
Hsp90 in Human Diseases: Molecular Mechanisms to Therapeutic Approaches


Abstract
:
1. Introduction
2. Structure and Functions of Hsp90
3. Conformation Dynamics of Hsp90
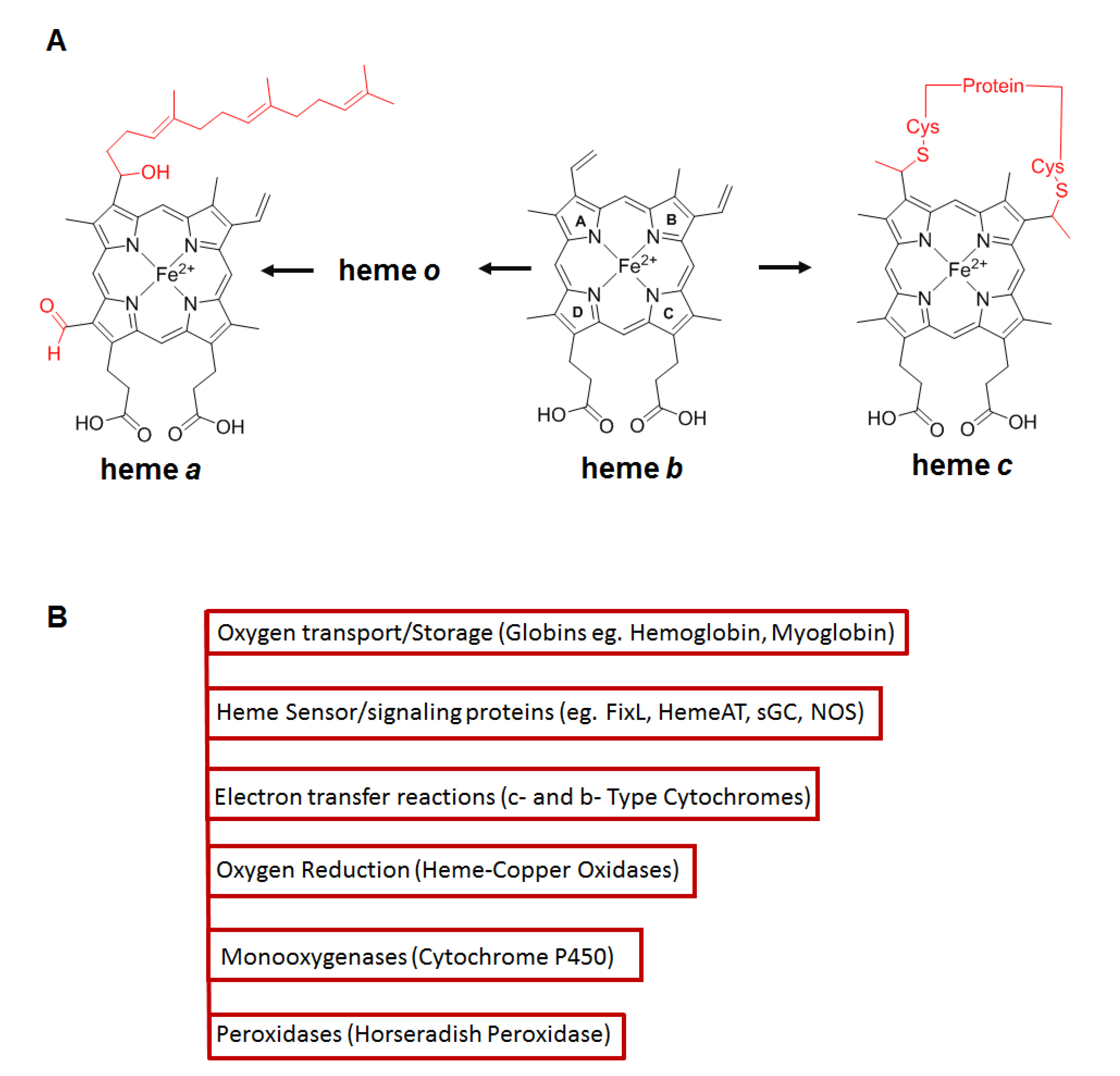
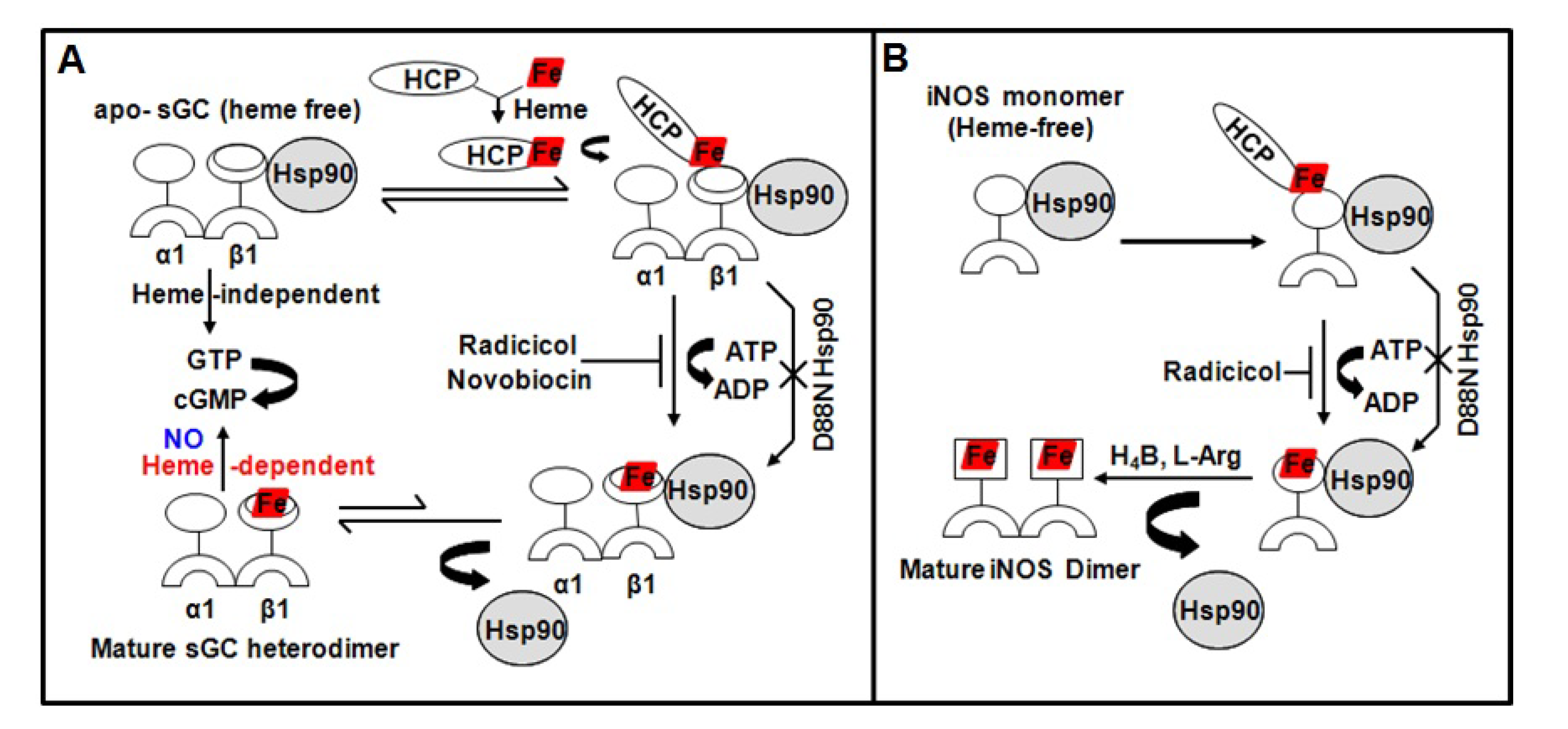
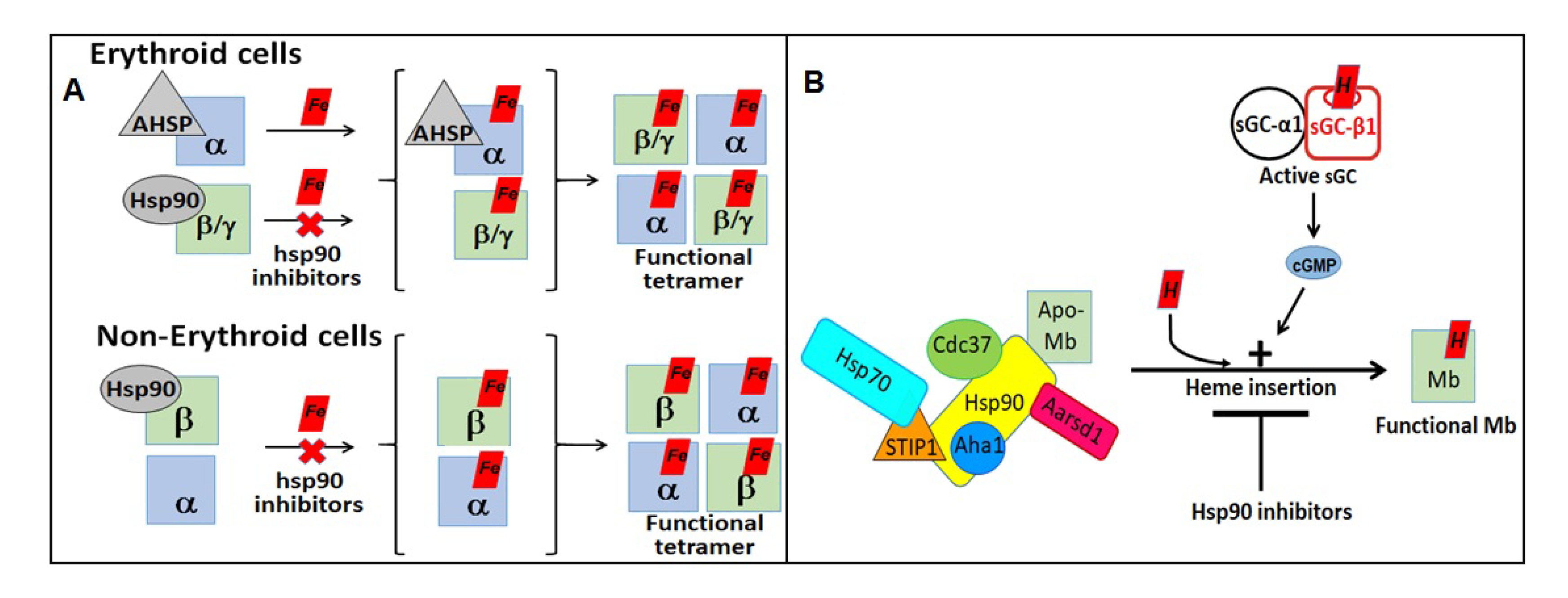
4. Role of Hsp90 in Client Hemeprotein Maturation
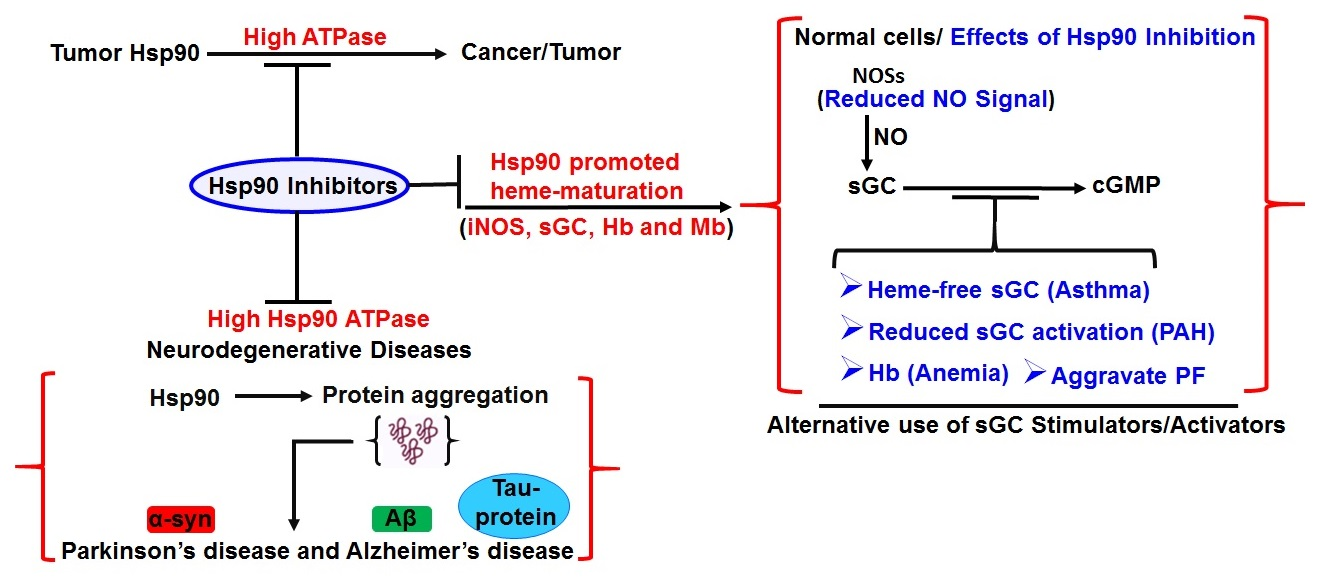
5. Hsp90 and Human Diseases
6. Role of Hsp90 in Cancer/Tumorogenesis
7. Role of HSP90 in Neurodegenerative Disorders
8. Role of Hsp90 in Asthma and Pulmonary Diseases
9. Therapeutics Facets of HSP90 Inhibition
10. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- De Maio, A.; Santoro, M.G.; Tanguay, R.M.; Hightower, L.E. Ferruccio Ritossa’s scientific legacy 50 years after his discovery of the heat shock response: A new view of biology, a new society, and a new journal. Cell Stress Chaperones 2012, 17, 139–143. [Google Scholar] [CrossRef] [Green Version]
- Schlesinger, M.J. Heat shock proteins. J. Biol. Chem. 1990, 265, 12111–12114. [Google Scholar] [CrossRef]
- Fu, X. Chaperone function and mechanism of small heat-shock proteins. Acta Biochim. Biophys. Sin. 2014, 46, 347–356. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Millar, N.L.; Murrell, G.A. Heat shock proteins in tendinopathy: Novel molecular regulators. Mediat. Inflamm. 2012, 2012, 436203. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dukay, B.; Csoboz, B.; Tóth, M.E. Heat-Shock Proteins in Neuroinflammation. Front. Pharmacol. 2019, 10, 920. [Google Scholar] [CrossRef] [Green Version]
- Taipale, M.; Jarosz, D.F.; Lindquist, S. HSP90 at the hub of protein homeostasis: Emerging mechanistic insights. Nat. Rev. Mol. Cell Biol. 2010, 11, 515–528. [Google Scholar] [CrossRef]
- Picard, D. Heat-shock protein 90, a chaperone for folding and regulation. Cell. Mol. Life Sci. 2002, 59, 1640–1648. [Google Scholar] [CrossRef]
- Schopf, F.H.; Biebl, M.M.; Buchner, J. The HSP90 chaperone machinery. Nat. Rev. Mol. Cell Biol. 2017, 18, 345–360. [Google Scholar] [CrossRef] [PubMed]
- Radli, M.; Rüdiger, S.G.D. Picky Hsp90—Every Game with Another Mate. Mol. Cell 2017, 67, 899–900. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Buchner, J. Structure, function and regulation of the Hsp90 machinery. Biomed. J. 2013, 36, 106–117. [Google Scholar] [CrossRef] [PubMed]
- Gupta, A.; Bansal, A.; Hashimoto-Torii, K. HSP70 and HSP90 in neurodegenerative diseases. Neurosci. Lett. 2020, 716, 134678. [Google Scholar] [CrossRef]
- Ghosh, A.; Koziol-White, C.J.; Asosingh, K.; Cheng, G.; Ruple, L.; Groneberg, D.; Friebe, A.; Comhair, S.A.; Stasch, J.P.; Panettieri, R.A., Jr.; et al. Soluble guanylate cyclase as an alternative target for bronchodilator therapy in asthma. Proc. Natl. Acad. Sci. USA 2016, 113, E2355–E2362. [Google Scholar] [CrossRef] [Green Version]
- Wang, G.K.; Li, S.H.; Zhao, Z.M.; Liu, S.X.; Zhang, G.X.; Yang, F.; Wang, Y.; Wu, F.; Zhao, X.X.; Xu, Z.Y. Inhibition of heat shock protein 90 improves pulmonary arteriole remodeling in pulmonary arterial hypertension. Oncotarget 2016, 7, 54263–54273. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Street, T.O.; Lavery, L.A.; Agard, D.A. Substrate binding drives large-scale conformational changes in the Hsp90 molecular chaperone. Mol. Cell 2011, 42, 96–105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ali, M.M.; Roe, S.M.; Vaughan, C.K.; Meyer, P.; Panaretou, B.; Piper, P.W.; Prodromou, C.; Pearl, L.H. Crystal structure of an Hsp90-nucleotide-p23/Sba1 closed chaperone complex. Nature 2006, 440, 1013–1017. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tatu, U.; Neckers, L. Chaperoning parasitism: The importance of molecular chaperones in pathogen virulence. Parasitology 2014, 141, 1123–1126. [Google Scholar] [CrossRef] [Green Version]
- Huai, Q.; Wang, H.; Liu, Y.; Kim, H.Y.; Toft, D.; Ke, H. Structures of the N-terminal and middle domains of E. coli Hsp90 and conformation changes upon ADP binding. Structure 2005, 13, 579–590. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Soroka, J.; Buchner, J. The Hsp90 chaperone machinery: Conformational dynamics and regulation by co-chaperones. Biochim. Biophys. Acta 2012, 1823, 624–635. [Google Scholar] [CrossRef] [Green Version]
- Penna, C.; Sorge, M.; Femminò, S.; Pagliaro, P.; Brancaccio, M. Redox Aspects of Chaperones in Cardiac Function. Front. Physiol. 2018, 9, 216. [Google Scholar] [CrossRef]
- Ghosh, A.; Stasch, J.P.; Papapetropoulos, A.; Stuehr, D.J. Nitric oxide and heat shock protein 90 activate soluble guanylate cyclase by driving rapid change in its subunit interactions and heme content. J. Biol. Chem. 2014, 289, 15259–15271. [Google Scholar] [CrossRef] [Green Version]
