
Reduced Sarcolemmal Membrane Repair Exacerbates Striated Muscle Pathology in a Mouse Model of Duchenne Muscular Dystrophy



Abstract
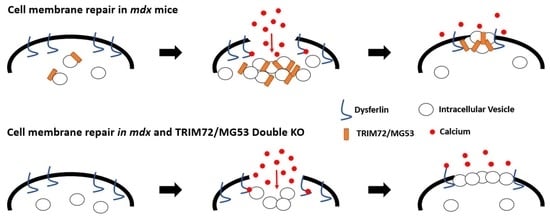
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Mouse Model Breeding
2.2. Tissue Preparation for Histological Procedures
2.3. Myocyte Size Measurements
2.4. Central Nuclei Measurements
2.5. IgG Staining and Analysis
2.6. Masson’s Trichrome Analysis
2.7. Ex Vivo Assessment of Skeletal Muscle Contractility
2.8. Western Blotting and ELISA Measurements
2.9. Membrane Repair Assessment following Laser Injury
2.10. Statistical Analysis
3. Results
3.1. Membrane Repair Is Compromised in the Six-Week-Old DKO Mouse
3.2. Compensatory Changes in Membrane Repair Proteins in the DKO Mouse Skeletal Muscle
3.3. Deletion of TRIM72/MG53 Does Not Alter Skeletal Muscle Histological Pathology in Six-Week-Old DKO Mice
3.4. Decreased Skeletal Muscle Force Production in Aged DKO Mice
3.5. Increased Fibrosis in the Striated Muscle of Aged DKO Mice
3.6. Sarcolemmal Membrane Integrity and Repair Is Compromised in Aged DKO Mice
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Bi, G.Q.; Alderton, J.M.; Steinhardt, R.A. Calcium-regulated exocytosis is required for cell membrane resealing. J. Cell Biol. 1995, 131, 1747–1758. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McNeil, P.L.; Ito, S. Gastrointestinal cell plasma membrane wounding and resealing in vivo. Gastroenterology 1989, 96, 1238–1248. [Google Scholar] [CrossRef]
- McNeil, P.L.; Khakee, R. Disruptions of muscle fiber plasma membranes. Role in exercise-induced damage. Am. J. Pathol. 1992, 140, 1097–1109. [Google Scholar]
- Steinhardt, R.A.; Bi, G.; Alderton, J.M. Cell membrane resealing by a vesicular mechanism similar to neurotransmitter release. Science 1994, 263, 390–393. [Google Scholar] [CrossRef]
- Miyake, K.; McNeil, P.L. Vesicle accumulation and exocytosis at sites of plasma membrane disruption. J. Cell Biol. 1995, 131, 1737–1745. [Google Scholar] [CrossRef]
- Reddy, A.; Caler, E.V.; Andrews, N.W. Plasma membrane repair is mediated by Ca2+-regulated exocytosis of lysosomes. Cell 2001, 106, 157–169. [Google Scholar] [CrossRef] [Green Version]
- Idone, V.; Tam, C.; Goss, J.W.; Toomre, D.; Pypaert, M.; Andrews, N.W. Repair of injured plasma membrane by rapid Ca2+-dependent endocytosis. J. Cell Biol. 2008, 180, 905–914. [Google Scholar] [CrossRef] [Green Version]
- Bement, W.M.; Forscher, P.; Mooseker, M.S. A novel cytoskeletal structure involved in purse string wound closure and cell polarity maintenance. J. Cell Biol. 1993, 121, 565–578. [Google Scholar] [CrossRef]
- Babiychuk, E.B.; Monastyrskaya, K.; Potez, S.; Draeger, A. Intracellular Ca2+ operates a switch between repair and lysis of streptolysin O-perforated cells. Cell Death Differ. 2009, 16, 1126–1134. [Google Scholar] [CrossRef] [Green Version]
- Jimenez, A.J.; Maiuri, P.; Lafaurie-Janvore, J.; Divoux, S.; Piel, M.; Perez, F. ESCRT machinery is required for plasma membrane repair. Science 2014, 343, 1247136. [Google Scholar] [CrossRef]
- Keyel, P.A.; Loultcheva, L.; Roth, R.; Salter, R.D.; Watkins, S.C.; Yokoyama, W.M.; Heuser, J.E. Streptolysin O clearance through sequestration into blebs that bud passively from the plasma membrane. J. Cell Sci. 2011, 124, 2414–2423. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Friden, J.; Sjostrom, M.; Ekblom, B. Myofibrillar damage following intense eccentric exercise in man. Int. J. Sports Med. 1983, 4, 170–176. [Google Scholar] [CrossRef] [PubMed]
- Lin, P.; Zhu, H.; Cai, C.; Wang, X.; Cao, C.; Xiao, R.; Pan, Z.; Weisleder, N.; Takeshima, H.; Ma, J. Nonmuscle myosin IIA facilitates vesicle trafficking for MG53-mediated cell membrane repair. FASEB J. 2012, 26, 1875–1883. [Google Scholar] [CrossRef] [PubMed]
- Weisleder, N.; Takeshima, H.; Ma, J. Mitsugumin 53 (MG53) facilitates vesicle trafficking in striated muscle to contribute to cell membrane repair. Commun. Integr. Biol. 2009, 2, 225–226. [Google Scholar] [CrossRef] [PubMed]
