
Deciphering Haplotypic Variation and Gene Expression Dynamics Associated with Nutritional and Cooking Quality in Rice

 , , ,
, , ,  ,
, 
Abstract
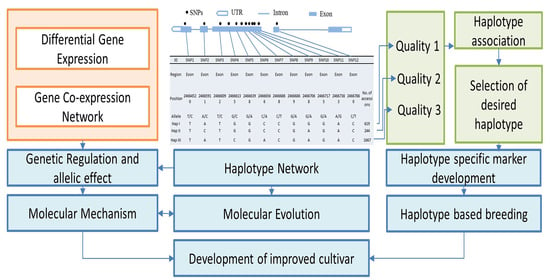
:
1. Introduction
2. Materials and Methods
2.1. Selection of Genes Governing Cooking Quality and Nutritional Value Related Traits
2.2. SNP Variation and Effect Prediction
2.3. Haplotype Variation in Nutritional Quality-Related Genes
2.4. Haplotype Network Analysis
2.5. Gene Expression Dynamics and Co-Expression Network for Grain Quality-Related Traits in Rice
2.6. Quantitative Real-Time PCR Analysis
2.7. Transcription Factors and Their Interaction with Nutritional Quality-Related Genes
2.8. Phenotyping of Rice Grains for Chalkiness Trait
3. Results
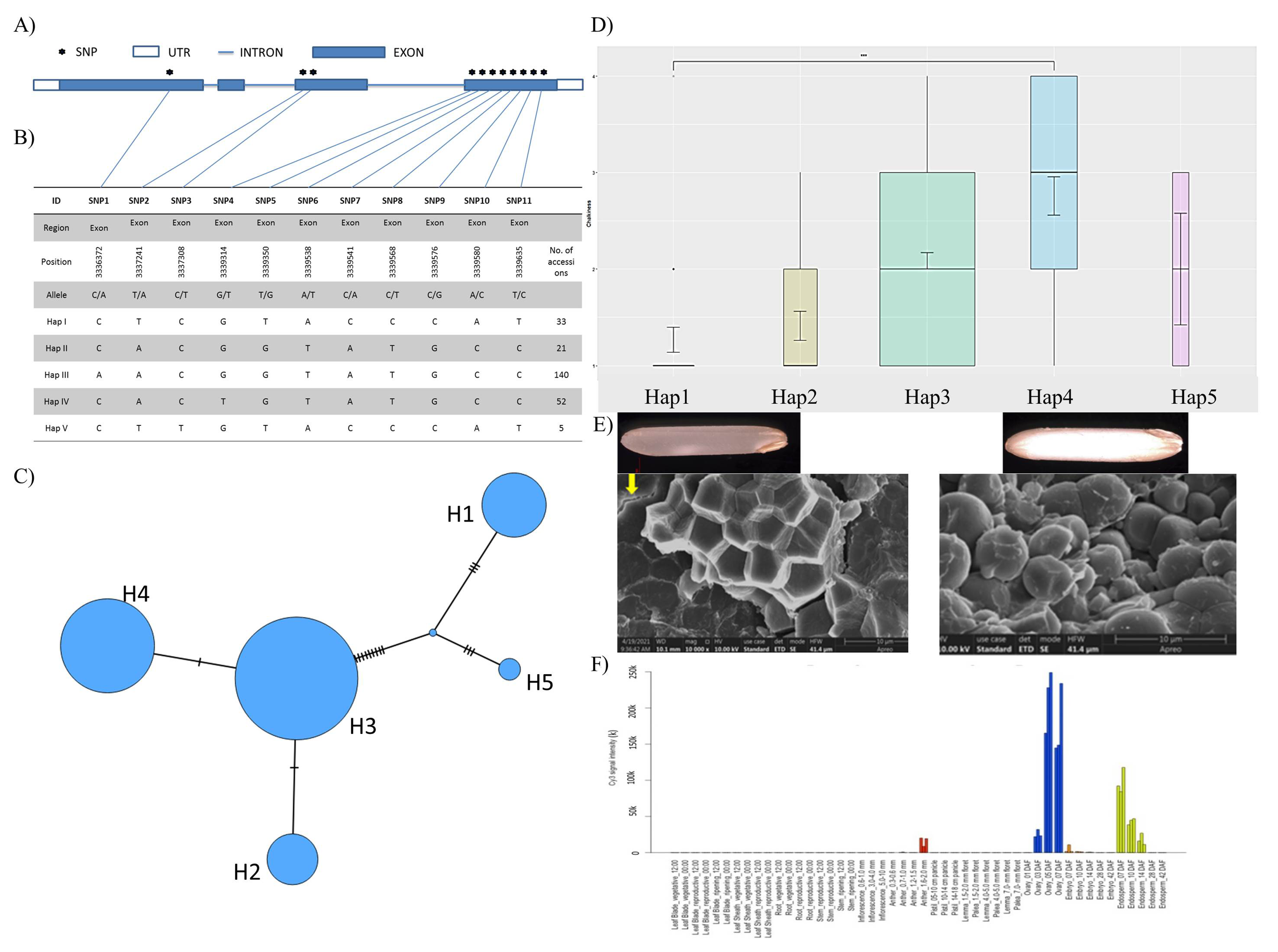
3.1. Haplotypic Diversity in Nutritional and Cooking Quality-Related Genes in Rice
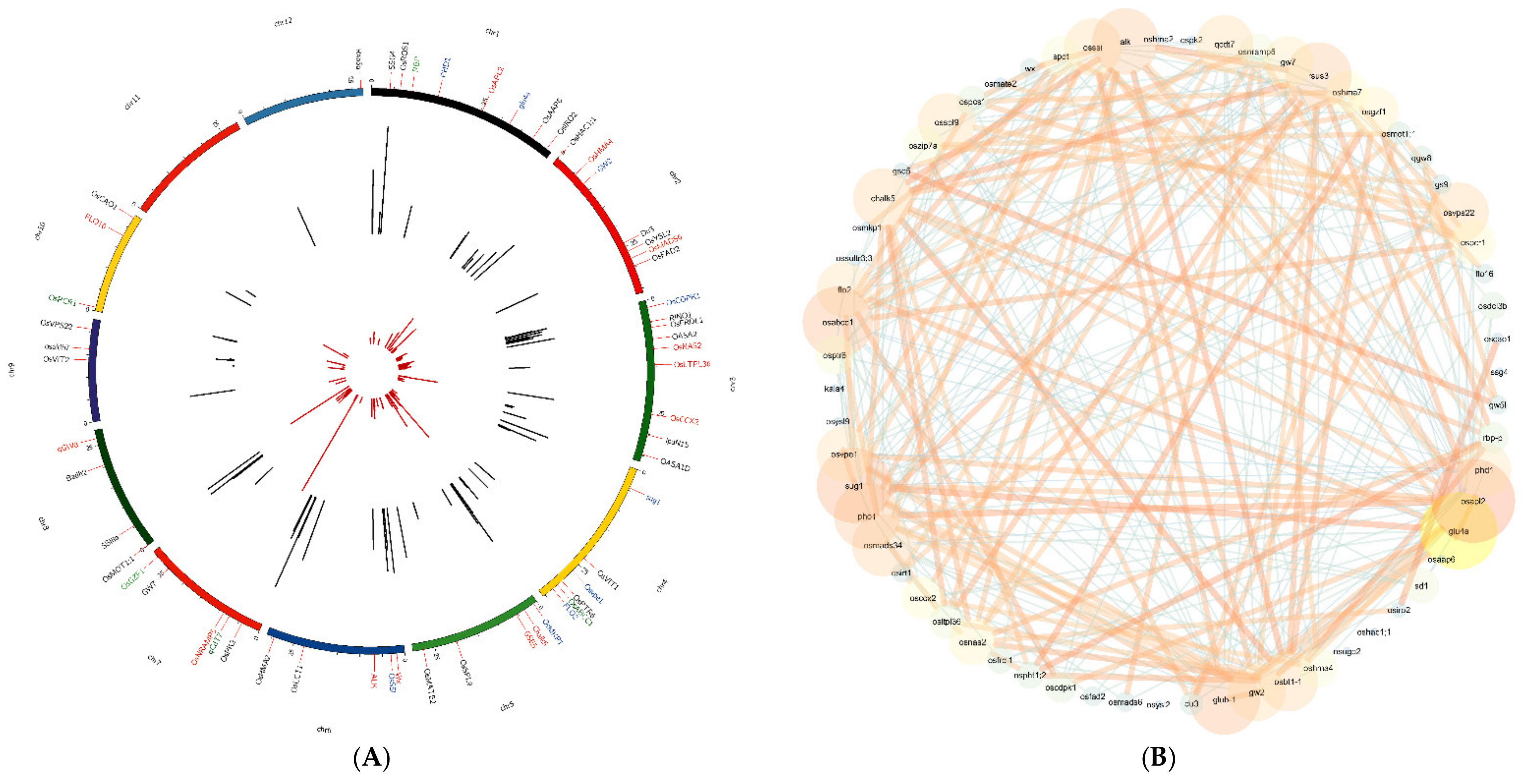
3.2. Haplotype Network Defining the Evolution of Important Rice Genes
Diversity Analysis
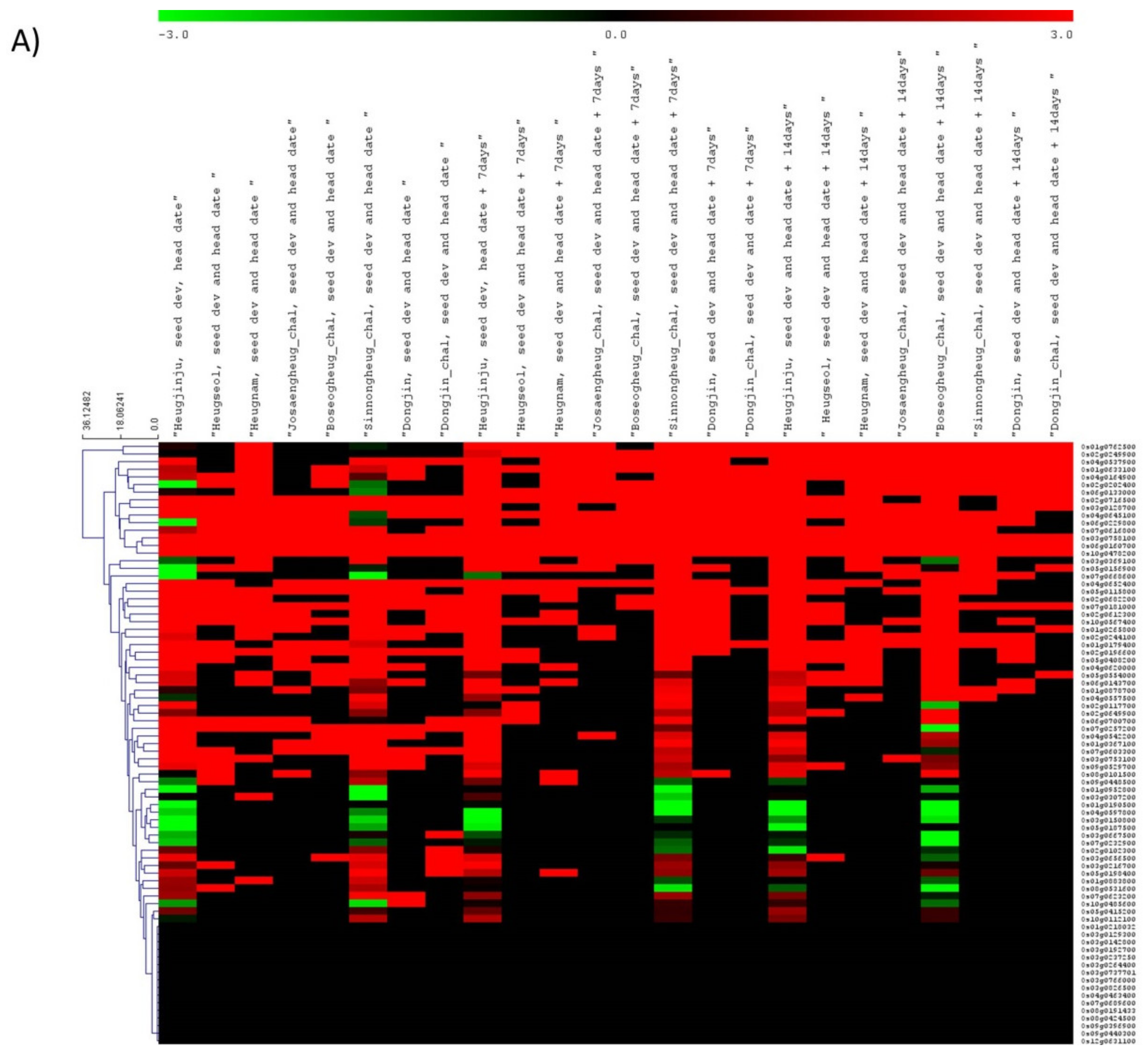
3.3. Gene Expression Dynamics for Grain Quality-Related Traits in Rice
3.4. Hub Genes Identified through Gene Co-Expression Network Analysis in Rice
3.5. Interaction of Transcription Factors and Nutritional Quality-Related Genes
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Rana, N.; Rahim, M.S.; Kaur, G.; Bansal, R.; Kumawat, S.; Roy, J.; Deshmukh, R.; Sonah, H.; Sharma, T.R. Applications and challenges for efficient exploration of omics interventions for the enhancement of nutritional quality in rice (Oryza sativa L.). Crit. Rev. Food Sci. Nutr. 2020, 60, 3304–3320. [Google Scholar] [CrossRef] [PubMed]
- Awika, J.M. Major cereal grains production and use around the world. In Advances in Cereal Science: Implications to Food Processing and Health Promotion; ACS Publications: Washington, DC, USA, 2011; pp. 1–13. [Google Scholar]
- Das, P.; Adak, S.; Majumder, A.L. Genetic manipulation for improved nutritional quality in rice. Front. Genet. 2020, 11, 776. [Google Scholar] [CrossRef] [PubMed]
- Kasote, D.; Sreenivasulu, N.; Acuin, C.; Regina, A. Enhancing health benefits of milled rice: Current status and future perspectives. Crit. Rev. Food Sci. Nutr. 2021, 1–21. [Google Scholar] [CrossRef] [PubMed]
- Hedden, P. The genes of the Green Revolution. Trends Genet. 2003, 19, 5–9. [Google Scholar] [CrossRef]
- Li, N.; Xu, R.; Duan, P.; Li, Y. Control of grain size in rice. Plant Reprod. 2018, 31, 237–251. [Google Scholar] [CrossRef]
- Wang, S.; Wu, K.; Yuan, Q.; Liu, X.; Liu, Z.; Lin, X.; Zeng, R.; Zhu, H.; Dong, G.; Qian, Q.; et al. Control of grain size, shape and quality by OsSPL16 in rice. Nat. Genet. 2012, 44, 950. [Google Scholar] [CrossRef]
- Si, L.; Chen, J.; Huang, X.; Gong, H.; Luo, J.; Hou, Q.; Zhou, T.; Lu, T.; Zhu, J.; Shangguan, Y.; et al. OsSPL13 controls grain size in cultivated rice. Nat. Genet. 2016, 48, 447–456. [Google Scholar] [CrossRef]
- Che, R.; Tong, H.; Shi, B.; Liu, Y.; Fang, S.; Liu, D.; Xiao, Y.; Hu, B.; Liu, L.; Wang, H.; et al. Control of grain size and rice yield by GL2-mediated brassinosteroid responses. Nat. Plants 2015, 2, 15195. [Google Scholar] [CrossRef]