- Ghosh, A.; Garee, G.; Sweeny, E.A.; Nakamura, Y.; Stuehr, D.J. Hsp90 chaperones hemoglobin maturation in erythroid and nonerythroid cells. Proc. Natl. Acad. Sci. USA 2018, 115, E1117–E1126. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martinez-Ruiz, A.; Villanueva, L.; Gonzalez de Orduna, C.; Lopez-Ferrer, D.; Higueras, M.A.; Tarin, C.; Rodriguez-Crespo, I.; Vazquez, J.; Lamas, S. S-nitrosylation of Hsp90 promotes the inhibition of its ATPase and endothelial nitric oxide synthase regulatory activities. Proc. Natl. Acad. Sci. USA 2005, 102, 8525–8530. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nedvetsky, P.I.; Meurer, S.; Opitz, N.; Nedvetskaya, T.Y.; Muller, H.; Schmidt, H.H. Heat shock protein 90 regulates stabilization rather than activation of soluble guanylate cyclase. FEBS Lett. 2008, 582, 327–331. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quintana-Gallardo, L.; Martin-Benito, J.; Marcilla, M.; Espadas, G.; Sabido, E.; Valpuesta, J.M. The cochaperone CHIP marks Hsp70- and Hsp90-bound substrates for degradation through a very flexible mechanism. Sci. Rep. 2019, 9, 5102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Theodoraki, M.A.; Caplan, A.J. Quality control and fate determination of Hsp90 client proteins. Biochim. Biophys. Acta 2012, 1823, 683–688. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, P.; Weaver, V.M.; Werb, Z. The extracellular matrix: A dynamic niche in cancer progression. J. Cell Biol. 2012, 196, 395–406. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.S.; Alarcon, S.V.; Lee, S.; Lee, M.J.; Giaccone, G.; Neckers, L.; Trepel, J.B. Update on Hsp90 inhibitors in clinical trial. Curr. Top. Med. Chem. 2009, 9, 1479–1492. [Google Scholar] [CrossRef] [PubMed]
- Graf, C.; Lee, C.T.; Eva Meier-Andrejszki, L.; Nguyen, M.T.; Mayer, M.P. Differences in conformational dynamics within the Hsp90 chaperone family reveal mechanistic insights. Front. Mol. Biosci. 2014, 1, 4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hessling, M.; Richter, K.; Buchner, J. Dissection of the ATP-induced conformational cycle of the molecular chaperone Hsp90. Nat. Struct. Mol. Biol. 2009, 16, 287–293. [Google Scholar] [CrossRef] [PubMed]
- Genest, O.; Wickner, S.; Doyle, S.M. Hsp90 and Hsp70 chaperones: Collaborators in protein remodeling. J. Biol. Chem. 2019, 294, 2109–2120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, J.; Richter, K.; Reinstein, J.; Buchner, J. Integration of the accelerator Aha1 in the Hsp90 co-chaperone cycle. Nat. Struct. Mol. Biol. 2013, 20, 326–331. [Google Scholar] [CrossRef] [PubMed]
- Retzlaff, M.; Stahl, M.; Eberl, H.C.; Lagleder, S.; Beck, J.; Kessler, H.; Buchner, J. Hsp90 is regulated by a switch point in the C-terminal domain. EMBO Rep. 2009, 10, 1147–1153. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Severance, S.; Hamza, I. Trafficking of heme and porphyrins in metazoa. Chem. Rev. 2009, 109, 4596–4616. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paoli, M.; Marles-Wright, J.; Smith, A. Structure—Function relationships in heme-proteins. DNA Cell Biol. 2002, 21, 271–280. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ponka, P. Cell biology of heme. Am. J. Med. Sci. 1999, 318, 241–256. [Google Scholar] [CrossRef] [PubMed]
- Smith, A.T.; Veitch, N.C. Substrate binding and catalysis in heme peroxidases. Curr. Opin. Chem. Biol. 1998, 2, 269–278. [Google Scholar] [CrossRef]
- Taylor, B.L.; Zhulin, I.B. PAS domains: Internal sensors of oxygen, redox potential, and light. Microbiol. Mol. Biol. Rev. 1999, 63, 479–506. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taketani, S. Aquisition, mobilization and utilization of cellular iron and heme: Endless findings and growing evidence of tight regulation. Tohoku J. Exp. Med. 2005, 205, 297–318. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Steiner, H.; Kispal, G.; Zollner, A.; Haid, A.; Neupert, W.; Lill, R. Heme binding to a conserved Cys-Pro-Val motif is crucial for the catalytic function of mitochondrial heme lyases. J. Biol. Chem. 1996, 271, 32605–32611. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dumont, M.E.; Cardillo, T.S.; Hayes, M.K.; Sherman, F. Role of cytochrome c heme lyase in mitochondrial import and accumulation of cytochrome c in Saccharomyces cerevisiae. Mol. Cell. Biol. 1991, 11, 5487–5496. [Google Scholar]
- Wang, X.; Dumont, M.E.; Sherman, F. Sequence requirements for mitochondrial import of yeast cytochrome c. J. Biol. Chem. 1996, 271, 6594–6604. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Richard-Fogal, C.L.; Frawley, E.R.; Bonner, E.R.; Zhu, H.; San Francisco, B.; Kranz, R.G. A conserved haem redox and trafficking pathway for cofactor attachment. EMBO J. 2009, 28, 2349–2359. [Google Scholar] [CrossRef] [PubMed]
- West, A.R.; Oates, P.S. Mechanisms of heme iron absorption: Current questions and controversies. World J. Gastroenterol. 2008, 14, 4101–4110. [Google Scholar] [CrossRef] [PubMed]
- Yi, L.; Jenkins, P.M.; Leichert, L.I.; Jakob, U.; Martens, J.R.; Ragsdale, S.W. Heme regulatory motifs in heme oxygenase-2 form a thiol/disulfide redox switch that responds to the cellular redox state. J. Biol. Chem. 2009, 284, 20556–20561. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fleming, M.D.; Hamza, I. Mitochondrial heme: An exit strategy at last. J. Clin. Investig. 2012, 122, 4328–4330. [Google Scholar] [CrossRef] [PubMed]
