubMed]
- Labedz-Maslowska, A.; Szkaradek, A.; Mirzwinski, T.; Madeja, Z.; Zuba-Surma, E. Processing and ex vivo expansion of adipose tissue-derived mesenchymal stem/stromal cells for the development of an advanced therapy medicinal product for use in humans. Cells 2021, 10, 1908. [Google Scholar] [CrossRef] [PubMed]
- Mansnérus, J. Encountering challenges with the EU regulation on Advance Therapy Medical Products. Eur. J. Health Law 2015, 22, 426–461. [Google Scholar] [CrossRef] [PubMed]
- De Wilde, S.; Veltrop-Duits, L.; Hoozemans-Strik, M.; Ras, T.; Blom-Veenman, J.; Guchelaar, H.-J.; Zandvliet, M.; Meij, P. Hurdles in clinical implementation of academic advanced therapy medicinal products: A national evaluation. Cytotherapy 2016, 18, 797–805. [Google Scholar] [CrossRef]
- Pearce, K.F.; Hildebrandt, M.; Greinix, H.; Scheding, S.; Koehl, U.; Worel, N.; Apperley, J.; Edinger, M.; Hauser, A.; Mischak-Weissinger, E.; et al. Regulation of advanced therapy medicinal products in Europe and the role of academia. Cytotherapy 2014, 16, 289–297. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johnson, P.C.; Bertram, T.A.; Tawil, B.; Hellman, K.B. Hurdles in tissue engineering/regenerative medicine product commercialization: A survey of North American academia and industry. Tissue Eng. Part A 2011, 17, 5–15. [Google Scholar] [CrossRef]
- Bertram, T.A.; Tentoff, E.; Johnson, P.C.; Tawil, B.; Van Dyke, M.; Hellman, K.B. Hurdles in tissue engineering/regenerative medicine product commercialization: A pilot survey of governmental funding agencies and the financial industry. Tissue Eng. Part A 2012, 18, 2187–2194. [Google Scholar] [CrossRef] [PubMed]
- Dimitropoulos, G.; Jafari, P.; de Buys Roessingh, A.; Hirt-Burri, N.; Raffoul, W.; Applegate, L.A. Burn patient care lost in good manufacturing practices? Ann. Burn. Fire Disasters 2016, 29, 111–115. [Google Scholar]
- Detela, G.; Lodge, A. EU regulatory pathways for ATMPs: Standard, accelerated and adaptive pathways to marketing authorisation. Mol. Ther. Methods Clin. Dev. 2019, 13, 205–232. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Green, H. The birth of therapy with cultured cells. BioEssays 2008, 30, 897–903. [Google Scholar] [CrossRef] [PubMed]
- Abdel-Sayed, P.; Michetti, M.; Scaletta, C.; Flahaut, M.; Hirt-Burri, N.; Roessingh, A.d.B.; Raffoul, W.; Applegate, L.A. Cell therapies for skin regeneration: An overview of 40 years of experience in burn units. Swiss Med. Wkly. 2019, 149, w20079. [Google Scholar] [CrossRef] [PubMed]
- Marks, P.; Gottlieb, S. Balancing safety and innovation for cell-based regenerative medicine. N. Engl. J. Med. 2018, 378, 954–959. [Google Scholar] [CrossRef] [PubMed]
- Rajabzadeh, N.; Fathi, E.; Farahzadi, R. Stem cell-based regenerative medicine. Stem Cell Investig. 2019, 6, 19. [Google Scholar] [CrossRef] [PubMed]
- Hartmann-Fritsch, F.; Marino, D.; Reichmann, E. About ATMPs, SOPs and GMP: The hurdles to produce novel skin grafts for clinical use. Transfus. Med. Hemother. 2016, 43, 344–352. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Viganò, M.; Giordano, R.; Lazzari, L. Challenges of running a GMP facility for regenerative medicine in a public hospital. Regen. Med. 2017, 12, 803–813. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iancu, E.M.; Kandalaft, L.E. Challenges and advantages of cell therapy manufacturing under Good Manufacturing Practices within the hospital setting. Curr. Opin. Biotechnol. 2020, 65, 233–241. [Google Scholar] [CrossRef] [PubMed]