- Wang, Y.; Xiong, G.; Hu, J.; Jiang, L.; Yu, H.; Xu, J.; Fang, Y.; Zeng, L.; Xu, E.; Xu, J.; et al. Copy number variation at the GL7 locus contributes to grain size diversity in rice. Nat. Genet. 2015, 47, 944–948. [Google Scholar] [CrossRef]
- Kashiwagi, T.; Munakata, J. Identification and characteristics of quantitative trait locus for grain protein content, TGP12, in rice (Oryza sativa L.). Euphytica 2018, 214, 165. [Google Scholar] [CrossRef]
- Peng, B.; Kong, H.; Li, Y.; Wang, L.; Zhong, M.; Sun, L.; Gao, G.; Zhang, Q.; Luo, L.; Wang, G.; et al. OsAAP6 functions as an important regulator of grain protein content and nutritional quality in rice. Nat. Commun. 2014, 5, 4847. [Google Scholar] [CrossRef] [Green Version]
- Yang, Y.; Guo, M.; Sun, S.; Zou, Y.; Yin, S.; Liu, Y.; Tang, S.; Gu, M.; Yang, Z.; Yan, C. Natural variation of OsGluA2 is involved in grain protein content regulation in rice. Nat. Commun. 2019, 10, 1949. [Google Scholar] [CrossRef]
- 3000 Rice Genomes Project. The 3000 rice genomes project. GigaScience 2014, 3, 2047-217X. [Google Scholar]
- Lachagari, V.; Gupta, R.; Lekkala, S.P.; Mahadevan, L.; Kuriakose, B.; Chakravartty, N.; Mohan Katta, A.; Santhosh, S.; Reddy, A.R.; Thomas, G. Whole genome sequencing and comparative genomic analysis reveal allelic variations unique to a purple colored rice landrace (Oryza sativa ssp. indica cv. Purpleputtu). Front. Plant Sci. 2019, 10, 513. [Google Scholar] [CrossRef] [Green Version]
- Zhao, H.; Yao, W.; Ouyang, Y.; Yang, W.; Wang, G.; Lian, X.; Xing, Y.; Chen, L.; Xie, W. RiceVarMap: A comprehensive database of rice genomic variations. Nucleic Acids Res. 2015, 43, D1018–D1022. [Google Scholar] [CrossRef]
- Huang, X.; Zhao, Y.; Wei, X.; Li, C.; Wang, A.; Zhao, Q.; Li, W.; Guo, Y.; Deng, L.; Zhu, C.; et al. Genome-wide association study of flowering time and grain yield traits in a worldwide collection of rice germplasm. Nat. Genet. 2012, 44, 32–39. [Google Scholar] [CrossRef]
- Chen, W.; Gao, Y.; Xie, W.; Gong, L.; Lu, K.; Wang, W.; Li, Y.; Liu, X.; Zhang, H.; Dong, H.; et al. Genome-wide association analyses provide genetic and biochemical insights into natural variation in rice metabolism. Nat. Genet. 2014, 46, 714–721. [Google Scholar] [CrossRef]
- Xu, X.; Liu, X.; Ge, S.; Jensen, J.D.; Hu, F.; Dong, Y.; Gutenkunst, R.N.; Fang, L.; Huang, L.; Li, J.; et al. Resequencing 50 accessions of cultivated and wild rice yields markers for identifying agronomically important genes. Nat. Biotechnol. 2012, 30, 105–111. [Google Scholar] [CrossRef]
- Qin, P.; Lu, H.; Du, H.; Wang, H.; Chen, W.; Chen, Z.; He, Q.; Ou, S.; Zhang, H.; Li, X.; et al. Pan-genome analysis of 33 genetically diverse rice accessions reveals hidden genomic variations. Cell 2021, 184, 3542–3558. [Google Scholar] [CrossRef]
- Wei, X.; Qiu, J.; Yong, K.; Fan, J.; Zhang, Q.; Hua, H.; Liu, J.; Wang, Q.; Olsen, K.M.; Han, B.; et al. A quantitative genomics map of rice provides genetic insights and guides breeding. Nat. Genet. 2021, 53, 243–253. [Google Scholar] [CrossRef]