- Tsiftsoglou, A.S.; Tsamadou, A.I.; Papadopoulou, L.C. Heme as key regulator of major mammalian cellular functions: Molecular, cellular, and pharmacological aspects. Pharmacol. Ther. 2006, 111, 327–345. [Google Scholar] [CrossRef] [PubMed]
- Wijayanti, N.; Katz, N.; Immenschuh, S. Biology of heme in health and disease. Curr. Med. Chem. 2004, 11, 981–986. [Google Scholar] [CrossRef] [PubMed]
- Chiabrando, D.; Marro, S.; Mercurio, S.; Giorgi, C.; Petrillo, S.; Vinchi, F.; Fiorito, V.; Fagoonee, S.; Camporeale, A.; Turco, E.; et al. The mitochondrial heme exporter FLVCR1b mediates erythroid differentiation. J. Clin. Investig. 2012, 122, 4569–4579. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Byon, J.C.; Chen, J.; Doty, R.T.; Abkowitz, J.L. FLVCR is necessary for erythroid maturation, may contribute to platelet maturation, but is dispensable for normal hematopoietic stem cell function. Blood 2013, 122, 2903–2910. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ghosh, A.; Chawla-Sarkar, M.; Stuehr, D.J. Hsp90 interacts with inducible NO synthase client protein in its heme-free state and then drives heme insertion by an ATP-dependent process. FASEB J. 2011, 25, 2049–2060. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ghosh, A.; Stuehr, D.J. Soluble guanylyl cyclase requires heat shock protein 90 for heme insertion during maturation of the NO-active enzyme. Proc. Natl. Acad. Sci. USA 2012, 109, 12998–13003. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Billecke, S.S.; Draganov, D.I.; Morishima, Y.; Murphy, P.J.; Dunbar, A.Y.; Pratt, W.B.; Osawa, Y. The role of Hsp90 in heme-dependent activation of apo-neuronal nitric-oxide synthase. J. Biol. Chem. 2004, 279, 30252–30258. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dai, Y.; Faul, E.M.; Ghosh, A.; Stuehr, D.J. NO rapidly mobilizes cellular heme to trigger assembly of its own receptor. Proc. Natl. Acad. Sci. USA 2022, 119, e2115774119. [Google Scholar] [CrossRef] [PubMed]
- Ignarro, L.J.; Adams, J.B.; Horwitz, P.M.; Wood, K.S. Activation of soluble guanylate cyclase by NO-hemoproteins involves NO-heme exchange. Comparison of heme-containing and heme-deficient enzyme forms. J. Biol. Chem. 1986, 261, 4997–5002. [Google Scholar] [CrossRef]
- Chen, F.; Pandey, D.; Chadli, A.; Catravas, J.D.; Chen, T.; Fulton, D.J. Hsp90 regulates NADPH oxidase activity and is necessary for superoxide but not hydrogen peroxide production. Antioxid. Redox Signal. 2011, 14, 2107–2119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cai, H.; Harrison, D.G. Endothelial dysfunction in cardiovascular diseases: The role of oxidant stress. Circ. Res. 2000, 87, 840–844. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Griendling, K.K.; Sorescu, D.; Ushio-Fukai, M. NAD(P)H oxidase: Role in cardiovascular biology and disease. Circ. Res. 2000, 86, 494–501. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sweeny, E.A.; Schlanger, S.; Stuehr, D.J. Dynamic regulation of NADPH oxidase 5 by intracellular heme levels and cellular chaperones. Redox Biol. 2020, 36, 101656. [Google Scholar] [CrossRef]
- Ghosh, A.; Dai, Y.; Biswas, P.; Stuehr, D.J. Myoglobin maturation is driven by the Hsp90 chaperone machinery and by soluble guanylyl cyclase. FASEB J. 2019, 33, 9885–9896. [Google Scholar] [CrossRef] [Green Version]
- Tupta, B.; Stuehr, E.; Sumi, M.P.; Sweeny, E.A.; Smith, B.; Stuehr, D.J.; Ghosh, A. GAPDH is involved in the heme-maturation of myoglobin and hemoglobin. FASEB J. 2022, 36, e22099. [Google Scholar] [CrossRef]
- Zheng, Y.; Miyamoto, D.T.; Wittner, B.S.; Sullivan, J.P.; Aceto, N.; Jordan, N.V.; Yu, M.; Karabacak, N.M.; Comaills, V.; Morris, R.; et al. Expression of beta-globin by cancer cells promotes cell survival during blood-borne dissemination. Nat. Commun. 2017, 8, 14344. [Google Scholar] [CrossRef] [PubMed]
- Maman, S.; Sagi-Assif, O.; Yuan, W.; Ginat, R.; Meshel, T.; Zubrilov, I.; Keisari, Y.; Lu, W.; Lu, W.; Witz, I.P. The Beta Subunit of Hemoglobin (HBB2/HBB) Suppresses Neuroblastoma Growth and Metastasis. Cancer Res. 2017, 77, 14–26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kato, G.J.; Steinberg, M.H.; Gladwin, M.T. Intravascular hemolysis and the pathophysiology of sickle cell disease. J. Clin. Investig. 2017, 127, 750–760. [Google Scholar] [CrossRef] [PubMed]
- Sundd, P.; Gladwin, M.T.; Novelli, E.M. Pathophysiology of Sickle Cell Disease. Annu. Rev. Pathol. Mech. Dis. 2019, 14, 263–292. [Google Scholar] [CrossRef] [PubMed]
- Taher, A.T.; Weatherall, D.J.; Cappellini, M.D. Thalassaemia. Lancet 2018, 391, 155–167. [Google Scholar] [CrossRef]
- Wu, J.; Liu, T.; Rios, Z.; Mei, Q.; Lin, X.; Cao, S. Heat Shock Proteins and Cancer. Trends Pharmacol. Sci. 2017, 38, 226–256. [Google Scholar] [CrossRef] [PubMed]