- Bicudo, E.; Brass, I.; Carmichael, P.; Farid, S. The UKs emerging regulatory framework for point-of-care manufacture: Insights from a workshop on advanced therapies. Cell Gene Ther. Insights 2021, 7, 1005–1015. [Google Scholar] [CrossRef]
- Haeusner, S.; Herbst, L.; Bittorf, P.; Schwarz, T.; Henze, C.; Mauermann, M.; Ochs, J.; Schmitt, R.; Blanche, U.; Wixmerten, A.; et al. From single batch to mass production-automated platform design concept for a Phase II clinical trial tissue engineered cartilage product. Front. Med. 2021, 8, 712917. [Google Scholar] [CrossRef] [PubMed]
- Greenberg-Worisek, A.J.; Runge, B.K.; Solyntjes, S.A.; St Helene-Kraft, J.; Glass, S.L.; Waltzki, B.E.; Herrice, J.L.; Miller, A.L.; Yaszemski, M.J.; Windebank, A.J.; et al. Establishing a current good manufacturing practice facility for biomaterials and biomolecules in an academic medical center. Tissue Eng. Part B 2018, 24, 493–498. [Google Scholar] [CrossRef] [PubMed]
- Digiusto, D.L.; Messop, K.; Srvastava, R.; Tran, C.-A.T. Proceedings of the first academic symposium on developing, qualifying and operating a cell and gene therapy manufacturing facility. Cytotherapy 2018, 20, 1486–1494. [Google Scholar] [CrossRef] [PubMed]
- Bedford, P.; Jy, J.; Collins, L.; Keizer, S. Considering cell therapy product “Good manufacturing practice” status. Front. Med. 2018, 5, 118. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brittberg, M.; Lindahl, A.; Nilsson, A.; Ohlsson, C.; Isaksson, O.; Peterson, L. Treatment of deep cartilage defects in the knee with autologous chondrocyte transplantation. N. Engl. J. Med. 1994, 331, 889–895. [Google Scholar] [CrossRef] [PubMed]
- Horas, U.; Pelinkovic, D.; Herr, G.; Aigner, T.; Schnettler, R. Autologous chondrocyte implantation and osteochondral cylinder transplantation in cartilage repair of the knee joint. A prospective, comparative trial. J. Bone Jt. Surg. 2003, 85, 185–192. [Google Scholar] [CrossRef] [PubMed]
- Urlic, I.; Ivkovic, A. Cell sources for cartilage repair-biological and clinical perspective. Cells 2021, 10, 2496. [Google Scholar] [CrossRef] [PubMed]
- Minas, T.; Peterson, L. Autologous chondrocyte transplantation. Oper. Tech. Sports Med. 2012, 20, 72–86. [Google Scholar] [CrossRef]
- Mumme, M.; Barbero, A.; Miot, S.; Wixmerten, A.; Feliciano, S.; Wolf, F.; Asnaghi, A.M.; Baumhoer, D.; Bieri, O.; Kretzcchmar, M.; et al. Nasal chondrocyte-based engineered autologous cartilage tissue for repair of articular cartilage defects: An observational first-in-human trial. Lancet 2016, 388, 1985–1994. [Google Scholar] [CrossRef]
- Smith, A. Survival of frozen chondrocytes isolated from cartilage of adult mammals. Nature 1965, 205, 782–784. [Google Scholar] [CrossRef]
- Kawiak, J.; Moskalewski, S.; Darzynkiewicz, Z. Isolation of chondrocytes from calf cartilage. Exp. Cell Res. 1965, 39, 59–68. [Google Scholar] [CrossRef]
