
Immunogenic Cell Death, DAMPs and Prothymosin α as a Putative Anticancer Immune Response Biomarker

 ,
,  , ,
, ,  , ,
, ,  ,
,  , ,
, ,  and
and
Abstract
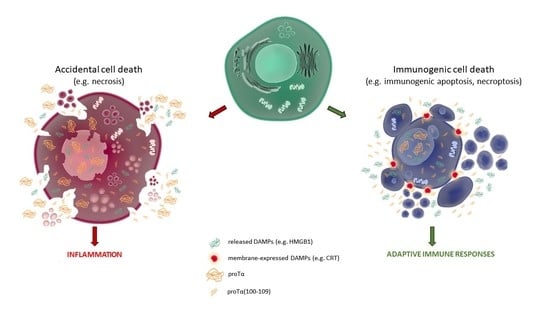
:
1. Introduction
2. Immunogenic Cell Death (ICD): Moving beyond the Classic Death Dipole Apoptosis/Necrosis
2.1. Apoptosis
2.2. Necroptosis
2.3. Pyroptosis
2.4. Ferroptosis
2.5. Parthanatos
3. Danger-Associated Molecular Patterns (DAMPs) Are Major Players in ICD
4. Inducers of ICD and Their Types
4.1. Type I ICD Inducers
4.2. Type II ICD Inducers
4.3. Other ICD Inducers
5. The Novel DAMP proΤα: Characteristics and Role
5.1. Intracellular Role of proTα
5.2. Extracellular Role of proTα
5.3. Evidence Supporting That proTα Acts as Alarmin—Proposed Role of proTα(100–109)
5.4. Detecting and Quantifying proTα and proTα(100–109)
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
References
- Bezu, L.; Gomes-de-Silva, L.C.; Dewitte, H.; Breckpot, K.; Fucikova, J.; Spisek, R.; Galluzzi, L.; Kepp, O.; Kroemer, G. Com-binatorial strategies for the induction of immunogenic cell death. Front. Immunol. 2015, 6, 187. [Google Scholar] [PubMed]
- Pentimalli, F.; Grelli, S.; Di Daniele, N.; Melino, G.; Amelio, I. Cell death pathologies: Targeting death pathways and the immune system for cancer therapy. Genes Immun. 2018, 20, 539–554. [Google Scholar] [CrossRef] [PubMed]
- Galluzzi, L.; Buqué, A.; Kepp, O.; Zitvogel, L.; Kroemer, G. Immunogenic cell death in cancer and infectious disease. Nat. Rev. Immunol. 2017, 17, 97–111. [Google Scholar] [CrossRef]
- Pavlopoulou, A.; Karaca, E.; Balestrazzi, A.; Georgakilas, A.G. In silico phylogenetic and structural analyses of plant endogenous danger signaling molecules upon stress. Oxid. Med. Cell Longev. 2019, 14, 8683054. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Samara, P.; Karachaliou, C.E.; Ioannou, K.; Papaioannou, N.E.; Voutsas, I.F.; Zikos, C.; Pirmettis, I.; Papadopoulos, M.; Kalbacher, H.; Livaniou, E.; et al. Prothymosin alpha: An alarmin and more. Curr. Med. Chem. 2017, 24, 1747–1760. [Google Scholar] [CrossRef]
- Krysko, D.V.; Garg, A.D.; Kaczmarek, A.; Krysko, O.; Agostinis, P.; Vandenabeele, P. Immunogenic cell death and DAMPs in cancer therapy. Nat. Rev. Cancer 2012, 12, 860–875. [Google Scholar] [CrossRef] [PubMed]
- Adkins, I.; Sadilkova, L.; Hradilova, N.; Tomala, J.; Kovar, M.; Spisek, R. Severe, but not mild heat-shock treatment induces immunogenic cell death in cancer cells. Oncoimmunology 2017, 6, e1311433. [Google Scholar] [CrossRef] [Green Version]
- Serrano-Del Valle, A.; Anel, A.; Naval, J.; Marzo, I. Immunogenic cell death and immunotherapy of multiple myeloma. Front. Cell Dev. Biol. 2019, 7, 50. [Google Scholar] [CrossRef]
- Galluzzi, L.; Vitale, I.; Aaronson, S.A.; Abrams, J.M.; Adam, D.; Agostinis, P.; Alnemri, E.S.; Altucci, L.; Amelio, I.; Andrews, D.W.; et al. Molecular mechanisms of cell death: Recommendations of the Nomenclature Committee on Cell Death 2018. Cell Death Differ. 2018, 25, 486–541. [Google Scholar] [CrossRef]
- Poon, I.K.H.; Lucas, C.D.; Rossi, A.G.; Ravichandran, K.S. Apoptotic cell clearance: Basic biology and therapeutic potential. Nat. Rev. Immunol. 2014, 14, 166–180. [Google Scholar] [CrossRef] [Green Version]
- Yatim, N.; Jusforgues-Saklani, H.; Orozco, S.; Schulz, O.; Barreira da Silva, R.; Reis e Sousa, C.; Green, D.R.; Oberst, A.; Albert, M.L. RIPK1 and NF-κB signaling in dying cells determines cross-priming of CD8 + T cells. Science 2015, 350, 328–334. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krysko, D.V.; Vandenabeele, P. Clearance of dead cells: Mechanisms, immune responses and implication in the development of diseases. Apoptosis 2010, 15, 995–997. [Google Scholar] [CrossRef] [PubMed]
- Stuart, L.M.; Lucas, M.; Simpson, C.; Lamb, J.; Savill, J.; Lacy-Hulbert, A. Inhibitory effects of apoptotic cell ingestion upon endotoxin-driven myeloid dendritic cell maturation. J. Immunol. 2002, 168, 1627–1635. [Google Scholar] [CrossRef] [PubMed]
- Voll, R.E.; Herrmann, M.; Roth, E.A.; Stach, C.; Kalden, J.R.; Girkontaite, I. Immunosuppressive effects of apoptotic cells. Nature 1997, 390, 350–351. [Google Scholar] [CrossRef]
- Garg, A.D.; Nowis, D.; Golab, J.; Vandenabeele, P.; Krysko, D.V.; Agostinis, P. Immunogenic cell death, DAMPs and anticancer therapeutics: An emerging amalgamation. Biochim. Biophys. Acta Rev. Cancer 2010, 1805, 53–71. [Google Scholar] [CrossRef]