- Luo, W.; Sun, W.; Taldone, T.; Rodina, A.; Chiosis, G. Heat shock protein 90 in neurodegenerative diseases. Mol. Neurodegener. 2010, 5, 24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trepel, J.; Mollapour, M.; Giaccone, G.; Neckers, L. Targeting the dynamic HSP90 complex in cancer. Nat. Rev. Cancer 2010, 10, 537–549. [Google Scholar] [CrossRef] [Green Version]
- Calderwood, S.K.; Neckers, L. Hsp90 in Cancer: Transcriptional Roles in the Nucleus. Adv. Cancer Res. 2016, 129, 89–106. [Google Scholar] [CrossRef] [PubMed]
- Franco, M.C.; Ricart, K.C.; Gonzalez, A.S.; Dennys, C.N.; Nelson, P.A.; Janes, M.S.; Mehl, R.A.; Landar, A.; Estévez, A.G. Nitration of Hsp90 on Tyrosine 33 Regulates Mitochondrial Metabolism. J. Biol. Chem. 2015, 290, 19055–19066. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miyata, Y.; Nakamoto, H.; Neckers, L. The therapeutic target Hsp90 and cancer hallmarks. Curr. Pharm. Des. 2013, 19, 347–365. [Google Scholar] [CrossRef] [PubMed]
- Calderwood, S.K.; Khaleque, M.A.; Sawyer, D.B.; Ciocca, D.R. Heat shock proteins in cancer: Chaperones of tumorigenesis. Trends Biochem. Sci. 2006, 31, 164–172. [Google Scholar] [CrossRef] [PubMed]
- Whitesell, L.; Lindquist, S.L. HSP90 and the chaperoning of cancer. Nat. Rev. Cancer 2005, 5, 761–772. [Google Scholar] [CrossRef]
- Ciocca, D.R.; Calderwood, S.K. Heat shock proteins in cancer: Diagnostic, prognostic, predictive, and treatment implications. Cell Stress Chaperones 2005, 10, 86–103. [Google Scholar] [CrossRef] [PubMed]
- Pandey, P.; Saleh, A.; Nakazawa, A.; Kumar, S.; Srinivasula, S.M.; Kumar, V.; Weichselbaum, R.; Nalin, C.; Alnemri, E.S.; Kufe, D.; et al. Negative regulation of cytochrome c-mediated oligomerization of Apaf-1 and activation of procaspase-9 by heat shock protein 90. EMBO J. 2000, 19, 4310–4322. [Google Scholar] [CrossRef] [Green Version]
- Zhang, R.; Luo, D.; Miao, R.; Bai, L.; Ge, Q.; Sessa, W.C.; Min, W. Hsp90—Akt phosphorylates ASK1 and inhibits ASK1-mediated apoptosis. Oncogene 2005, 24, 3954–3963. [Google Scholar] [CrossRef] [Green Version]
- Fulda, S.; Galluzzi, L.; Kroemer, G. Targeting mitochondria for cancer therapy. Nat. Rev. Drug Discov. 2010, 9, 447–464. [Google Scholar] [CrossRef] [PubMed]
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2018. CA Cancer J. Clin. 2018, 68, 7–30. [Google Scholar] [CrossRef]
- Shafi, A.A.; Yen, A.E.; Weigel, N.L. Androgen receptors in hormone-dependent and castration-resistant prostate cancer. Pharmacol. Ther. 2013, 140, 223–238. [Google Scholar] [CrossRef]
- Centenera, M.M.; Fitzpatrick, A.K.; Tilley, W.D.; Butler, L.M. Hsp90: Still a viable target in prostate cancer. Biochim. Biophys. Acta 2013, 1835, 211–218. [Google Scholar] [CrossRef] [PubMed]
- Hoter, A.; Rizk, S.; Naim, H.Y. The Multiple Roles and Therapeutic Potential of Molecular Chaperones in Prostate Cancer. Cancers 2019, 11, 1194. [Google Scholar] [CrossRef] [Green Version]
- Zagouri, F.; Sergentanis, T.N.; Chrysikos, D.; Papadimitriou, C.A.; Dimopoulos, M.A.; Psaltopoulou, T. Hsp90 inhibitors in breast cancer: A systematic review. Breast 2013, 22, 569–578. [Google Scholar] [CrossRef] [PubMed]
- Powers, M.V.; Workman, P. Targeting of multiple signalling pathways by heat shock protein 90 molecular chaperone inhibitors. Endocr. Relat. Cancer 2006, 13 (Suppl. 1), S125–S135. [Google Scholar] [CrossRef] [PubMed]
- Pick, E.; Kluger, Y.; Giltnane, J.M.; Moeder, C.; Camp, R.L.; Rimm, D.L.; Kluger, H.M. High HSP90 expression is associated with decreased survival in breast cancer. Cancer Res. 2007, 67, 2932–2937. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, W.; Wu, Y.; Wang, L.; Gao, L.; Wang, Y.; Liu, X.; Zhang, K.; Song, J.; Wang, H.; Bayer, T.A.; et al. Protein signature for non-small cell lung cancer prognosis. Am. J. Cancer Res. 2014, 4, 256–269. [Google Scholar] [PubMed]
- Senju, M.; Sueoka, N.; Sato, A.; Iwanaga, K.; Sakao, Y.; Tomimitsu, S.; Tominaga, M.; Irie, K.; Hayashi, S.; Sueoka, E. Hsp90 inhibitors cause G2/M arrest associated with the reduction of Cdc25C and Cdc2 in lung cancer cell lines. J. Cancer Res. Clin. Oncol. 2006, 132, 150–158. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.H.; Ji, J.H.; Park, K.T.; Lee, J.H.; Kang, K.W.; Park, J.H.; Hwang, S.W.; Lee, E.H.; Cho, Y.J.; Jeong, Y.Y.; et al. High-level expression of Hsp90beta is associated with poor survival in resectable non-small-cell lung cancer patients. Histopathology 2015, 67, 509–519. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Huang, B.; Liu, Q.; Liu, Y. Heat shock protein 90-beta over-expression is associated with poor survival in stage I lung adenocarcinoma patients. Int. J. Clin. Exp. Pathol. 2015, 8, 8252–8259. [Google Scholar] [PubMed]
- Hanahan, D.; Folkman, J. Patterns and emerging mechanisms of the angiogenic switch during tumorigenesis. Cell 1996, 86, 353–364. [Google Scholar] [CrossRef] [Green Version]