- Manning, W.K.; Bonner, W.M. Isolation and culture of chondrocytes from human adult articular cartilage. Arthritis Rheum. 1967, 10, 235–239. [Google Scholar] [CrossRef] [PubMed]
- Oseni, A.O.; Butler, P.E.; Seifalian, A.M. Optimization of chondrocyte isolation and characterization for large-scale cartilage tissue engineering. J. Surg. Res. 2013, 181, 41–48. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mandl, E.W.; van der Veen, S.W.; Verhaar, J.A.; van Osch, G.J. Serum-free medium supplemented with high-concentration FGF2 for cell expansion culture of human ear chondrocytes promotes redifferentiation capacity. Tissue Eng. 2002, 8, 573–580. [Google Scholar] [CrossRef]
- Olney, R.C.; Wang, J.; Sylvester, J.E.; Mougey, E.B. Growth factor regulation of human growth plate chondrocyte proliferation in vitro. Biochem. Biophys. Res. Commun. 2004, 317, 1171–1182. [Google Scholar] [CrossRef]
- Chua, K.H.; Aminuddin, B.S.; Fuzina, N.H.; Ruszymah, B.H. Insulin-transferrin-selenium prevent human chondrocyte dedifferentiation and promote the formation of high quality tissue engineered human hyaline cartilage. Eur. Cell Mater. 2005, 9, 58–67. [Google Scholar] [CrossRef] [PubMed]
- Heidari, M.; Tahmasebi, M.N.; Etemad, S.; Salehkhou, S.; Heidari-Vala, H.; Akhondi, M.A. In vitro human chondrocyte culture; A modified protocol. Middle-East J. Sci. Res. 2011, 9, 102–109. [Google Scholar]
- Brittberg, M. Autologous chondrocyte transplantation. Clin. Orthop. Relat. Res. 1999, S147–S155. [Google Scholar] [CrossRef] [PubMed]
- van den Borne, M.P.; Raijmakers, N.J.; Vanlauwe, J.; Victor, J.; de Jong, S.N.; Bellemans, J.; Saris, D.B.; International Cartilage Repair Society. International Cartilage Repair Society (ICRS) and Oswestry macroscopic cartilage evaluation scores validated for use in Autologous Chondrocyte Implantation (ACI) and microfracture. Osteoarthr. Cartil. 2007, 15, 1397–1402. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Philippe, V.; Laurent, A.; Abdel-Sayed, P.; Hirt-Burri, N.; Applegate, L.A.; Martin, R. Human platelet lysate as an alternative to autologous serum for human chondrocyte clinical use. Cartilage 2021, 13, 509S–518S. [Google Scholar] [CrossRef] [PubMed]
- Tallheden, T.; van der Lee, J.; Brantsing, C.; Månsson, J.E.; Sjögren-Jansson, E.; Lindahl, A. Human serum for culture of articular chondrocytes. Cell Transplant. 2005, 14, 469–479. [Google Scholar] [CrossRef] [Green Version]
- Doucet, C.; Ernou, I.; Zhang, Y.; Llense, J.R.; Begot, L.; Holy, X.; Lataillade, J.J. Platelet lysates promote mesenchymal stem cell expansion: A safety substitute for animal serum in cell-based therapy applications. J. Cell. Physiol. 2005, 205, 228–236. [Google Scholar] [CrossRef] [PubMed]
- Bieback, K. Platelet lysate as replacement for fetal bovine serum in mesenchymal stromal cell cultures. Transfus. Med. Hemotherapy 2013, 40, 326–335. [Google Scholar] [CrossRef] [Green Version]
- Spreafico, A.; Chellini, F.; Frediani, B.; Bernardini, G.; Niccolini, S.; Serchi, T.; Collodel, G.; Paffetti, A.; Fossombroni, V.; Galeazzi, M.; et al. Biochemical investigation of the effects of human platelet releasates on human articular chondrocytes. J. Cell. Biochem. 2009, 108, 1153–1165. [Google Scholar] [CrossRef] [PubMed]