- Obeid, M.; Tesniere, A.; Ghiringhelli, F.; Fimia, G.M.; Apetoh, L.; Perfettini, J.L.; Castedo, M.; Mignot, G.; Panaretakis, T.; Casares, N.; et al. Calreticulin exposure dictates the immunogenicity of cancer cell death. Nat. Med. 2007, 13, 54–61. [Google Scholar] [CrossRef]
- Galluzzi, L.; Humeau, J.; Buqué, A.; Zitvogel, L.; Kroemer, G. Immunostimulation with chemotherapy in the era of immune checkpoint inhibitors. Nat. Rev. Clin. Oncol. 2020, 17, 725–741. [Google Scholar] [CrossRef]
- Kroemer, G.; Galassi, C.; Zitvogel, L.; Galluzzi, L. Immunogenic cell stress and death. Nat. Immunol. 2022, 23, 487–500. [Google Scholar] [CrossRef]
- Bedognetti, D.; Ceccarelli, M.; Galluzzi, L.; Lu, R.; Karolina Palucka, K.; Samayoa, J.; Spranger, S.; Warren, S.; Wong, K.K.; Ziv, E.; et al. Toward a comprehensive view of cancer immune responsiveness: A synopsis from the SITC workshop. J. Immunother. Cancer 2019, 7, 167. [Google Scholar] [CrossRef] [Green Version]
- Fucikova, J.; Kepp, O.; Kasikova, L.; Petroni, G.; Yamazaki, T.; Liu, P.; Zhao, L.; Spisek, R.; Kroemer, G.; Galluzzi, L. Detection of immunogenic cell death and its relevance for cancer therapy. Cell Death Dis. 2020, 11, 1013. [Google Scholar] [CrossRef]
- Fulda, S.; Debatin, K.M. Apoptosis signaling in tumor therapy. Ann. N. Y. Acad. Sci. 2004, 1028, 150–156. [Google Scholar] [CrossRef] [PubMed]
- Fulda, S.; Debatin, K.M. Extrinsic versus intrinsic apoptosis pathways in anticancer chemotherapy. Oncogene 2006, 25, 4798–4811. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walczak, H.; Krammer, P.H. The CD95 (APO-1/Fas) and the TRAIL (APO-2L) apoptosis systems. Exp. Cell Res. 2000, 256, 58–66. [Google Scholar] [CrossRef] [PubMed]
- Saelens, X.; Festjens, N.; Vande Walle, L.; van Gurp, M.; van Loo, G.; Vandenabeele, P. Toxic proteins released from mitochondria in cell death. Oncogene 2004, 23, 2861–2874. [Google Scholar] [CrossRef] [Green Version]
- Yatim, N.; Cullen, S.; Albert, M.L. Dying cells actively regulate adaptive immune responses. Nat. Rev. Immunol. 2017, 17, 262–275. [Google Scholar] [CrossRef]
- Albert, M.L.; Sauter, B.; Bhardwaj, N. Dendritic cells acquire antigen from apoptotic cells and induce class I-restricted CTLs. Nature 1998, 392, 86–89. [Google Scholar] [CrossRef]
- Rawson, P.M.; Molette, C.; Videtta, M.; Altieri, L.; Franceschini, D.; Donato, T.; Finocchi, L.; Propato, A.; Paroli, M.; Meloni, F.; et al. Cross-presentation of caspase-cleaved apoptotic self antigens in HIV infection. Nat. Med. 2007, 13, 1431–1439. [Google Scholar] [CrossRef]
- Green, D.R.; Ferguson, T.; Zitvogel, L.; Kroemer, G. Immunogenic and tolerogenic cell death. Nat. Rev. Immunol. 2009, 9, 353–363. [Google Scholar] [CrossRef]
- Casares, N.; Pequignot, M.O.; Tesniere, A.; Ghiringhelli, F.; Roux, S.; Chaput, N.; Schmitt, E.; Hamai, A.; Hervas-Stubbs, S.; Obeid, M.; et al. Caspase-dependent immunogenicity of doxorubicin-induced tumor cell death. J. Exp. Med. 2005, 202, 1691–1701. [Google Scholar] [CrossRef]
- Gamrekelashvili, J.; Kapanadze, T.; Han, M.; Wissing, J.; Ma, C.; Jaensch, L.; Manns, M.P.; Armstrong, T.; Jaffee, E.; White, A.O.; et al. Peptidases released by necrotic cells control CD8+ T cell cross-priming. J. Clin. Investig. 2013, 123, 4755–4768. [Google Scholar] [CrossRef] [Green Version]
- Vercammen, B.D.; Brouckaert, G.; Denecker, G.; Van De Craen, M.; Declercq, W.; Fiers, W.; Vandenabeele, P. Dual signaling of the Fas receptor: Initiation of both apoptotic and necrotic cell death pathways. J. Exp. Med. 1998, 188, 919–930. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaiser, W.J.; Upton, J.W.; Mocarski, E.S. Viral modulation of programmed necrosis. Curr. Opin. Virol. 2013, 3, 296–306. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vande Walle, L.; Lamkanfi, M. Pyroptosis. Curr. Biol. 2016, 26, 568–572. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Franchi, L.; Eigenbrod, T.; Muñoz-Planillo, R.; Nuñez, G. The inflammasome: A caspase-1 activation platform regulating im-mune responses and disease pathogenesis. Nat. Immunol. 2009, 10, 241–247. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Sun, B. Neutrophil pyroptosis: New perspectives on sepsis. Cell Mol. Life Sci. 2019, 76, 2031–2042. [Google Scholar] [CrossRef] [Green Version]
- Stockwell, B.R.; Angeli, P.F.; Bayir, H.; Bush, A.I.; Conrad, M.; Dixon, S.J.; Linkermann, A.; Murphy, M.E.; Fulda, S.; Gascon, S.; et al. Primer ferroptosis: A regulated cell death nexus linking metabolism, redox biology, and disease. Cell 2017, 171, 273–285. [Google Scholar] [CrossRef] [Green Version]