- Flonta, S.E.; Arena, S.; Pisacane, A.; Michieli, P.; Bardelli, A. Expression and functional regulation of myoglobin in epithelial cancers. Am. J. Pathol. 2009, 175, 201–206. [Google Scholar] [CrossRef] [Green Version]
- Bian, K.; Ghassemi, F.; Sotolongo, A.; Siu, A.; Shauger, L.; Kots, A.; Murad, F. NOS-2 signaling and cancer therapy. IUBMB Life 2012, 64, 676–683. [Google Scholar] [CrossRef] [PubMed]
- Fukumura, D.; Kashiwagi, S.; Jain, R.K. The role of nitric oxide in tumour progression. Nat. Rev. Cancer 2006, 6, 521–534. [Google Scholar] [CrossRef] [PubMed]
- Castro-Giner, F.; Aceto, N. Tracking cancer progression: From circulating tumor cells to metastasis. Genome Med. 2020, 12, 31. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ponzetti, M.; Capulli, M.; Angelucci, A.; Ventura, L.; Monache, S.D.; Mercurio, C.; Calgani, A.; Sanita, P.; Teti, A.; Rucci, N. Non-conventional role of haemoglobin beta in breast malignancy. Br. J. Cancer 2017, 117, 994–1006. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paul, S.; Mahanta, S. Association of heat-shock proteins in various neurodegenerative disorders: Is it a master key to open the therapeutic door? Mol. Cell. Biochem. 2014, 386, 45–61. [Google Scholar] [CrossRef] [PubMed]
- Selkoe, D.J.; Schenk, D. Alzheimer’s disease: Molecular understanding predicts amyloid-based therapeutics. Annu. Rev. Pharmacol. Toxicol. 2003, 43, 545–584. [Google Scholar] [CrossRef] [PubMed]
- Mandelkow, E.M.; Mandelkow, E. Biochemistry and cell biology of tau protein in neurofibrillary degeneration. Cold Spring Harb. Perspect. Med. 2012, 2, a006247. [Google Scholar] [CrossRef] [PubMed]
- Takalo, M.; Salminen, A.; Soininen, H.; Hiltunen, M.; Haapasalo, A. Protein aggregation and degradation mechanisms in neurodegenerative diseases. Am. J. Neurodegener. Dis. 2013, 2, 1–14. [Google Scholar] [PubMed]
- Sahara, N.; Murayama, M.; Mizoroki, T.; Urushitani, M.; Imai, Y.; Takahashi, R.; Murata, S.; Tanaka, K.; Takashima, A. In vivo evidence of CHIP up-regulation attenuating tau aggregation. J. Neurochem. 2005, 94, 1254–1263. [Google Scholar] [CrossRef] [PubMed]
- Evans, C.G.; Wisen, S.; Gestwicki, J.E. Heat shock proteins 70 and 90 inhibit early stages of amyloid beta-(1–42) aggregation in vitro. J. Biol. Chem. 2006, 281, 33182–33191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peterson, L.B.; Blagg, B.S. To fold or not to fold: Modulation and consequences of Hsp90 inhibition. Future Med. Chem. 2009, 1, 267–283. [Google Scholar] [CrossRef] [Green Version]
- Hu, S.; Tan, J.; Qin, L.; Lv, L.; Yan, W.; Zhang, H.; Tang, B.; Wang, C. Molecular chaperones and Parkinson’s disease. Neurobiol. Dis. 2021, 160, 105527. [Google Scholar] [CrossRef]
- Schneider, S.A.; Obeso, J.A. Clinical and pathological features of Parkinson’s disease. Behav. Neurobiol. Huntingt. Dis. Parkinson’s Dis. 2015, 22, 205–220. [Google Scholar] [CrossRef]
- Poewe, W.; Seppi, K.; Tanner, C.M.; Halliday, G.M.; Brundin, P.; Volkmann, J.; Schrag, A.E.; Lang, A.E. Parkinson disease. Nat. Rev. Dis. Primers 2017, 3, 17013. [Google Scholar] [CrossRef] [PubMed]
- Falsone, S.F.; Kungl, A.J.; Rek, A.; Cappai, R.; Zangger, K. The molecular chaperone Hsp90 modulates intermediate steps of amyloid assembly of the Parkinson-related protein alpha-synuclein. J. Biol. Chem. 2009, 284, 31190–31199. [Google Scholar] [CrossRef] [Green Version]
- Lee, C.T.; Graf, C.; Mayer, F.J.; Richter, S.M.; Mayer, M.P. Dynamics of the regulation of Hsp90 by the co-chaperone Sti1. EMBO J. 2012, 31, 1518–1528. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pratt, W.B.; Gestwicki, J.E.; Osawa, Y.; Lieberman, A.P. Targeting Hsp90/Hsp70-based protein quality control for treatment of adult onset neurodegenerative diseases. Annu. Rev. Pharmacol. Toxicol. 2015, 55, 353–371. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiong, R.; Zhou, W.; Siegel, D.; Kitson, R.R.; Freed, C.R.; Moody, C.J.; Ross, D. A Novel Hsp90 Inhibitor Activates Compensatory Heat Shock Protein Responses and Autophagy and Alleviates Mutant A53T alpha-Synuclein Toxicity. Mol. Pharmacol. 2015, 88, 1045–1054. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neckers, L.; Workman, P. Hsp90 molecular chaperone inhibitors: Are we there yet? Clin. Cancer Res. 2012, 18, 64–76. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ghosh, S.; Erzurum, S.C. Nitric oxide metabolism in asthma pathophysiology. Biochim. Biophys. Acta Gen. Subj. 2011, 1810, 1008–1016. [Google Scholar] [CrossRef] [Green Version]
- Mora, A.L.; Rojas, M.; Pardo, A.; Selman, M. Emerging therapies for idiopathic pulmonary fibrosis, a progressive age-related disease. Nat. Rev. Drug Discov. 2017, 16, 755–772. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stasch, J.P.; Pacher, P.; Evgenov, O.V. Soluble guanylate cyclase as an emerging therapeutic target in cardiopulmonary disease. Circulation 2011, 123, 2263–2273. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bryan, N.S.; Bian, K.; Murad, F. Discovery of the nitric oxide signaling pathway and targets for drug development. Front. Biosci. 2009, 14, 1–18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murad, F. Shattuck Lecture. Nitric oxide and cyclic GMP in cell signaling and drug development. N. Engl. J. Med. 2006, 355, 2003–2011. [Google Scholar] [CrossRef] [PubMed]