- Abbai, R.; Singh, V.K.; Nachimuthu, V.V.; Sinha, P.; Selvaraj, R.; Vipparla, A.K.; Singh, A.K.; Singh, U.M.; Varshney, R.K.; Kumar, A. Haplotype analysis of key genes governing grain yield and quality traits across 3K RG panel reveals scope for the development of tailor-made rice with enhanced genetic gains. Plant Biotechnol. J. 2019, 17, 1612–1622. [Google Scholar] [CrossRef] [Green Version]
- Rasheed, A.; Hao, Y.; Xia, X.; Khan, A.; Xu, Y.; Varshney, R.K.; He, Z. Crop breeding chips and genotyping platforms: Progress, challenges, and perspectives. Mol. Plant 2017, 10, 1047–1064. [Google Scholar] [CrossRef] [Green Version]
- Yao, W.; Li, G.; Yu, Y.; Ouyang, Y. funRiceGenes dataset for comprehensive understanding and application of rice functional genes. Gigascience 2018, 7, gix119. [Google Scholar] [CrossRef] [Green Version]
- Yamamoto, E.; Yonemaru, J.-I.; Yamamoto, T.; Yano, M. OGRO: The Overview of functionally characterized Genes in Rice online database. Rice 2012, 5, 26. [Google Scholar] [CrossRef] [Green Version]
- Bradbury, P.J.; Zhang, Z.; Kroon, D.E.; Casstevens, T.M.; Ramdoss, Y.; Buckler, E.S. TASSEL: Software for association mapping of complex traits in diverse samples. Bioinformatics 2007, 23, 2633–2635. [Google Scholar] [CrossRef]
- McLaren, W.; Gil, L.; Hunt, S.E.; Riat, H.S.; Ritchie, G.R.; Thormann, A.; Flicek, P.; Cunningham, F. The ensembl variant effect predictor. Genome Biol. 2016, 17, 122. [Google Scholar] [CrossRef] [Green Version]
- Choi, Y.; Chan, A.P. PROVEAN web server: A tool to predict the functional effect of amino acid substitutions and indels. Bioinformatics 2015, 31, 2745–2747. [Google Scholar] [CrossRef] [Green Version]
- Zhao, H.; Li, J.; Yang, L.; Qin, G.; Xia, C.; Xu, X.; Su, Y.; Liu, Y.; Ming, L.; Chen, L.-L. An inferred functional impact map of genetic variants in rice. Mol. Plant 2021, 14, 1584–1599. [Google Scholar] [CrossRef]
- Wang, C.; Yu, H.; Huang, J.; Wang, W.-S.; Faruquee, M.; Zhang, F.; Zhao, X.-Q.; Fu, B.-Y.; Chen, K.; Zhang, H.-L.; et al. Towards a deeper haplotype mining of complex traits in rice with RFGB v2. 0. Plant Biotechnol. J. 2019, 18, 14. [Google Scholar] [CrossRef]
- Hu, Z.; Lu, S.-J.; Wang, M.-J.; He, H.; Sun, L.; Wang, H.; Liu, X.-H.; Jiang, L.; Sun, J.-L.; Xin, X.; et al. A novel QTL qTGW3 encodes the GSK3/SHAGGY-like kinase OsGSK5/OsSK41 that interacts with OsARF4 to negatively regulate grain size and weight in rice. Mol. Plant 2018, 11, 736–749. [Google Scholar] [CrossRef] [Green Version]
- Davidson, R.M.; Gowda, M.; Moghe, G.; Lin, H.; Vaillancourt, B.; Shiu, S.H.; Jiang, N.; Buell, C.R. Comparative transcriptomics of three Poaceae species reveals patterns of gene expression evolution. Plant J. 2012, 71, 492–502. [Google Scholar] [CrossRef] [PubMed]
- Matvienko, M. CLC Genomics Workbench. In Plant and Animal Genome. Sr. Field Application Scientist; CLC Bio: Aarhus, Denmark, 2015. [Google Scholar]