- Sykes, J.G.; Kuiper, J.H.; Richardson, J.B.; Roberts, S.; Wright, K.T.; Kuiper, N.J. Impact of human platelet lysate on the expansion and chondrogenic capacity of cultured human chondrocytes for cartilage cell therapy. Eur. Cell Mater. 2018, 35, 255–267. [Google Scholar] [CrossRef] [PubMed]
- Hildner, F.; Eder, M.J.; Hofer, K.; Aberl, J.; Redl, H.; van Griensven, M.; Gabriel, C.; Peterbauer-Scherb, A. Human platelet lysate successfully promotes proliferation and subsequent chondrogenic differentiation of adipose-derived stem cells: A comparison with articular chondrocytes. J. Tissue Eng. Regen. Med. 2015, 9, 808–818. [Google Scholar] [CrossRef] [PubMed]
- Kachroo, U.; Zachariah, S.M.; Thambaiah, A.; Tabasum, A.; Livingston, A.; Rebekah, G.; Srivastava, A.; Vinod, E. Comparison of human platelet lysate versus fetal bovine serum for expansion of human articular cartilage-derived chondroprogenitors. Cartilage 2021, 13, 107S–116S. [Google Scholar] [CrossRef] [PubMed]
- Burnouf, T.; Strunk, D.; Koh, M.B.; Schallmoser, K. Human platelet lysate: Replacing fetal bovine serum as a gold standard for human cell propagation? Biomaterials 2016, 76, 371–387. [Google Scholar] [CrossRef] [PubMed]
- Kaps, C.; Loch, A.; Haisch, A.; Smolian, H.; Burmester, G.R.; Häupl, T.; Sittinger, M. Human platelet supernatant promotes proliferation but not differentiation of articular chondrocytes. Med. Biol. Eng. Comput. 2002, 40, 485–490. [Google Scholar] [CrossRef] [PubMed]
- Liau, L.L.; bin Hassan, M.N.F.; Tang, Y.L.; Ng, M.H.; Law, J.X. Feasibility of human platelet lysate as an alternative to fetal bovine serum for in vitro expansion of chondrocytes. Int. J. Mol. Sci. 2021, 22, 1269. [Google Scholar] [CrossRef] [PubMed]
- Rikkers, M.; Levato, R.; Malda, J.; Vonk, L.A. Importance of timing of platelet lysate-supplementation in expanding or redifferentiating human chondrocytes for chondrogenesis. Front. Bioeng. Biotechnol. 2020, 8, 804. [Google Scholar] [CrossRef] [PubMed]
- Tritschler, H.; Fischer, K.; Seissler, J.; Fiedler, J.; Halbgebauer, R.; Huber-Lang, M.; Schnieke, A.; Brenner, R.E. New insights into xenotransplantation for cartilage repair: Porcine multi-genetically modified chondrocytes as a promising cell source. Cells 2021, 10, 2152. [Google Scholar] [CrossRef] [PubMed]
- Perry, J.; Roelofs, A.J.; Mennan, C.; McCarthy, H.S.; Richmond, A.; Clark, S.M.; Rieman, A.N.K.; Wright, K.; De Bari, C.; Roberts, S. Human mesenchymal stromal cells enhance c.cartilage healing in a murine joint surface injury model. Cells 2021, 10, 1999. [Google Scholar] [CrossRef]
- Kondo, M.; Kameishi, S.; Metzler, N.F.; Maak, T.G.; Hutchinson, D.T.; Wang, A.A.; Maehra, M.; Sato, M.; Grainger, D.W.; Okano, T. Safety and efficacy of human juvenile chondrocyte-derived cell sheets for osteochondral defect treatment. NPJ Regen. Med. 2021, 6, 65. [Google Scholar] [CrossRef]
- Makris, E.A.; Gomoll, A.H.; Malizos, K.N.; Hu, J.C.; Athanasiou, K.A. Repair and tissue engineering techniques for articular cartilage. Nat. Rev. Rheumatol. 2015, 11, 21–34. [Google Scholar] [CrossRef] [PubMed]
- Park, J.; Gelse, K.; Frank, S.; von der Mark, K.; Aigner, T.; Schneider, H. Transgene-activated mesenchymal cells for articular cartilage repair: A comparison of primary bone marrow-, perichondrium/periosteum- and fat-derived cells. J. Gene Med. 2006, 8, 112–125. [Google Scholar] [CrossRef]