- Guo, F.H.; Uetani, K.; Haque, S.J.; Williams, B.R.; Dweik, R.A.; Thunnissen, F.B.; Calhoun, W.; Erzurum, S.C. Interferon gamma and interleukin 4 stimulate prolonged expression of inducible nitric oxide synthase in human airway epithelium through synthesis of soluble mediators. J. Clin. Investig. 1997, 100, 829–838. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pankey, E.A.; Bhartiya, M.; Badejo, A.M., Jr.; Haider, U.; Stasch, J.P.; Murthy, S.N.; Nossaman, B.D.; Kadowitz, P.J. Pulmonary and systemic vasodilator responses to the soluble guanylyl cyclase activator, BAY 60–2770, are not dependent on endogenous nitric oxide or reduced heme. Am. J. Physiol. Heart Circ. Physiol. 2011, 300, H792–H802. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dong, H.M.; Le, Y.Q.; Wang, Y.H.; Zhao, H.J.; Huang, C.W.; Hu, Y.H.; Luo, L.S.; Wan, X.; Wei, Y.L.; Chu, Z.Q.; et al. Extracellular heat shock protein 90alpha mediates HDM-induced bronchial epithelial barrier dysfunction by activating RhoA/MLC signaling. Respir. Res. 2017, 18, 111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pezzulo, A.A.; Tudas, R.A.; Stewart, C.G.; Buonfiglio, L.G.V.; Lindsay, B.D.; Taft, P.J.; Gansemer, N.D.; Zabner, J. HSP90 inhibitor geldanamycin reverts IL-13–and IL-17–induced airway goblet cell metaplasia. J. Clin. Investig. 2019, 129, 744–758. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Intapad, S.; Dimitropoulou, C.; Snead, C.; Piyachaturawat, P.; Catravas, J.D. Regulation of asthmatic airway relaxation by estrogen and heat shock protein 90. J. Cell. Physiol. 2012, 227, 3036–3043. [Google Scholar] [CrossRef]
- Dai, Z.; Zhu, M.M.; Peng, Y.; Machireddy, N.; Evans, C.E.; Machado, R.; Zhang, X.; Zhao, Y.Y. Therapeutic Targeting of Vascular Remodeling and Right Heart Failure in Pulmonary Arterial Hypertension with a HIF-2alpha Inhibitor. Am. J. Respir. Crit. Care Med. 2018, 198, 1423–1434. [Google Scholar] [CrossRef]
- Kang, B.H.; Plescia, J.; Song, H.Y.; Meli, M.; Colombo, G.; Beebe, K.; Scroggins, B.; Neckers, L.; Altieri, D.C. Combinatorial drug design targeting multiple cancer signaling networks controlled by mitochondrial Hsp90. J. Clin. Investig. 2009, 119, 454–464. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hopkins, R.B.; Burke, N.; Fell, C.; Dion, G.; Kolb, M. Epidemiology and survival of idiopathic pulmonary fibrosis from national data in Canada. Eur. Respir. J. 2016, 48, 187–195. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Overgaard, D.; Kaldan, G.; Marsaa, K.; Nielsen, T.L.; Shaker, S.B.; Egerod, I. The lived experience with idiopathic pulmonary fibrosis: A qualitative study. Eur. Respir. J. 2016, 47, 1472–1480. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Froese, A.R.; Shimbori, C.; Bellaye, P.S.; Inman, M.; Obex, S.; Fatima, S.; Jenkins, G.; Gauldie, J.; Ask, K.; Kolb, M. Stretch-induced Activation of Transforming Growth Factor-beta1 in Pulmonary Fibrosis. Am. J. Respir. Crit. Care Med. 2016, 194, 84–96. [Google Scholar] [CrossRef] [PubMed]
- Sibinska, Z.; Tian, X.; Korfei, M.; Kojonazarov, B.; Kolb, J.S.; Klepetko, W.; Kosanovic, D.; Wygrecka, M.; Ghofrani, H.A.; Weissmann, N.; et al. Amplified canonical transforming growth factor-beta signalling via heat shock protein 90 in pulmonary fibrosis. Eur. Respir. J. 2017, 49, 1501941. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Solopov, P.; Biancatelli, R.; Marinova, M.; Dimitropoulou, C.; Catravas, J.D. The HSP90 Inhibitor, AUY-922, Ameliorates the Development of Nitrogen Mustard-Induced Pulmonary Fibrosis and Lung Dysfunction in Mice. Int. J. Mol. Sci. 2020, 21, 4740. [Google Scholar] [CrossRef] [PubMed]
- Marinova, M.; Solopov, P.; Dimitropoulou, C.; Colunga Biancatelli, R.M.L.; Catravas, J.D. Post-treatment with a heat shock protein 90 inhibitor prevents chronic lung injury and pulmonary fibrosis, following acute exposure of mice to HCl. Exp. Lung Res. 2020, 46, 203–216. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Wu, D.; Deng, S.; Li, J.; Zhang, F.; Zou, Y.; Zhang, T.; Xu, Y. NVP-AUY922 alleviates radiation-induced lung injury via inhibition of autophagy-dependent ferroptosis. Cell. Death Discov. 2022, 8, 86. [Google Scholar] [CrossRef] [PubMed]
- Sontake, V.; Wang, Y.; Kasam, R.K.; Sinner, D.; Reddy, G.B.; Naren, A.P.; McCormack, F.X.; White, E.S.; Jegga, A.G.; Madala, S.K. Hsp90 regulation of fibroblast activation in pulmonary fibrosis. JCI Insight 2017, 2, e91454. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sandner, P.; Berger, P.; Zenzmaier, C. The Potential of sGC Modulators for the Treatment of Age-Related Fibrosis: A Mini-Review. Gerontology 2017, 63, 216–227. [Google Scholar] [CrossRef] [PubMed]