- Howe, E.; Holton, K.; Nair, S.; Schlauch, D.; Sinha, R.; Quackenbush, J. Mev: Multiexperiment viewer. In Biomedical Informatics for Cancer Research; Springer: Berlin/Heidelberg, Germany, 2010; pp. 267–277. [Google Scholar]
- Sato, Y.; Takehisa, H.; Kamatsuki, K.; Minami, H.; Namiki, N.; Ikawa, H.; Ohyanagi, H.; Sugimoto, K.; Antonio, B.A.; Nagamura, Y. RiceXPro version 3.0: Expanding the informatics resource for rice transcriptome. Nucleic Acids Res. 2013, 41, D1206–D1213. [Google Scholar] [PubMed] [Green Version]
- Yu, Y.; Zhang, H.; Long, Y.; Shu, Y.; Zhai, J. PPRD: A comprehensive online database for expression analysis of ~45,000 plant public RNA-Seq libraries. bioRxiv 2022. [Google Scholar] [CrossRef]
- Tzfadia, O.; Diels, T.; De Meyer, S.; Vandepoele, K.; Aharoni, A.; Van de Peer, Y. CoExpNetViz: Comparative co-expression networks construction and visualization tool. Front. Plant Sci. 2016, 6, 1194. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef] [PubMed]
- Langfelder, P.; Horvath, S. WGCNA: An R package for weighted correlation network analysis. BMC Bioinform. 2008, 9, 559. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jin, J.; Tian, F.; Yang, D.-C.; Meng, Y.-Q.; Kong, L.; Luo, J.; Gao, G. PlantTFDB 4.0: Toward a central hub for transcription factors and regulatory interactions in plants. Nucleic Acids Res. 2016, 45, gkw982. [Google Scholar] [CrossRef] [Green Version]
- Smedley, D.; Haider, S.; Durinck, S.; Pandini, L.; Provero, P.; Allen, J.; Arnaiz, O.; Awedh, M.H.; Baldock, R.; Barbiera, G.; et al. The BioMart community portal: An innovative alternative to large, centralized data repositories. Nucleic Acids Res. 2015, 43, W589–W598. [Google Scholar] [CrossRef] [Green Version]
- Guo, L.; Chen, W.; Tao, L.; Hu, B.; Qu, G.; Tu, B.; Yuan, H.; Ma, B.; Wang, Y.; Zhu, X.; et al. GWC1 is essential for high grain quality in rice. Plant. Sci. 2020, 296, 110497. [Google Scholar] [CrossRef]
- Zhang, Y.; Lan, H.; Shao, Q.; Wang, R.; Chen, H.; Tang, H.; Zhang, H.; Huang, J. An A20/AN1-type zinc finger protein modulates gibberellins and abscisic acid contents and increases sensitivity to abiotic stress in rice (Oryza sativa). J. Exp. Bot. 2016, 67, 315–326. [Google Scholar] [CrossRef] [Green Version]
- Angira, B.; Addison, C.K.; Cerioli, T.; Rebong, D.B.; Wang, D.R.; Pumplin, N.; Ham, J.H.; Oard, J.H.; Linscombe, S.D.; Famoso, A.N. Haplotype characterization of the sd1 Semidwarf gene in United States Rice. Plant. Genome 2019, 12, 190010. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Fan, C.; Xing, Y.; Yun, P.; Luo, L.; Yan, B.; Peng, B.; Xie, W.; Wang, G.; Li, X.; et al. Chalk5 encodes a vacuolar H+-translocating pyrophosphatase influencing grain chalkiness in rice. Nat. Genet. 2014, 46, 398–404. [Google Scholar] [CrossRef]
- Zhao, D.-S.; Li, Q.-F.; Zhang, C.-Q.; Zhang, C.; Yang, Q.-Q.; Pan, L.-X.; Ren, X.-Y.; Lu, J.; Gu, M.-H.; Liu, Q.-Q. GS9 acts as a transcriptional activator to regulate rice grain shape and appearance quality. Nat. Commun. 2018, 9, 1240. [Google Scholar] [CrossRef]