- Fearnhead, H.O.; Vandenabeele, P.; Vanden Berghe, T. How do we fit ferroptosis in the family of regulated cell death? Cell Death Differ. 2017, 24, 1991–1998. [Google Scholar] [CrossRef] [Green Version]
- Sun, Y.; Chen, P.; Zhai, B.; Zhang, M.; Xiang, Y.; Fang, J.; Xu, S.; Gao, Y.; Chen, X.; Sui, X.; et al. The emerging role of ferroptosis in inflammation. Biomed. Pharmacother. 2020, 127, 110108. [Google Scholar] [CrossRef]
- Peng, J.J.; Song, W.T.; Yao, F.; Zhang, X.; Peng, J.; Luo, X.J.; Xia, X.B. Involvement of regulated necrosis in blinding diseases: Focus on necroptosis and ferroptosis. Exp. Eye Res. 2020, 191, 107922. [Google Scholar] [CrossRef]
- David, K.K.; Andrabi, S.A.; Dawson, T.M.; Lynn, V. Parthanatos, a messenger of death. Front. Biosci. Landmark 2009, 14, 1116–1128. [Google Scholar] [CrossRef] [Green Version]
- Robinson, N.; Ganesan, R.; Hegedus, C.; Kovács, K.; Kufer, T.A. Programmed necrotic cell death of macro-phages: Focus on pyroptosis, necroptosis, and parthanatos. Redox Biol. 2019, 26, 101239. [Google Scholar] [CrossRef] [PubMed]
- Kroemer, G.; El-Deiry, W.S.; Golstein, P.; Peter, M.E.; Vaux, D.; Vandenabeele, P.; Zhivotovsky, B.; Blagosklonny, M.V.; Malorni, W.; Knight, R.A.; et al. Classification of cell death: Recommendations of the Nomenclature Committee on Cell Death. Cell Death Differ. 2005, 12, 1463–1467. [Google Scholar] [CrossRef] [Green Version]
- Yan, G.E.; Elbadawi, M.; Efferth, T. Multiple cell death modalities and their key features. World Acad. Sci. J. 2020, 2, 39–48. [Google Scholar] [CrossRef] [Green Version]
- Tang, D.; Kang, R.; Berghe, T.V.; Vandenabeele, P.; Kroemer, G. The molecular machinery of regulated cell death. Cell Res. 2019, 29, 347–364. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yousefi, S.; Stojkov, D.; Germic, N.; Simon, D.; Wang, X.; Benarafa, C.; Simon, H.U. Untangling “NETosis” from NETs. Eur. J. Immunol. 2019, 49, 221–227. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Y.; Levine, B.L. Autosis and autophagic cell death: The dark side of autophagy. Cell Death Differ. 2015, 22, 367–376. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vandenabeele, P.; Vandecasteele, K.; Bachert, C.; Krysko, O.; Krysko, D.V. Immunogenic apoptotic cell death and anticancer immunity. Adv. Exp. Med. Biol. 2016, 930, 133–149. [Google Scholar] [CrossRef]
- Showalter, A.; Limaye, A.; Oyer, J.L.; Igarashi, R.; Kittipatarin, C.; Copik, A.J.; Khaled, A.R. Cytokines in immunogenic cell death: Applications for cancer immunotherapy. Cytokine 2017, 97, 123–132. [Google Scholar] [CrossRef]
- Yu, M.; Wang, H.; Ding, A.; Golenbock, D.T.; Latz, E.; Czura, C.J.; Fenton, M.J.; Tracey, K.J.; Yang, H. HMGB1 signals through Toll-like receptor (TLR) 4 and TLR2. Shock 2006, 26, 174–179. [Google Scholar] [CrossRef]
- Zhou, J.; Wang, G.; Chen, Y.; Wang, H.; Hua, Y.; Cai, Z. Immunogenic cell death in cancer therapy: Present and emerging inducers. J. Cell. Mol. Med. 2019, 23, 4854–4865. [Google Scholar] [CrossRef]
- Wang, Y.J.; Fletcher, R.; Yu, J.; Zhang, L. Immunogenic effects of chemotherapy-induced tumor cell death. Genes Dis. 2018, 5, 194–203. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.Y.; Chiang, S.F.; Ke, T.W.; Chen, T.W.; Lan, Y.C.; You, Y.S.; Shiau, A.C.; Chen, W.T.; Chao, K.S.C. Cytosolic high-mobility group box protein 1 (HMGB1) and/or PD-1+ TILs in the tumor microenvironment may be contributing prognostic biomarkers for patients with locally advanced rectal cancer who have undergone neoadjuvant chemoradiotherapy. Cancer Immunol. Immunother. 2018, 67, 551–562. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, Y.; Mimura, K.; Yoshimoto, Y.; Watanabe, M.; Ohkubo, Y.; Izawa, S.; Murata, K.; Fujii, H.; Nakano, T.; Kono, K. Immunogenic tumor cell death induced by chemoradiotherapy in patients with esophageal squamous cell carcinoma. Cancer Res. 2012, 72, 3967–3976. [Google Scholar] [CrossRef] [Green Version]
- Lämmer, F.; Delbridge, C.; Würstle, C.; Neff, F.; Meyer, B.; Schlegel, J.; Kessel, K.A.; Schmid, T.E.; Schilling, D.; Combs, S.E. Cytosolic Hsp70 as a biomarker to predict clinical outcome in patients with glioblastoma. PLoS ONE 2019, 14, e0221502. [Google Scholar]