- Toh, W.S.; Lee, E.H.; Guo, X.M.; Chan, J.K.; Yeow, C.H.; Choo, A.B.; Cao, T. Cartilage repair using hyaluronan hydrogel-encapsulated human embryonic stem cell-derived chondrogenic cells. Biomaterials 2010, 31, 6968–6980. [Google Scholar] [CrossRef] [PubMed]
- Gupta, P.K.; Das, A.K.; Chullikana, A.; Majumdar, A.S. Mesenchymal stem cells for cartilage repair in osteoarthritis. Stem Cell Res. Ther. 2012, 3, 25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Darwiche, S.E.; Scaletta, C.; Raffoul, W.; Pioletti, D.P.; Applegate, L.A. Epiphyseal chondroprogenitors provide a stable cell source for cartilage cell therapy. Cell Med. 2012, 4, 23–32. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cherian, J.J.; Parvizi, J.; Bramlet, D.; Lee, K.H.; Romness, D.W.; Mont, M.A. Preliminary results of a phase II randomized study to determine the efficacy and safety of genetically engineered allogeneic human chondrocytes expressing TGF-B1 in patients with grade 3 chronic degenerative joint disease of the knee. Osteoarthr. Cartil. 2015, 23, 2109–2118. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chemali, M.; Laurent, A.; Scaletta, C.; Waselle, L.; Simon, J.P.; Michetti, M.; Brunet, J.F.; Flahaut, M.; Hirt-Burri, N.; Raffoul, W.; et al. Burn center organization and cellular therapy integration: Managing risks and costs. J. Burn Care Res. 2021, 42, 911–924. [Google Scholar] [CrossRef] [PubMed]
- European Parliament and Council. Directive 2004/23/EC on setting standards of quality and safety for the donation, procurement, testing, processing, preservation, storage and distribution of human tissues and cells. Off. J. Eur. Union 2004, L102, 48–58. Available online: http://data.europa.eu/eli/dir/2004/23/oj (accessed on 1 December 2021).
- European Commission. Commission Directive (EC) No 2006/17/EC of 8 February 2006 implementing Directive 2004/23/EC of the European parliament and of the Council as regards certain technical requirements for the donation, procurement and testing of human tissues and cells. Off. J. Eur. Union 2006, L38, 40–52. Available online: http://data.europa.eu/eli/dir/2006/17/oj (accessed on 1 December 2021).
- European Parliament and Council. Directive 2001/83/EC on the Community code relating to medicinal products for human use. Off. J. Eur. Union 2001, L311, 67–128. Available online: http://data.europa.eu/eli/dir/2001/83/oj (accessed on 1 December 2021).
- European Commission. Commission Directive (EC) No 2003/94/EC of 8 October 2003 laying down the principles and guidelines of good manufacturing practice in respect of medicinal products for human use and investigational medicinal products for human use. Off. J. Eur. Union 2003, L262, 22–26. Available online: http://data.europa.eu/eli/dir/2003/94/oj (accessed on 1 December 2021).
- European Parliament and Council. Regulation (EC) No 1394/2007 on Advanced Therapy Medicinal Products and amending Directive 2001/83/EC and Regulation (EC) No 726/2004. Off. J. Eur. Union 2007, L324, 121–137. Available online: http://data.europa.eu/eli/reg/2007/1394/oj (accessed on 1 December 2021).