- Deshmukh, R.K.; Vivancos, J.; Ramakrishnan, G.; Guérin, V.; Carpentier, G.; Sonah, H.; Labbé, C.; Isenring, P.; Belzile, F.J.; Bélanger, R.R. A precise spacing between the NPA domains of aquaporins is essential for silicon permeability in plants. Plant. J. 2015, 83, 489–500. [Google Scholar] [CrossRef]
- Du, Q.; Campbell, M.; Yu, H.; Liu, K.; Walia, H.; Zhang, Q.; Zhang, C. Network-based feature selection reveals substructures of gene modules responding to salt stress in rice. Plant. Direct 2019, 3, e00154. [Google Scholar] [CrossRef] [Green Version]
- Shahan, R.; Zawora, C.; Wight, H.; Sittmann, J.; Wang, W.; Mount, S.M.; Liu, Z. Consensus coexpression network analysis identifies key regulators of flower and fruit development in wild strawberry. Plant. Physiol. 2018, 178, 202–216. [Google Scholar] [CrossRef] [Green Version]
- Pan, Y.; Li, Q.; Wang, Z.; Wang, Y.; Ma, R.; Zhu, L.; He, G.; Chen, R. Genes associated with thermosensitive genic male sterility in rice identified by comparative expression profiling. BMC Genom. 2014, 15, 1114. [Google Scholar] [CrossRef] [Green Version]
- Sun, S.; Wang, D.; Li, J.; Lei, Y.; Li, G.; Cai, W.; Zhao, X.; Liang, W.; Zhang, D. Transcriptome Analysis Reveals Photoperiod-Associated Genes Expressed in Rice Anthers. Front. Plant. Sci. 2021, 12, 90. [Google Scholar] [CrossRef]
- Wang, Q.; Zeng, X.; Song, Q.; Sun, Y.; Feng, Y.; Lai, Y. Identification of key genes and modules in response to cadmium stress in different rice varieties and stem nodes by weighted gene co-expression network analysis. Sci. Rep. 2020, 10, 9525. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene Name | RAP ID | Feature | Total SNPs | Number of Haplotypes | Number of Missense Mutations | Functional Impact of Missense Mutations | Number of InDels |
|---|---|---|---|---|---|---|---|
| GW5L | Os01g0190500 | Grain weight | 82 | 12 | 3 | V58G, A70S, H235R*, V58A | 70 |
| PHD1 | Os01g0367100 | Galactolipid biosynthesis | 72 | 12 | 6 | D63V, F335I*, S321C*, G305V*, D288N*, F273V* | 186 |
| GLUa | Os01g0762500 | Glutelin content | 29 | 8 | 8 | Y5H* | 7 |
| SD1| GA20ox2 | Os01g0883800 | Grain protein content | 66 | 11 | 9 | A82N*, A82N*, E100G, H101D*, H101D*, C193S*, P240L*, L266F*, Q340R, D349H | 105 |
| OsHMA4 | Os02g0196600 | Copper accumulation | 47 | 12 | 8 | I55M, F303L, T316M, A553V, S660A, I704M, G818S, V914A* | 64 |
| OsBT1-1|OsBT1 | Os02g0202400 | Endosperm granule formation | 65 | 9 | 17 | V25A, C46R, R102K, M107I, S115A, H127Q, R130Q, R152C*, R170H, M173I, R183H*, G184D*, T205R*, Y209C, P217L, V224I, Y209F | 70 |
| DU3 | Os02g0612300 | Grain amylose content | 107 | 21 | 7 | S96F*, Y237F, R189K | 116 |
| OsCDPK1| OsCDPK13| OsCDPK11| OsCPK11| OsCDPK12 | Os03g0128700 | Grain starch structure | 67 | 8 | 8 | I143T*, E259Q*, A265S*, I268V*, Q284P, F288Y*, R355W*, V474I | 54 |
| XS-LPA2-1 | Os03g0142800 | Seed phytic acid | 41 | 10 | 6 | N6T, A19T, R32L, A350T, N645K*, L1469F* | 28 |
| OsPht1;2|OsPT2 | Os03g0150800 | Selenite uptake | 20 | 9 | 5 | H398R, N335D, P269S, S258C*, V185I | 6 |
| OASA2 | Os03g0264400 | Grain tryptophan content | 78 | 17 | 6 | E585D, G527R*, P446S, R303P, E79K, R68P | 85 |