- Kasikova, L.; Truxova, I.; Cremer, I.; Sautes-Fridman, C.; Keep, O.; Kroemer, G.; Spisek, R.; Fucikova, J. Side-by-side comparison of flow cytometry and immunohistochemistry for detection of calreticulin exposure in the course of immunogenic cell death. Methods Enzymol. 2020, 632, 15–25. [Google Scholar]
- Sukkurwala, A.Q.; Adjemian, S.; Senovilla, L.; Michaud, M.; Spaggiari, S.; Vacchelli, E.; Baracco, E.E.; Galluzzi, L.; Zitvogel, L.; Kepp, O.; et al. Screening of novel immunogenic cell death inducers within the NCI Mechanistic Diversity Set. Oncoimmunology 2014, 3, e28473. [Google Scholar] [CrossRef] [Green Version]
- Golden, E.B.; Apetoh, L. Radiotherapy and immunogenic cell death. Semin. Radiat. Oncol. 2015, 25, 11–17. [Google Scholar] [CrossRef] [Green Version]
- Golden, E.B.; Frances, D.; Pellicciotta, I.; Demaria, S.; Barcellos-Hoff, M.H.; Formenti, S.C. Radiation fosters dose-dependent and chemotherapy-induced immunogenic cell death. Oncoimmunology 2014, 25, e28518. [Google Scholar] [CrossRef] [Green Version]
- Golden, E.B.; Pellicciotta, I.; Demaria, S.; Barcellos-Hoff, M.H.; Formenti, S.C. The convergence of radiation and immunogenic cell death signaling pathways. Front. Oncol. 2012, 2, 88. [Google Scholar] [CrossRef] [Green Version]
- Fucikova, J.; Kralikova, P.; Fialová, A.; Brtnicky, T.; Rob, L.; Bartunkova, J.; Špíšek, R. Human tumor cells killed by anthracyclines induce a tumor-specific immune response. Cancer Res. 2011, 71, 4821–4833. [Google Scholar] [CrossRef] [Green Version]
- Tesniere, A.; Schlemmer, F.; Boige, V.; Kepp, O.; Martins, I.; Ghiringhelli, F.; Aymeric, L.; Michaud, M.; Apetoh, L.; Barault, L.; et al. Immunogenic death of colon cancer cells treated with oxaliplatin. Oncogene 2010, 29, 482–491. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gandolfi, S.; Laubach, J.P.; Hideshima, T.; Chauhan, D.; Anderson, K.C.; Richardson, P.G. The proteasome and proteasome inhibitors in multiple myeloma. Cancer Metastasis Rev. 2017, 36, 561–584. [Google Scholar] [CrossRef] [PubMed]
- Spisek, R.; Charalambous, A.; Mazumder, A.; Vesole, D.H.; Jagannath, S.; Dhodapkar, M.V. Bortezomib enhances dendritic cell (DC)–mediated induction of immunity to human myeloma via exposure of cell surface heat shock protein 90 on dying tumor cells: Therapeutic implications. Blood 2007, 109, 4839–4845. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.M.; Wang, P.H.; Chen, S.S.; Wen, C.C.; Chen, Y.H.; Yang, W.C.; Yang, N.S. Shikonin induces immunogenic cell death in tumor cells and enhances dendritic cell-based cancer vaccine. Cancer Immunol. Immunother. 2012, 61, 1989–2002. [Google Scholar] [CrossRef] [PubMed]
- Mavragani, I.V.; Nikitaki, Z.; Kalospyros, S.A.; Georgakilas, A.G. Ionizing radiation and complex DNA damage: From pre-diction to detection challenges and biological significance. Cancers 2019, 11, 1789. [Google Scholar] [CrossRef] [Green Version]
- Garg, A.; Dudek, A.M.; Ferreira, G.B.; Verfaillie, T.; Vandenabeele, P.; Krysko, D.; Mathieu, C.; Agostinis, P. ROS-induced autophagy in cancer cells assists in evasion from determinants of immunogenic cell death. Autophagy 2013, 9, 1292–1307. [Google Scholar] [CrossRef]
- Garg, A.D.; Krysko, D.V.; Verfaillie, T.; Kaczmarek, A.; Ferreira, G.B.; Marysael, T.; Rubio, N.; Firczuk, M.; Mathieu, C.; Roebroek, A.J.; et al. A novel pathway combining calreticulin exposure and ATP secretion in immunogenic cancer cell death. EMBO J. 2012, 31, 1062–1079. [Google Scholar] [CrossRef] [Green Version]
- Bugaut, H.; Bruchard, M.; Berger, H.; Derangère, V.; Odoul, L.; Euvrard, R.; Ladoire, S.; Chalmin, F.; Végran, F.; Rébé, C.; et al. Bleomycin exerts ambivalent antitumor immune effect by triggering both immunogenic cell death and proliferation of regu-latory T cells. PLoS ONE 2013, 8, e65181. [Google Scholar] [CrossRef] [Green Version]
- Haritos, A.A.; Goodall, G.J.; Horecker, B.L. Prothymosin alpha: Isolation and properties of the major immunoreactive form of thymosin alpha 1 in rat thymus. Proc. Natl. Acad. Sci. USA 1984, 81, 1008–1011. [Google Scholar] [CrossRef] [Green Version]
- Manrow, R.E.; Sburlati, A.R.; Hanover, J.A.; Berger, S.L. Nuclear targeting of prothymosin alpha. J. Biol. Chem. 1991, 266, 3916–3924. [Google Scholar] [CrossRef]
- Rubtsov, Y.P.; Zolotukhin, A.S.; Vorobjev, I.A.; Chichkova, N.V.; Pavlov, N.A.; Karger, E.M.; Evstafieva, A.G.; Felber, B.K.; Vartapetian, A.B. Mutational analysis of human prothymosin α reveals a bipartite nuclear localization signal. FEBS Lett. 1997, 413, 135–141. [Google Scholar] [CrossRef] [Green Version]