- Verbeke, P.; Fonager, J.; Clark, B.F.; Rattan, S.I. Heat shock response and ageing: Mechanisms and applications. Cell Biol. Int. 2001, 25, 845–857. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- DeBoer, C.; Meulman, P.A.; Wnuk, R.J.; Peterson, D.H. Geldanamycin, a new antibiotic. J. Antibiot. 1970, 23, 442–447. [Google Scholar] [CrossRef] [PubMed]
- Chiosis, G.; Tao, H. Purine-scaffold Hsp90 inhibitors. Curr. Top. Med. Chem. 2006, 9, 778–782. [Google Scholar]
- Eccles, S.A.; Massey, A.; Raynaud, F.I.; Sharp, S.Y.; Box, G.; Valenti, M.; Patterson, L.; de Haven Brandon, A.; Gowan, S.; Boxall, F.; et al. NVP-AUY922: A novel heat shock protein 90 inhibitor active against xenograft tumor growth, angiogenesis, and metastasis. Cancer Res. 2008, 68, 2850–2860. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fuhrmann-Stroissnigg, H.; Ling, Y.Y.; Zhao, J.; McGowan, S.J.; Zhu, Y.; Brooks, R.W.; Grassi, D.; Gregg, S.Q.; Stripay, J.L.; Dorronsoro, A.; et al. Identification of HSP90 inhibitors as a novel class of senolytics. Nat. Commun. 2017, 8, 422. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Chen, M.; Zhou, J.; Zhang, X. HSP27, 70 and 90, anti-apoptotic proteins, in clinical cancer therapy (Review). Int. J. Oncol. 2014, 45, 18–30. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koay, Y.C.; Wahyudi, H.; McAlpine, S.R. Reinventing Hsp90 Inhibitors: Blocking C-Terminal Binding Events to Hsp90 by Using Dimerized Inhibitors. Chem. A Eur. J. 2016, 22, 18572–18582. [Google Scholar] [CrossRef]
- Donnelly, A.; Blagg, B.S. Novobiocin and additional inhibitors of the Hsp90 C-terminal nucleotide-binding pocket. Curr. Med. Chem. 2008, 15, 2702–2717. [Google Scholar] [CrossRef] [Green Version]
- Solarova, Z.; Mojzis, J.; Solar, P. Hsp90 inhibitor as a sensitizer of cancer cells to different therapies (review). Int. J. Oncol. 2015, 46, 907–926. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Allan, R.K.; Mok, D.; Ward, B.K.; Ratajczak, T. Modulation of chaperone function and cochaperone interaction by novobiocin in the C-terminal domain of Hsp90: Evidence that coumarin antibiotics disrupt Hsp90 dimerization. J. Biol. Chem. 2006, 281, 7161–7171. [Google Scholar] [CrossRef] [Green Version]
- Eskew, J.D.; Sadikot, T.; Morales, P.; Duren, A.; Dunwiddie, I.; Swink, M.; Zhang, X.; Hembruff, S.; Donnelly, A.; Rajewski, R.A.; et al. Development and characterization of a novel C-terminal inhibitor of Hsp90 in androgen dependent and independent prostate cancer cells. BMC Cancer 2011, 11, 468. [Google Scholar] [CrossRef] [Green Version]
- Neckers, L.; Kern, A.; Tsutsumi, S. Hsp90 inhibitors disrupt mitochondrial homeostasis in cancer cells. Chem. Biol. 2007, 14, 1204–1206. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiao, Y.; Liu, Y. Recent Advances in the Discovery of Novel HSP90 Inhibitors: An Update from 2014. Curr. Drug Targets 2020, 21, 302–317. [Google Scholar] [CrossRef] [PubMed]
- Chatterjee, S.; Burns, T.F. Targeting Heat Shock Proteins in Cancer: A Promising Therapeutic Approach. Int. J. Mol. Sci. 2017, 18, 1978. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, H.K.; Yoon, N.G.; Lee, J.E.; Hu, S.; Yoon, S.; Kim, S.Y.; Hong, J.H.; Nam, D.; Chae, Y.C.; Park, J.B.; et al. Unleashing the full potential of Hsp90 inhibitors as cancer therapeutics through simultaneous inactivation of Hsp90, Grp94, and TRAP1. Exp. Mol. Med. 2020, 52, 79–91. [Google Scholar] [CrossRef] [Green Version]
- Fadden, P.; Huang, K.H.; Veal, J.M.; Steed, P.M.; Barabasz, A.F.; Foley, B.; Hu, M.; Partridge, J.M.; Rice, J.; Scott, A.; et al. Application of chemoproteomics to drug discovery: Identification of a clinical candidate targeting Hsp90. Chem. Biol. 2010, 17, 686–694. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Socinski, M.A.; Goldman, J.; El-Hariry, I.; Koczywas, M.; Vukovic, V.; Horn, L.; Paschold, E.; Salgia, R.; West, H.; Sequist, L.V.; et al. A multicenter phase II study of ganetespib monotherapy in patients with genotypically defined advanced non-small cell lung cancer. Clin. Cancer Res. 2013, 19, 3068–3077. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Modi, S.; Stopeck, A.; Linden, H.; Solit, D.; Chandarlapaty, S.; Rosen, N.; D’Andrea, G.; Dickler, M.; Moynahan, M.E.; Sugarman, S.; et al. HSP90 inhibition is effective in breast cancer: A phase II trial of tanespimycin (17-AAG) plus trastuzumab in patients with HER2-positive metastatic breast cancer progressing on trastuzumab. Clin. Cancer Res. 2011, 17, 5132–5139. [Google Scholar] [CrossRef] [Green Version]
- Do, K.; Speranza, G.; Chang, L.C.; Polley, E.C.; Bishop, R.; Zhu, W.; Trepel, J.B.; Lee, S.; Lee, M.J.; Kinders, R.J.; et al. Phase I study of the heat shock protein 90 (Hsp90) inhibitor onalespib (AT13387) administered on a daily for 2 consecutive days per week dosing schedule in patients with advanced solid tumors. Investig. New Drugs 2015, 33, 921–930. [Google Scholar] [CrossRef] [PubMed]