| OsCCX2 | Os03g0656500 | Grain cadmium content | 61 | 7 | 5 | H90N, D292N, F412V*, V448L*, L532V | 12 |
| OsIRT1 | Os03g0667500 | Grain iron and zinc content | 243 | 5 | 20 | V369I, R307K, R304K, R281K, I227M, V213A, R189W, H180R, V174I, N122S, N121S, G93R, L82F, V71A, A58V*, D49E, I29L, I27F, L21V, P9A | 217 |
| OsMADS34|PAP2 | Os03g0753100 | Grain yield | 90 | 8 | 5 | Q89H*, Q106K*, Q71K*, T20A*, R10P* | 140 |
| OsPho1 | Os03g0758100 | Starch structure in endosperm | 33 | 9 | 11 | T268N*, V165I, E153K, R550H*, R501C*, P391S*, T268N*, I254F*, S203L*, L60F*, M1del* | 37 |
| OsVIT1 | Os04g0463400 | Iron translocation | 72 | 10 | 5 | A170T*, V136A*, Q105K | 70 |
| OsVPE1 | Os04g0537900 | Seed glutelin | 33 | 5 | 3 | E384G*, Q90R, Y86C* | 27 |
| OsYSL9 | Os04g0542200 | Iron distribution | 48 | 8 | 4 | S511N, T368R, L256F, R90L* | 22 |
| Kala4| OsS1 | Os04g0557500 | Grain pericarp colour | 1140 | 14 | 6 | E308D, D173N, L140V, D101H, P84L, A29V* | 448 |
| OsABCC1| MRP1 | Os04g0620000 | Arsenic accumulation | 245 | 23 | 40 | S1468T, R1398G*, R1300Q, E1231V*, R990Q, L933F, K892R, Q879L, V814L, R712H*, P642H*, A524S, R518C*, A449V, L285I, N283S, R276Q, T216S, P206L*, L176V, A156S, I150S, I134M, A107V, A92V, R90Q, T61A, G47S, T33S, N23Y*, V21L, S1468N, F708L*, A409T*, R383H*, S266I*, F252T*, A233V*, C228F*, C83F*, T29A* | 135 |
| FLO2 | Os04g0645100 | Grain size | 117 | 9 | 30 | A274T, L399P, I466T, R579K, P599L, G804D, S1203L, N1319D, A1608T, F195L*, H200N*, P204T*, S306P*, N323D*, M348R*, M348I*, L369F*, A378S*, G452S*, P515T*, W589C*, R725K*, A748V*, A789S*, N829Y*, R892Q*, G926C*, R987H*, A1060V*, L1107F*, Y1146F* | 217 |
| chalk5 | Os05g0156900 | Chalkiness | 212 | 11 | 19 | R525L, I497V, V412I, K401M, A379T, A364V, G237R*, A215V, A139G, R137S, T125P, V62D, G61V, G59V, D58G, S52N, E49D, V48G, M30V | 143 |
| OsSPL9 | Os05g0408200 | Grain copper accumulation | 79 | 8 | 7 | A132V, A200T, P309S, I576V, S751T, I789V, S248F* | 114 |
| WX | Os06g0133000 | Grain characteristics | 137 | 14 | 4 | D166G*, Y224S*, P415S, D528Y*, D528N | 205 |
| SPDT | Os06g0143700 | Phosphorus accumulation | 281 | 7 | 6 | Q385L*, I251V, L247F, V71G*, A47V, A21V | 308 |
| OsSSI | Os06g0160700 | Grain starch content | 197 | 10 | 12 | S596L, K438E*, H420Y*, S319G, D214N*, L86F, A78S, T74A, L60M, R29L, R343S*, C251Y* | 138 |
| ALK|SSIIa | Os06g0229800 | Grain starch quality | 98 | 16 | 9 | P56A, T117P, A148S, D161E, E208D, D283E, S604G, M737V, L781F* | 65 |
| OsLCT1 | LOC_Os06g38120 | Cadmium in grains | 236 | 4 | 31 | E15D, D26A, E35Q, P43L, P48S, A54S, I60T, L67H, L70Q, A71D, G73D, A77T, A80S, N84K, E87K, V95I, L101F, T147S, R152S, V183A, V211L, L215F, K223N, M241V, Q246H, E258L, M310V, L380F, T480S, V494M, L495F* | 218 |
| OsHMA2|OsHMA2v | Os06g0700700 | Grain zinc and cadmium content | 104 | 8 | 3 | C19R*, R7W | 261 |