- Piñeiro, A.; Cordero, O.J.; Nogueira, M. Fifteen years of prothymosin alpha: Contradictory past and new horizons. Peptides 2000, 21, 1433–1446. [Google Scholar] [CrossRef]
- Samara, P.; Ioannou, K.; Tsitsilonis, O.E. Prothymosin alpha and immune responses: Are we close to potential clinical ap-plications? Vitam. Horm. 2016, 102, 179–207. [Google Scholar]
- Karetsou, Z.; Sandaltzopoulos, R.; Frangou-Lazaridis, M.; Lai, C.Y.; Tsolas, O.; Becker, P.B.; Papamarcaki, T. Prothymosin alpha modulates the interaction of histone H1 with chromatin. Nucleic Acids Res. 1998, 26, 3111–3118. [Google Scholar] [CrossRef] [Green Version]
- George, E.; Brown, D.T. Prothymosin α is a component of a linker histone chaperone. FEBS Lett. 2011, 584, 2833–2836. [Google Scholar] [CrossRef] [Green Version]
- Karetsou, Z.; Kretsovali, A.; Murphy, C.; Tsolas, O.; Papamarcaki, T. Prothymosin α interacts with the CREB-binding protein and potentiates transcription. EMBO Rep. 2002, 3, 361–366. [Google Scholar] [CrossRef] [Green Version]
- Qi, X.; Wang, L.; Du, F. Novel small molecules relieve prothymosin α-mediated inhibition of apoptosome formation by blocking its interaction with Apaf-1. Biochemistry 2010, 49, 1923–1930. [Google Scholar] [CrossRef] [Green Version]
- Niture, S.K.; Kaspar, J.W.; Shen, J.; Jaiswal, A.K. Nrf2 signaling and cell survival. Toxicol. Appl. Pharmacol. 2010, 244, 37–42. [Google Scholar] [CrossRef] [Green Version]
- Tang, D.; Kang, R. HMGB1 in Cell Death. In Cell Death-Autophagy, Apoptosis and Necrosis; Ntuli, M.T., Ed.; IntechOpen: London, UK, 2015; pp. 397–418. [Google Scholar]
- Štros, M.; Ozaki, T.; Bačíková, A.; Kageyama, H.; Nakagawara, A. HMGB1 and HMGB2 cell-specifically down-regulate the p53- and p73-dependent sequence-specific transactivation from the human Bax gene promoter. J. Biol. Chem. 2002, 277, 7157–7164. [Google Scholar] [CrossRef] [Green Version]
- Song, M.J.; Hwang, S.; Wong, W.; Round, J.; Martinez-Guzman, D.; Turpaz, Y.; Liang, J.; Wong, B.; Johnson, R.C.; Carey, M.; et al. The DNA architectural protein HMGB1 facilitates RTA-mediated viral gene expression in gamma-2 herpesviruses. J. Virol. 2004, 78, 12940–12950. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Prasad, R.; Wilson, S.H. HMGB1: Roles in base excision repair and related function. Biochim. Biophys. Acta 2010, 1799, 119–130. [Google Scholar] [CrossRef] [Green Version]
- Livesey, K.M.; Kang, R.; Vernon, P.; Buchser, W.; Loughran, P.; Watkins, S.C.; Zhang, L.; Manfredi, J.J.; Zeh, H.J.; Li, L.; et al. p53/HMGB1 complexes regulate autophagy and apoptosis. Cancer Res. 2012, 72, 1996–2005. [Google Scholar] [CrossRef] [Green Version]
- Skopeliti, M.; Voutsas, I.F.; Klimentzou, P.; Tsiatas, M.L.; Beck, A.; Bamias, A.; Moraki, M.; Livaniou, E.; Neagu, M.; Voelter, W.; et al. The immunologically active site of prothymosin alpha is located at the carboxy-terminus of the polypeptide. Evaluation of its in vitro effects in cancer patients. Cancer Immunol. Immunother. 2006, 55, 1247–1257. [Google Scholar] [CrossRef]
- Skopeliti, M.; Iconomidou, V.A.; Derhovanessian, E.; Pawelec, G.; Voelter, W.; Kalbacher, H.; Hamodrakas, S.J.; Tsitsilonis, O.E. Prothymosin alpha immunoactive carboxyl-terminal peptide TKKQKTDEDD stimulates lymphocyte reactions, induces den-dritic cell maturation and adopts a beta-sheet conformation in a sequence-specific manner. Mol. Immunol. 2009, 46, 784–792. [Google Scholar] [CrossRef]
- Pan, L.X.; Haritos, A.A.; Wideman, J.; Komiyama, T.; Chang, M.; Stein, S.; Salvin, S.; Horecker, B.L. Human prothymosin α: Amino acid sequence and immunologic properties. Arch. Biochem. Biophys. 1986, 250, 197–201. [Google Scholar] [CrossRef]
- Baxevanis, C.N.; Frillingos, S.; Seferiadis, K.; Reclos, G.J.; Arsenis, P.; Katsiyiannis, A.; Anastasopoulos, E.; Tsolas, O.; Papamichail, M. Enhancement of human T lymphocyte function by prothymosin alpha: Increased production of interleukin-2 and expression of interleukin-2 receptors in normal human peripheral blood T lymphocytes. Immunopharmacol. Immunotoxicol. 1990, 12, 595–617. [Google Scholar] [CrossRef]
- Cordero, O.J.; Sarandeses, C.S.; López, J.L.; Cancio, E.; Regueiro, B.J.; Nogueira, M. Prothymosin α enhances interleukin 2 receptor expression in normal human T-lymphocytes. Int. J. Immunopharmacol. 1991, 13, 1059–1065. [Google Scholar] [CrossRef]
- Baxevanis, C.N.; Thanos, D.; Reclos, G.J.; Anastasopoulos, E.; Tsokos, G.C.; Papamatheakis, J.; Papamichail, M. Prothymosin alpha enhances human and murine MHC class II surface antigen expression and messenger RNA accumulation. J. Immunol. 1992, 148, 1979–1984. [Google Scholar]