- European Commission. EudraLex, The Rules Governing Medicinal Products in the European Union, Volume 4, Good Manufacturing Practice, Guidelines of 22 November 2017, Good Manufacturing Practice for Advanced Therapy Medicinal Products. 2017. Available online: https://ec.europa.eu/health/documents/eudralex/vol-4_en (accessed on 1 December 2021).
- Federal Assembly of the Swiss Confederation. Federal Act on Medicinal Products and Medical Devices (Therapeutic Products Act, TPA). 2000, SR 812: 21. Available online: https://fedlex.data.admin.ch/eli/cc/2001/422 (accessed on 1 December 2021).
- Federal Assembly of the Swiss Confederation. Federal Act on the Transplantation of Organs, Tissues and Cells (Transplantation Act). 2004, SR 810: 21. Available online: https://fedlex.data.admin.ch/eli/cc/2007/279 (accessed on 1 September 2021).
- Ramezankhani, R.; Torabi, S.; Minaei, N.; Madani, H.; Rezaeiani, S.; Hassani, S.N.; Gee, A.P.; Dominici, M.; Silva, D.N.; Baharvand, H.; et al. Two decades of global progress in authorized advanced therapy medicinal products: An emerging revolution in therapeutic strategies. Front. Cell Dev. Biol. 2020, 8, 547653. [Google Scholar] [CrossRef]
- Rapko, S.; Zhang, M.; Richards, B.; Hutto, E.; Dethlefsen, S.; Duguay, S. Identification of the chondrocyte lineage using microfibril-associated glycoprotein-2, a novel marker that distinguishes chondrocytes from synovial cells. Tissue Eng. Part C 2010, 16, 1367–1375. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Asnaghi, M.A.; Power, L.; Barbero, A.; Haug, M.; Köppl, R.; Wendt, D.; Martin, I. Biomarker signatures of quality for engineering nasal chondrocyte-derived cartilage. Front. Bioeng. Biotechnol. 2020, 8, 283. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Teo, A.Q.A.; Wong, K.L.; Shen, L.; Lim, J.Y.; Wei, S.T.; Lee, H.; Hui, J.H.P. Equivalent 10-year outcomes after implantation of autologous bone marrow-derived mesenchymal stem cells versus autologous chondrocyte implantation for chondral defects of the knee. Am. J. Sports Med. 2019, 47, 2881–2887. [Google Scholar] [CrossRef] [PubMed]
- Benthien, J.P.; Behrens, P. The treatment of chondral and osteochondral defects of the knee with autologous matrix-induced chondrogenesis (AMIC): Method description and recent developments. Knee Surg. Sports Traumatol. Arthrosc. 2011, 19, 1316–1319. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gille, J.; Behrens, P.; Volpi, P.; de Girolamo, L.; Reiss, E.; Zoch, W.; Anders, S. Outcome of Autologous Matrix Induced Chondrogenesis (AMIC) in cartilage knee surgery: Data of the AMIC registry. Arch. Orthop. Trauma Surg. 2013, 133, 87–93. [Google Scholar] [CrossRef] [Green Version]
- Peterson, L.; Vasiliadis, H.S.; Brittberg, M.; Lindahl, A. Autologous chondrocyte implantation: A long-term follow-up. Am. J. Sports Med. 2010, 38, 1117–1124. [Google Scholar] [CrossRef]
- Laurent, A.; Abdel-Sayed, P.; Ducrot, A.; Hirt-Burri, N.; Scaletta, C.; Jaccoud, S.; Nuss, K.; de Buys Roessingh, A.S.; Raffoul, W.; Pioletti, D.P.; et al. Development of standardized fetal progenitor cell therapy for cartilage regenerative medicine: Industrial transposition and preliminary safety in xenogeneic transplantation. Biomolecules 2021, 11, 250. [Google Scholar] [CrossRef] [PubMed]