| qCdT7| OsHMA3 | Os07g0232900 | Grain zinc content | 142 | 10 | 56 | G990A, E975D, C960G, T953I, G930R, D926G, K912R, A908G, A908T, S873G, G787S, E775D, G770A, D768A, A759P, A758S, V752A, E733K, C678R, A638V, S614D, S614D, G595A, G594S*, S575T, D556H, V550I, T526I, S525T, R493Q, G490A, A381V*, S380R*, D338N*, T333M*, F299L, N298I*, Q269R, G268S*, V259I, E257K*, G256D*, G256S*, V250I, E238D, A234V, I233L, V229A, A184S, T134M, G130S*, A95V, E93A, P92S, A91T, R80H*, S614G, P92T, N725H*, L708F*, V697A*, N686K*, G642D*, E607A*, A341T*, V323G*, W293C*, P283L*, D267G*, D262N*, A252V*, R163C*, A99V*, D87N*, V82A* | 50 |
| GW7|GL7|SLG7 | Os07g0603300 | Grain quality and yield | 60 | 10 | 10 | I915M, S647A, S620G, N605K, P518S, A462S, R361H, R361C, L259F* | 75 |
| RSUS3| SUS3 | Os07g0616800 | Grain starch content | 45 | 12 | 7 | A26T, E541K*, L551S, S559N, N634D, E637K, S15G* | 47 |
| OsHMA7 | Os07g0623200 | Grain iron and zinc content | 75 | 10 | 4 | A32V, C37R, L147V*, R159C | 5 |
| OsGZF1 | Os07g0668600 | Seed storage protein | 26 | 7 | 9 | R255H, A219S, S179P, M174I, L169V, A111P, A102V, R100H, E47K*, A111T | 12 |
| SSIIIa | Os08g0191433 | Endosperm appearance | 186 | 14 | 35 | A33T, M38K, T43N, A62S, R109H, K116N, E142D, A184T, A195V, A217T, G226E, E231K, A268V, S350L, F401S, D427G, V480L, T486I, A503T, R576K, E641V, S681N, G686E, R702Q, R748H, E790V, G817D, V843E, L957M, Y964C*, K1006N*, R1118K, R1240H, A1528S, T1755I | 136 |
| BADH2 | Os08g0424500 | Grain aroma | 156 | 21 | 5 | A190V, K244I*, A316E, P458S*, G468V* | 123 |
| qGW8| OsSPL16|GW8 | Os08g0531600 | Grain quality and shape | 69 | 10 | 8 | P79L, A110V, D172N*, T274N, Q285K, G315S, M364I, A397T | 222 |
| OsDCL3b | Os10g0485600 | Seed quality | 59 | 11 | 14 | H89L*, A115V*, P129S*, N149D, S155A, T181A, F205L, I355M, G712S, R811S, V874L, R1063Q, F1101V, I1320L | 135 |
| OsCAO1| PGL | Os10g0567400 | Grain yield and quality | 28 | 5 | 4 | L394F*, T181P, V35L | 39 |
| Gene Name | Gene ID | Neighborhood Connectivity | Clustering Coefficient | Number of Directed Edges | WGCNA Module | WGCNA Module Description | Tissue Expression | Normalized Expression Value (RPKM) |
|---|---|---|---|---|---|---|---|---|
| OsCAO1 | Os10g0567400 | 9.80 | 0.24 | 20 | 5 | Iron, Phytate, Phosphorus | Seedling | 249.5 |
| OsSSI | Os06g0160700 | 11.84 | 0.39 | 19 | 2 | Grain size, Starch, Seed storage protein, Glutelin | Endsoperm | 397.9 |
| FLO2 | Os04g0645100 | 10.16 | 0.33 | 19 | 2 | Grain size, Starch, Seed storage protein, Glutelin | Developing seed | 491.1 |
| OsSULTR3;3 | Os04g0652400 | 10.67 | 0.39 | 18 | 5 | Iron, Phytate, Phosphorus | Seedling | 149.3 |
| OsGAPDHB | Os03g0129300 | 8.88 | 0.13 | 17 | 0 | NA | Seedling and shoot | 672.4 |
| OsPTR6 | Os04g0597800 | 12.63 | 0.48 | 16 | 0 | NA | Seedling | 57.5 |
| OsFRDL1 | Os03g0216700 | 12.64 | 0.48 | 14 | 0 | NA | Endosperm | 86.7 |
| SSIIIa | Os08g0191433 | 10.31 | 0.54 | 13 | 0 | NA | Endosperm | 363.9 |
| OsNRAMP5 | Os07g0257200 | 13.92 | 0.67 | 13 | 4 | Grain width, size, weight, Cadmium, Copper, | Caryopsis | 136.4 |
| GW2 | Os02g0244100 | 12.54 | 0.56 | 13 | 2 | Grain size, Starch, Seed storage protein, Glutelin | Seed | 492.1 |
| OsGZF1 | Os07g0668600 | 10.67 | 0.68 | 12 | 6 | Glutelin, Prolamine, Selenite | Seed | 264.4 |