- Ioannou, K.; Derhovanessian, E.; Tsakiri, E.; Samara, P.; Kalbacher, H.; Voelter, W.; Trougakos, I.P.; Pawelec, G.; Tsitsilonis, O.E. Prothymosin α and a prothymosin α-derived peptide enhance TH1-type immune responses against defined HER-2/neu epitopes. BMC Immunol. 2013, 14, 43. [Google Scholar] [CrossRef] [Green Version]
- Samara, P.; Ioannou, K.; Neagu, M.; Arnogiannaki, N.; Ardavanis, A.; Voelter, W.; Tsitsilonis, O. The C-terminal decapeptide of prothymosin α is responsible for its stimulatory effect on the functions of human neutrophils in vitro. Int. Immunopharmacol. 2013, 15, 50–57. [Google Scholar] [CrossRef]
- Birmpilis, A.I.; Karachaliou, C.E.; Samara, P.; Ioannou, K.; Selemenakis, P.; Kostopoulos, I.V.; Kavrochorianou, N.; Kalbacher, H.; Livaniou, E.; Haralambous, S.; et al. Antitumor reactive T-cell responses are enhanced in vivo by DAMP prothymosin alpha and its C-terminal decapeptide. Cancers 2019, 11, 1764. [Google Scholar] [CrossRef] [Green Version]
- Mosoian, A.; Teixeira, A.; Burns, C.S.; Sander, L.E.; Gusella, G.L.; He, C.; Blander, J.M.; Klotman, P.; Klotman, M.E. Prothymosin-alpha inhibits HIV-1 via Toll-like receptor 4-mediated type I interferon induction. Proc. Natl. Acad. Sci. USA 2010, 107, 10178–10183. [Google Scholar] [CrossRef] [Green Version]
- Karachaliou, C.E.; Triantis, C.; Liolios, C.; Palamaris, L.; Zikos, H.; Tsitsilonis, O.E.; Kalbacher, H.; Voelter, W.; Loudos, G.; Papadopoulos, M.; et al. In vivo biodistribution and imaging studies with a 99mTc-radiolabeled derivative of the C-terminus of prothymosin alpha in mice bearing experimentally-induced inflammation. Eur. J. Pharm. Biopharm. 2017, 113, 188–197. [Google Scholar] [CrossRef]
- Curtin, J.F.; Liu, N.; Candolfi, M.; Xiong, W.; Assi, H.; Yagiz, K.; Edwards, M.R.; Michelsen, K.S.; Kroeger, K.M.; Liu, C.; et al. HMGB1 mediates endogenous TLR2 activation and brain tumor regression. PLoS Med. 2009, 6, e1000010. [Google Scholar] [CrossRef]
- Fan, J.; Li, Y.; Levy, R.M.; Fan, J.J.; Hackam, D.J.; Vodovotz, Y.; Yang, H.; Tracey, K.J.; Billiar, T.R.; Wilson, M.A. Hemorrhagic shock induces NAD(P)H oxidase activation in neutrophils: Role of HMGB1-TLR4 signaling. J. Immunol. 2007, 178, 6573–6580. [Google Scholar] [CrossRef] [Green Version]
- Apetoh, L.; Ghiringhelli, F.; Tesniere, A.; Obeid, M.; Ortiz, C.; Criollo, A.; Mignot, G.; Maiuri, M.C.; Ullrich, E.; Saulnier, P.; et al. Toll-like receptor 4–dependent contribution of the immune system to anticancer chemotherapy and radiotherapy. Nat. Med. 2007, 13, 1050–1059. [Google Scholar] [CrossRef]
- Telusma, G.; Datta, S.; Mihajlov, I.; Ma, W.; Li, J.; Yang, H.; Newman, W.; Messmer, B.T.; Minev, B.; Schmidt-Wolf, I.G.; et al. Dendritic cell activating peptides induce distinct cytokine profiles. Int. Immunol. 2006, 18, 1563–1573. [Google Scholar] [CrossRef] [Green Version]
- Saenz, R.; Souza, C.D.S.; Huang, C.T.; Larsson, M.; Esener, S.; Messmer, D. HMGB1-derived peptide acts as adjuvant inducing immune responses to peptide and protein antigen. Vaccine 2010, 28, 7556–7562. [Google Scholar] [CrossRef] [Green Version]
- LeBlanc, P.M.; Doggett, T.A.; Choi, J.; Hancock, M.A.; Durocher, Y.; Frank, F.; Nagar, B.; Ferguson, T.A.; Saleh, M. An immunogenic peptide in the A-box of HMGB1 protein reverses apoptosis-induced tolerance through RAGE receptor. J. Biol. Chem. 2014, 289, 7777–7786. [Google Scholar] [CrossRef] [Green Version]
- Evstafieva, A.G.; Belov, G.A.; Rubtsov, Y.P.; Kalkum, M.; Joseph, B.; Chichkova, N.V.; Sukhacheva, E.A.; Bogdanov, A.A.; Pettersson, R.F.; Agol, V.I.; et al. Apoptosis-related fragmentation, translocation, and properties of human prothymosin alpha. Exp. Cell Res. 2003, 284, 211–223. [Google Scholar] [CrossRef]
- Samara, P.; Miriagou, V.; Zachariadis, M.; Mavrofrydi, O.; Promponas, V.J.; Dedos, S.G.; Papazafiri, P.; Kalbacher, H.; Voelter, W.; Tsitsilonis, O. A fragment of the alarmin prothymosin α as a novel biomarker in murine models of bacteria-induced sepsis. Oncotarget 2017, 8, 48635–48649. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Samara, P.; Kalbacher, H.; Ioannou, K.; Radu, D.L.; Livaniou, E.; Promponas, V.J.; Voelter, W.; Tsitsilonis, O. Development of an ELISA for the quantification of the C-terminal decapeptide prothymosin α(100–109) in sera of mice infected with bacteria. J. Immunol. Methods 2013, 395, 54–62. [Google Scholar] [CrossRef] [PubMed]