- Pillai, R.N.; Ramalingam, S.S. Hsp90 inhibitors. J. Thorac. Oncol. 2012, 7, S407–S408. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wiesgigl, M.; Clos, J. Heat shock protein 90 homeostasis controls stage differentiation in Leishmania donovani. Mol. Biol. Cell 2001, 12, 3307–3316. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Graefe, S.E.; Wiesgigl, M.; Gaworski, I.; Macdonald, A.; Clos, J. Inhibition of HSP90 in Trypanosoma cruzi induces a stress response but no stage differentiation. Eukaryot. Cell 2002, 1, 936–943. [Google Scholar] [CrossRef] [Green Version]
- Banumathy, G.; Singh, V.; Pavithra, S.R.; Tatu, U. Heat shock protein 90 function is essential for Plasmodium falciparum growth in human erythrocytes. J. Biol. Chem. 2003, 278, 18336–18345. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Angel, S.O.; Matrajt, M.; Echeverria, P.C. A review of recent patents on the protozoan parasite HSP90 as a drug target. Recent Pat. Biotechnol. 2013, 7, 2–8. [Google Scholar] [CrossRef] [PubMed]
- Pizarro, J.C.; Hills, T.; Senisterra, G.; Wernimont, A.K.; Mackenzie, C.; Norcross, N.R.; Ferguson, M.A.; Wyatt, P.G.; Gilbert, I.H.; Hui, R. Exploring the Trypanosoma brucei Hsp83 potential as a target for structure guided drug design. PLoS Negl. Trop. Dis. 2013, 7, e2492. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gillan, V.; O’Neill, K.; Maitland, K.; Sverdrup, F.M.; Devaney, E. A repurposing strategy for Hsp90 inhibitors demonstrates their potency against filarial nematodes. PLoS Negl. Trop. Dis. 2014, 8, e2699. [Google Scholar] [CrossRef] [Green Version]
- Guswanto, A.; Nugraha, A.B.; Tuvshintulga, B.; Tayebwa, D.S.; Rizk, M.A.; Batiha, G.E.; Gantuya, S.; Sivakumar, T.; Yokoyama, N.; Igarashi, I. 17-DMAG inhibits the multiplication of several Babesia species and Theileria equi on in vitro cultures, and Babesia microti in mice. Int. J. Parasitol. Drugs Drug Resist. 2018, 8, 104–111. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Liu, F.; Liu, J.; Wang, D.; Yan, Y.; Ji, S.; Zan, J.; Zhou, J. The co-chaperone Cdc37 regulates the rabies virus phosphoprotein stability by targeting to Hsp90AA1 machinery. Sci. Rep. 2016, 6, 27123. [Google Scholar] [CrossRef]
- Geller, R.; Andino, R.; Frydman, J. Hsp90 inhibitors exhibit resistance-free antiviral activity against respiratory syncytial virus. PLoS ONE 2013, 8, e56762. [Google Scholar] [CrossRef]
- Alsamman, A.M.; Zayed, H. The transcriptomic profiling of SARS-CoV-2 compared to SARS, MERS, EBOV, and H1N1. PLoS ONE 2020, 15, e0243270. [Google Scholar] [CrossRef] [PubMed]
- Gomez-Monterrey, I.; Sala, M.; Musella, S.; Campiglia, P. Heat shock protein 90 inhibitors as therapeutic agents. Recent Pat. Anticancer. Drug Discov. 2012, 7, 313–336. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taldone, T.; Ochiana, S.O.; Patel, P.D.; Chiosis, G. Selective targeting of the stress chaperome as a therapeutic strategy. Trends Pharmacol. Sci. 2014, 35, 592–603. [Google Scholar] [CrossRef] [PubMed] [Green Version]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Disease | Hsp90 Inhibitors | Mechanism | Results |
|---|---|---|---|
| Colorectal cancer | Cetuximab | VEGF/VEGFR signaling pathway | Block angiogenesis |
| Panitumumab | |||
| Bevacizumab | |||
| Regorafenib | |||
| Ziv-aflibercept | |||
| Prostate cancer | Ganetespid | PI3K/mTOR signaling pathway | Tumour cell death Growth inhibition |
| Brest cancer | 17 AAG (Demethoxygeldanamycin) (tanespimycin) | P378/MEPK, EGFR pathway | Growth inhibition |
| Lung cancer | AUY922 | AR and PI3K/mTOR RAF/MEK/ERK pathway | Antitumor activity |
| CS-6 | Targeting IKKβ/NF-κB pathway | Growth inhibition | |
| Rheumatoid arthiritis, Inflammatory bowel disease, Osteoarthritis | Celastrol | RAF/MEK/ERK and PI3K/AKT/mTOR signaling pathways | Anti-inflammatory effect, Induce apoptosis |
| Prostate cancer, colon, and ovarian cancer | Gedunin | Disruptor of Hsp 90-p23 interaction | Ant proliferative activity |
| Gynecological cancer, Gastrointestinal cancer, Thyroid cancers and other cancers | Withaferin A | Hsp90-Cdc 37 | Antitumor activity |
| Derrubone | Disruptor of Hsp 90-Cdc 37 interaction | Anticancerous activity | |
| Cruentaren A | Hsp90/F1F0 ATP synthase disruptor | Antitumor activity. Highly cytotoxic to different cell lines |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sumi, M.P.; Ghosh, A. Hsp90 in Human Diseases: Molecular Mechanisms to Therapeutic Approaches. Cells 2022, 11, 976. https://doi.org/10.3390/cells11060976
Sumi MP, Ghosh A. Hsp90 in Human Diseases: Molecular Mechanisms to Therapeutic Approaches. Cells. 2022; 11(6):976. https://doi.org/10.3390/cells11060976
Chicago/Turabian StyleSumi, Mamta P., and Arnab Ghosh. 2022. "Hsp90 in Human Diseases: Molecular Mechanisms to Therapeutic Approaches" Cells 11, no. 6: 976. https://doi.org/10.3390/cells11060976
APA StyleSumi, M. P., & Ghosh, A. (2022). Hsp90 in Human Diseases: Molecular Mechanisms to Therapeutic Approaches. Cells, 11(6), 976. https://doi.org/10.3390/cells11060976