| RSUS3 | Os07g0616800 | 10.67 | 0.68 | 12 | 1 | Starch, Chalkiness, Zinc bioavailability, Sucrose | Endosperm | 665.1 |
| qCdT7 | Os07g0232900 | 14.42 | 0.74 | 12 | 3 | Cd, Zn, Glumes, Phosphorus, Arsenic, Heavy metals, | Seedling | 47.9 |
| ALK | Os06g0229800 | 10.67 | 0.68 | 12 | 1 | Starch, Chalkiness, Zinc bioavailability, Sucrose | Seed | 444.2 |
| Ospho1 | Os03g0758100 | 11.92 | 0.64 | 12 | 1 | Starch, Chalkiness, Zinc bioavailability, Sucrose | Grain | 1237.6 |
| OsHMA4 | Os02g0196600 | 14.25 | 0.61 | 12 | 4 | Grain width, size, weight, Cadmium, Copper, | Seed | 127.9 |
| Osvpe1 | Os04g0537900 | 11.45 | 0.64 | 11 | 2 | Grain size, Starch, Seed Storage protein, Glutelin | Seed | 370.6 |
| OASA1D | Os03g0826500 | 12.80 | 0.58 | 10 | 0 | NA | Seed | 246.5 |
| OsAPL2 | Os01g0633100 | 11.60 | 0.80 | 10 | 1 | Starch, Chalkiness, Zinc bioavailability, Sucrose | Developing seed | 1138.6 |
| TF | Common Name | TF Family | Query_All # | Query_Bind $ | p-Value ¥ | q-Value |
|---|---|---|---|---|---|---|
| LOC_Os03g60630 | OJ1754_E06.26 | Dof | 80 | 33 | 3.18 × 10−6 | 3.75 × 10−4 |
| LOC_Os07g13260 | Os07g0236700 | Dof | 80 | 37 | 3.56 × 10−6 | 3.75 × 10−4 |
| LOC_Os01g53220 | Os01g0733200 | HSF | 80 | 6 | 5.58 × 10−5 | 3.93 × 10−3 |
| LOC_Os02g41510 | Os02g0624300 | MYB | 80 | 11 | 2.28 × 10−4 | 8.59 × 10−3 |
| LOC_Os04g43680 | Os04g0517100 | MYB | 80 | 11 | 2.28 × 10−4 | 8.59 × 10−3 |
| LOC_Os12g39400 | Os12g0583700 | C2H2 | 80 | 6 | 2.75 × 10−4 | 8.59 × 10−3 |
| LOC_Os02g47810 | OsJ_35953 | Dof | 80 | 26 | 2.85 × 10−4 | 8.59 × 10−3 |
| LOC_Os11g29870 | Os11g0490900 | WRKY | 80 | 6 | 4.24 × 10−4 | 1.12 × 10−2 |
| LOC_Os05g09020 | Os05g0183100 | WRKY | 80 | 8 | 4.79 × 10−4 | 1.12 × 10−2 |
| LOC_Os04g50770 | Os04g0594100 | MYB | 80 | 9 | 7.53 × 10−4 | 1.26 × 10−2 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rana, N.; Kumawat, S.; Kumar, V.; Bansal, R.; Mandlik, R.; Dhiman, P.; Patil, G.B.; Deshmukh, R.; Sharma, T.R.; Sonah, H. Deciphering Haplotypic Variation and Gene Expression Dynamics Associated with Nutritional and Cooking Quality in Rice. Cells 2022, 11, 1144. https://doi.org/10.3390/cells11071144
Rana N, Kumawat S, Kumar V, Bansal R, Mandlik R, Dhiman P, Patil GB, Deshmukh R, Sharma TR, Sonah H. Deciphering Haplotypic Variation and Gene Expression Dynamics Associated with Nutritional and Cooking Quality in Rice. Cells. 2022; 11(7):1144. https://doi.org/10.3390/cells11071144
Chicago/Turabian StyleRana, Nitika, Surbhi Kumawat, Virender Kumar, Ruchi Bansal, Rushil Mandlik, Pallavi Dhiman, Gunvant B. Patil, Rupesh Deshmukh, Tilak Raj Sharma, and Humira Sonah. 2022. "Deciphering Haplotypic Variation and Gene Expression Dynamics Associated with Nutritional and Cooking Quality in Rice" Cells 11, no. 7: 1144. https://doi.org/10.3390/cells11071144
APA StyleRana, N., Kumawat, S., Kumar, V., Bansal, R., Mandlik, R., Dhiman, P., Patil, G. B., Deshmukh, R., Sharma, T. R., & Sonah, H. (2022). Deciphering Haplotypic Variation and Gene Expression Dynamics Associated with Nutritional and Cooking Quality in Rice. Cells, 11(7), 1144. https://doi.org/10.3390/cells11071144