- Karachaliou, C.E.; Kostopoulos, I.V.; Vassilakopoulou, V.; Klimentzou, P.; Paravatou-Petsotas, M.; Voelter, W.; Kalbacher, H.; Zikos, C.; Tsitsilonis, O.; Livaniou, E. Development of a specific IgY-based ELISA for prothymosin alpha, a bioactive polypeptide with diagnostic and therapeutic potential. Heliyon 2019, 5, e02616. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baxevanis, C.N.; Gritzapis, A.D.; Dedoussis, G.V.; Papadopoulos, N.G.; Tsolas, O.; Papamichail, M. Induction of lymphokine-activated killer activity in mice by prothymosin alpha. Cancer Immunol. Immunother. 1994, 38, 281–286. [Google Scholar] [CrossRef]
- Eggleton, P.; Bremer, E.; Dudek, E.; Michalak, M. Calreticulin, a therapeutic target? Expert Opin. Ther. Targets 2016, 20, 1137–1147. [Google Scholar] [CrossRef] [PubMed]
- Rapaport, E.; Fishman, R.F.; Gercel, C. Growth inhibition of human tumor cells in soft-agar cultures by treatment with low levels of adenosine 5′-triphosphate. Cancer Res. 1983, 43, 4402–4406. [Google Scholar]
- Beijer, S.; Van Rossum, E.; Hupperets, P.S.; Spreeuwenberg, C.; van den Beuken, M.; Winkens, R.A.; Ars, L.; van den Borne, B.E.; de Graeff, A.; Dagnelie, P.C. Application of adenosine 5′-triphosphate (ATP) infusions in palliative home care: Design of a randomized clinical trial. BMC Public Health 2007, 7, 4. [Google Scholar] [CrossRef] [Green Version]
- Jiang, J.X.; Riquelme, M.A.; Zhou, J.Z. ATP, a double-edged sword in cancer. Oncoscience 2015, 2, 673–674. [Google Scholar] [CrossRef]
- Andersson, U.; Tracey, K.J. HMGB1 is a therapeutic target for sterile inflammation and infection. Annu. Rev. Immunol. 2011, 29, 139–162. [Google Scholar] [CrossRef] [Green Version]
- Vulczak, A.; Catalão, C.H.R.; De Freitas, L.A.P.; Rocha, M.J.A. HSP-Target of therapeutic agents in sepsis treatment. Int. J. Mol. Sci. 2019, 20, 4255. [Google Scholar] [CrossRef] [Green Version]
- Venereau, E.; De Leo, F.; Mezzapelle, R.; Careccia, G.; Musco, G.; Bianchi, M.E. HMGB1 as biomarker and drug target. Pharmacol. Res. 2016, 111, 534–544. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cell Death Modality | Classification | Morphological Characteristics | Immunologic Profile | Regulators |
|---|---|---|---|---|
| Necrosis | ACD | cell swelling; DNA fragmentation; membrane rupture; loss of cell organelles | Tolerogenic/immunogenic | None |
| Apoptosis | RCD | cell shrinkage/rounding; nuclear condensation/fragmentation; nuclear membrane rupture; membrane blebbing; apoptotic body formation | Tolerogenic/immunogenic | Death receptors, BAX, BAK, AIF, caspases 2, 3, 6, 7, 8, and 9 |
| Necroptosis | RCD | cell/mitochondrial swelling; membrane rupture; chromatin condensation; loss of cell organelles | Immunogenic | TLRs, TCR, RIPK1, RIPK3, MLKL |
| Pyroptosis | RCD | cell swelling; membrane permeabilization/rupture; DNA condensation/ fragmentation | Immunogenic | CASP1, CASP11, GSDMD, NLRs, ALRs |
| Ferroptosis | RCD | mitochondrial shrinkage; reduced mitochondrial cristae; mitochondrial membrane rupture | Immunogenic | System XC−, GPX4, TFRC, ACSL4, LPCAT3, ALOX15, GLS2, DPP4, NCOA4, BAP1, BECN1, PEBP1, CARS, VDAC2/3, RAB7A, HSP90, ALK4/5 |
| Parthanatos | RCD | chromatin condensation; DNA fragmentation; membrane rupture; inconsistent mitochondrial membrane; no apoptotic body formation | Immunogenic | PARP-1, AIFM1, MIF, OGG1 |
| Anoikis | RCD | cell shrinkage/rounding; nuclear condensation/fragmentation; nuclear membrane rupture; membrane blebbing; apoptotic body formation; detachment from substrate/other cells | Tolerogenic/immunogenic | Death receptors, BAX, BAK, AIF, caspases 2, 3, 6, 7, 8, and 9 |
| MPT-driven necrosis | RCD | similar to necrosis; loss of mitochondrial inner membrane impermeability; mitochondrial membrane dissipation/breakdown | Immunogenic | CYPD (PPIF) |
| Entotic cell death (Entosis) | RCD | cell-in-cell formation | Tolerogenic/immunogenic | RhoA, ROCKI/II, E-cadherin, α-catenin, actomyosin, LC3, ATGs |
| Neutrophil extracellular trap cell death (NETosis) | RCD | membrane rupture; nuclear membrane dissolvement; chromatin decondensation/release | Tolerogenic/immunogenic | NOX4, PAD4, ELANE, MMP, MPO, ELANE, MMP, MPO |
| Lysosome-dependent cell death (LDCD) | RCD | lysosome/plasma membrane rupture | Immunogenic | BECN1, Na+/K+-ATPase, AMPK, Ras-like protein A |