- Studer, D.; Cavalli, E.; Formica, F.A.; Kuhn, G.A.; Salzmann, G.; Mumme, M.; Steinwachs, M.R.; Applegate, L.A.; Maniura-Weber, K.; Zenobi-Wong, M. Human chondroprogenitors in alginate-collagen hybrid scaffolds produce stable cartilage in vivo. J. Tissue Eng. Regen. Med. 2017, 11, 3014–3026. [Google Scholar] [CrossRef] [PubMed]
- Cavalli, E.; Fisch, P.; Formica, F.A.; Gareus, R.; Linder, T.; Applegate, L.A.; Zenobi-Wong, M. A comparative study of cartilage engineered constructs in immunocompromised, humanized and immunocompetent mice. J. Immunol. Regen. Med. 2018, 2, 36–46. [Google Scholar] [CrossRef] [Green Version]
- Levinson, C.; Lee, M.; Applegate, L.A.; Zenobi-Wong, M. An injectable heparin-conjugated hyaluronan scaffold for local delivery of transforming growth factor β1 promotes successful chondrogenesis. Acta Biomater. 2019, 99, 168–180. [Google Scholar] [CrossRef]
- Almqvist, K.F.; Dhollander, A.A.; Verdonk, P.C.; Forsyth, R.; Verdonk, R.; Verbruggen, G. Treatment of cartilage defects in the knee using alginate beads containing human mature allogenic chondrocytes. Am. J. Sports Med. 2009, 37, 1920–1929. [Google Scholar] [CrossRef]
- Dhollander, A.A.; Verdonk, P.C.; Lambrecht, S.; Verdonk, R.; Elewaut, D.; Verbruggen, G.; Almqvist, K.F. Midterm results of the treatment of cartilage defects in the knee usi.ing alginate beads containing human mature allogenic chondrocytes. Am. J. Sports Med. 2012, 40, 75–82. [Google Scholar] [CrossRef] [PubMed]
- Brittberg, M. Cell carriers as the next generation of cell therapy for cartilage repair: A review of the matrix-induced autologous chondrocyte implantation procedure. Am. J. Sports Med. 2010, 38, 1259–1271. [Google Scholar] [CrossRef] [PubMed]
- Mhanna, R.; Kashyap, A.; Palazzolo, G.; Vallmajo-Martin, Q.; Becher, J.; Möller, S.; Schnabelrauch, M.; Zenobi-Wong, M. Chondrocyte culture in three-dimensional alginate sulfate hydrogels promotes proliferation while maintaining expression of chondrogenic markers. Tissue Eng. Part A 2014, 20, 1454–1464. [Google Scholar] [CrossRef] [Green Version]
- World Medical Association. Declaration of Helsinki: Ethical principles for medical research involving human subjects. JAMA 2013, 310, 2191–2194. [Google Scholar] [CrossRef] [Green Version]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Patient Parameters | Average Numerical Data | ||
|---|---|---|---|
| Male Patients | Female Patients | ||
| Number of Patients (n) | 29 | 18 | |
| Patient Age (years) | 24.0 ± 7.5 | 24.2 ± 8.1 | |
| Patient BMI (kg/m2) | 24.0 ± 4.3 | 22.0 ± 3.6 | |
| Lesion Size (cm2) | 5.1 ± 2.4 | 4.1 ± 2.4 | |
| Number of Chondral Lesions (n) | 12 | 13 | |
| Number of Osteochondral Lesions (n) | 17 | 5 | |
| Affected Limb (Right/Left) | 16/13 | 9/9 | |
| Anatomical Zone of the Lesion | Internal Femoral Condyle | 14 | 6 |
| External Femoral Condyle | 5 | 0 | |
| Rotula | 5 | 12 | |
| Trochlea | 3 | 0 | |
| External Tibial Plateau | 2 | 0 | |
| Deviation Type | Number of Deviations | Description/Comments | Corrective Actions Resulting in Eventual Lot Liberation |
|---|---|---|---|
| Deviation to cell culture TS | 8 | Low number of cells in the preliminary cell population; cell seeding density variability; accelerated or delayed harvest due to inhomogeneous growth or to calendar constraints; low cell harvest yield; low cell viability at thawing | Communication and coordination with research laboratory and clinical unit personnel; documentary reconciliation of manufacturing records for all subsequent steps to demonstrate the appropriate endpoint quality of the material lots. |