| Autophagy-dependent cell death (ADCD) | RCD | vacuolization (large intracellular vesicles); enlargement of cell organelles; depletion of cell organelles | Immunogenic | UKL1, PI3KIII, ATGs, LC3 |
| Autosis | RCD | enhanced cell-substrate adherence; ER fragmentation/breakdown; cell swelling; chromatin condensation | Immunogenic | Na+/K+-ATPase |
| Alkaliptosis | RCD | similar to necrosis | Immunogenic | IKBKB, NF-κB |
| Oxeiptosis | RCD | similar to apoptosis | Tolerogenic | KEAP1, PGAM5, AIFM1 |
| Identifier | Pathological Condition | DAMP(s) | Aim of Investigation | Status |
|---|---|---|---|---|
| NCT02921854 | Cancer/non-small cell lung cancer | HMGB1, HSP70, CRT, HSP90 | Detectability of ICD markers in the serum of patients post high-dose radiotherapy alone or concurrent cisplatin-doublet therapy and radiotherapy to access induction of anticancer immune responses. | Completed |
| NCT03581695 | Pediatric pulmonary hypertension | HMGB1 | HMGB1 levels in pediatric patients with pulmonary hypertension | Recruiting |
| NCT04837391 | Postoperative cognitive dysfunction | HMGB1 | Relationship between postoperative cognitive dysfunction and brain injury biomarkers in geriatric urologic oncology patients via measuring HMGB1 levels | Recruiting |
| NCT03986736 | Tissue injury and rhabdomyolysis after major trauma | HMGB1 | Correlation between the levels of HMGB1 and the degree of injury | Recruiting |
| NCT03741738 | Autoimmuno diseases/Vitiligo | HMGB1 | HMGB1 as a biomarker for predicting the severity of Vitiligo, by measuring apoptosis levels of melanocytes | Completed |
| NCT04080453 | Sepsis/septic shock | HMGB1 | Correlation of HMGB1 levels with platelet activation | Recruiting |
| NCT02914756 | Sepsis/ Severe sepsis or septic shock at the ICU | HMGB1 | HMGB1 levels in sepsis patients for weeks after recovery from severe sepsis/septic shock; association of prolonged HMGB1 levels in plasma with cognitive impairment in patients recovering from severe sepsis/septic shock | Completed |
| NCT03535441 | Hemorrhagic shock (HS) | HMGB1 | Determination of the levels of HMGB1-mediated inflammation in the serum of patients with HS | Completed |
| NCT03346018 | Tuberculosis/ Sarcoidosis | HSP70 | Establishment as a biomarker for the differential diagnosis of tuberculosis and sarcoidosis | Recruiting |
| NCT04614441 | Certain types of lung disease | HMGB1, HSP27 | Assessment of levels in patients with lung disease | Recruiting |
| NCT04787770 | Diabetic atherosclerosis | HSP90 | Assessment of the correlation between HSP90 levels and diabetic atherosclerosis | Completed |
| NCT05007444 | Cancer/ Breast cancer | HMGB1, CRT, ATP | Assessment of the efficacy of the P2Et extract in ICD induction | Not yet recruiting |
| NCT01637532 | Cancer/ Recurrent ovarian cancer | HMGB1, CRT | Assessment of the efficacy of carbo/doxorubicin/tocilizumab/ Peg-Intron combination in ICD induction | Completed |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Birmpilis, A.I.; Paschalis, A.; Mourkakis, A.; Christodoulou, P.; Kostopoulos, I.V.; Antimissari, E.; Terzoudi, G.; Georgakilas, A.G.; Armpilia, C.; Papageorgis, P.; et al. Immunogenic Cell Death, DAMPs and Prothymosin α as a Putative Anticancer Immune Response Biomarker. Cells 2022, 11, 1415. https://doi.org/10.3390/cells11091415
Birmpilis AI, Paschalis A, Mourkakis A, Christodoulou P, Kostopoulos IV, Antimissari E, Terzoudi G, Georgakilas AG, Armpilia C, Papageorgis P, et al. Immunogenic Cell Death, DAMPs and Prothymosin α as a Putative Anticancer Immune Response Biomarker. Cells. 2022; 11(9):1415. https://doi.org/10.3390/cells11091415
Chicago/Turabian StyleBirmpilis, Anastasios I., Antonios Paschalis, Apostolis Mourkakis, Panayiota Christodoulou, Ioannis V. Kostopoulos, Elina Antimissari, Georgia Terzoudi, Alexandros G. Georgakilas, Christina Armpilia, Panagiotis Papageorgis, and et al. 2022. "Immunogenic Cell Death, DAMPs and Prothymosin α as a Putative Anticancer Immune Response Biomarker" Cells 11, no. 9: 1415. https://doi.org/10.3390/cells11091415
APA StyleBirmpilis, A. I., Paschalis, A., Mourkakis, A., Christodoulou, P., Kostopoulos, I. V., Antimissari, E., Terzoudi, G., Georgakilas, A. G., Armpilia, C., Papageorgis, P., Kastritis, E., Terpos, E., Dimopoulos, M. A., Kalbacher, H., Livaniou, E., Christodoulou, M. -I., & Tsitsilonis, O. E. (2022). Immunogenic Cell Death, DAMPs and Prothymosin α as a Putative Anticancer Immune Response Biomarker. Cells, 11(9), 1415. https://doi.org/10.3390/cells11091415