| Deviation related to finished product TS | 0 | NA | NA |
| Deviations related to logistical process | 3 | Use of out-of-validity biopsy harvest and transport kit; biopsy transport in an alternative kit | Communication and coordination with research laboratory and clinical unit personnel resulting in conjoint validation of material use (risk analysis-based assessment, for sparing use of the valuable biospecimen); documentary reconciliation of manufacturing records for all subsequent steps to demonstrate the appropriate endpoint quality of the material lots; investigation of the origin and specifications of alternative and out-of-validity transport kits with clinical unit personnel. |
| Deviations related to manufacturing process controls 1 | 9 | Airflow microbiological monitoring (Methylobacterium radiotolerans); imprint microbiological monitoring (Bacillus spp.; Lysinibacillus fusiformis; Micrococcus luteus; Kocuria rhizophila; unspecified Gram+ bacteria) | Complementary microbiological investigation performed on available retention samples. |
| Deviations related to storage process | 0 | NA | NA |
| Deviations related to microbiological QC 2 | 2 | Cryotube microbiological testing (Bacillus spp.; Moraxella osloensis or Enhydrobacter aerosaccus) | Complementary microbiological QC & investigation performed on additional samples. |
| Deviations related to functional QC | 2 | Low RNA quantity | Repetition of the functional QC cell culture step for generation of appropriate biological material quantity. |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Philippe, V.; Laurent, A.; Hirt-Burri, N.; Abdel-Sayed, P.; Scaletta, C.; Schneebeli, V.; Michetti, M.; Brunet, J.-F.; Applegate, L.A.; Martin, R. Retrospective Analysis of Autologous Chondrocyte-Based Cytotherapy Production for Clinical Use: GMP Process-Based Manufacturing Optimization in a Swiss University Hospital. Cells 2022, 11, 1016. https://doi.org/10.3390/cells11061016
Philippe V, Laurent A, Hirt-Burri N, Abdel-Sayed P, Scaletta C, Schneebeli V, Michetti M, Brunet J-F, Applegate LA, Martin R. Retrospective Analysis of Autologous Chondrocyte-Based Cytotherapy Production for Clinical Use: GMP Process-Based Manufacturing Optimization in a Swiss University Hospital. Cells. 2022; 11(6):1016. https://doi.org/10.3390/cells11061016
Chicago/Turabian StylePhilippe, Virginie, Alexis Laurent, Nathalie Hirt-Burri, Philippe Abdel-Sayed, Corinne Scaletta, Valentine Schneebeli, Murielle Michetti, Jean-François Brunet, Lee Ann Applegate, and Robin Martin. 2022. "Retrospective Analysis of Autologous Chondrocyte-Based Cytotherapy Production for Clinical Use: GMP Process-Based Manufacturing Optimization in a Swiss University Hospital" Cells 11, no. 6: 1016. https://doi.org/10.3390/cells11061016
APA StylePhilippe, V., Laurent, A., Hirt-Burri, N., Abdel-Sayed, P., Scaletta, C., Schneebeli, V., Michetti, M., Brunet, J. -F., Applegate, L. A., & Martin, R. (2022). Retrospective Analysis of Autologous Chondrocyte-Based Cytotherapy Production for Clinical Use: GMP Process-Based Manufacturing Optimization in a Swiss University Hospital. Cells, 11(6), 1016. https://doi.org/10.3390/cells11061016