- Cai, C.; Weisleder, N.; Ko, J.K.; Komazaki, S.; Sunada, Y.; Nishi, M.; Takeshima, H.; Ma, J. Membrane repair defects in muscular dystrophy are linked to altered interaction between MG53, caveolin-3, and dysferlin. J. Biol. Chem. 2009, 284, 15894–15902. [Google Scholar] [CrossRef] [Green Version]
- Codding, S.J.; Marty, N.; Abdullah, N.; Johnson, C.P. Dysferlin Binds SNAREs (Soluble N-Ethylmaleimide-sensitive Factor (NSF) Attachment Protein Receptors) and Stimulates Membrane Fusion in a Calcium-sensitive Manner. J. Biol. Chem. 2016, 291, 14575–14584. [Google Scholar] [CrossRef] [Green Version]
- Hatakeyama, S. TRIM proteins and cancer. Nat. Rev. Cancer 2011, 11, 792–804. [Google Scholar] [CrossRef]
- Ozato, K.; Shin, D.M.; Chang, T.H.; Morse, H.C. TRIM family proteins and their emerging roles in innate immunity. Nat. Rev. Immunol. 2008, 8, 849–860. [Google Scholar] [CrossRef] [Green Version]
- Weisleder, N.; Takeshima, H.; Ma, J. Immuno-proteomic approach to excitation--contraction coupling in skeletal and cardiac muscle: Molecular insights revealed by the mitsugumins. Cell Calcium 2008, 43, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Lee, C.S.; Yi, J.S.; Jung, S.Y.; Kim, B.W.; Lee, N.R.; Choo, H.J.; Jang, S.Y.; Han, J.; Chi, S.G.; Park, M.; et al. TRIM72 negatively regulates myogenesis via targeting insulin receptor substrate-1. Cell Death Differ. 2010, 17, 1254–1265. [Google Scholar] [CrossRef]
- Lee, H.; Park, J.J.; Nguyen, N.; Park, J.S.; Hong, J.; Kim, S.H.; Song, W.Y.; Kim, H.J.; Choi, K.; Cho, S.; et al. MG53-IRS-1 (Mitsugumin 53-Insulin Receptor Substrate-1) Interaction Disruptor Sensitizes Insulin Signaling in Skeletal Muscle. J. Biol. Chem. 2016, 291, 26627–26635. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cai, C.; Masumiya, H.; Weisleder, N.; Matsuda, N.; Nishi, M.; Hwang, M.; Ko, J.K.; Lin, P.; Thornton, A.; Zhao, X.; et al. MG53 nucleates assembly of cell membrane repair machinery. Nat. Cell Biol. 2009, 11, 56–64. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cao, C.M.; Zhang, Y.; Weisleder, N.; Ferrante, C.; Wang, X.; Lv, F.; Song, R.; Hwang, M.; Jin, L.; Guo, J.; et al. MG53 constitutes a primary determinant of cardiac ischemic preconditioning. Circulation 2010, 121, 2565–2574. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duann, P.; Li, H.; Lin, P.; Tan, T.; Wang, Z.; Chen, K.; Zhou, X.; Gumpper, K.; Zhu, H.; Ludwig, T.; et al. MG53-mediated cell membrane repair protects against acute kidney injury. Sci. Transl. Med. 2015, 7, 279ra236. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.C.; Kellett, T.; Wang, S.; Nishi, M.; Nagre, N.; Zhou, B.; Flodby, P.; Shilo, K.; Ghadiali, S.N.; Takeshima, H.; et al. TRIM72 is required for effective repair of alveolar epithelial cell wounding. Am. J. Physiol. Lung Cell Mol. Physiol. 2014, 307, L449–L459. [Google Scholar] [CrossRef] [Green Version]
- Cai, C.; Masumiya, H.; Weisleder, N.; Pan, Z.; Nishi, M.; Komazaki, S.; Takeshima, H.; Ma, J. MG53 regulates membrane budding and exocytosis in muscle cells. J. Biol. Chem. 2009, 284, 3314–3322. [Google Scholar] [CrossRef] [Green Version]
- Bansal, D.; Miyake, K.; Vogel, S.S.; Groh, S.; Chen, C.C.; Williamson, R.; McNeil, P.L.; Campbell, K.P. Defective membrane repair in dysferlin-deficient muscular dystrophy. Nature 2003, 423, 168–172. [Google Scholar] [CrossRef]
- Defour, A.; Van der Meulen, J.H.; Bhat, R.; Bigot, A.; Bashir, R.; Nagaraju, K.; Jaiswal, J.K. Dysferlin regulates cell membrane repair by facilitating injury-triggered acid sphingomyelinase secretion. Cell Death Dis. 2014, 5, e1306. [Google Scholar] [CrossRef] [Green Version]
- Lennon, N.J.; Kho, A.; Bacskai, B.J.; Perlmutter, S.L.; Hyman, B.T.; Brown, R.H. Dysferlin interacts with annexins A1 and A2 and mediates sarcolemmal wound-healing. J. Biol. Chem. 2003, 278, 50466–50473. [Google Scholar] [CrossRef] [Green Version]
- Bouter, A.; Gounou, C.; Bérat, R.; Tan, S.; Gallois, B.; Granier, T.; d’Estaintot, B.L.; Pöschl, E.; Brachvogel, B.; Brisson, A.R. Annexin-A5 assembled into two-dimensional arrays promotes cell membrane repair. Nat. Commun. 2011, 2, 270. [Google Scholar] [CrossRef] [Green Version]
- Demonbreun, A.R.; Quattrocelli, M.; Barefield, D.Y.; Allen, M.V.; Swanson, K.E.; McNally, E.M. An actin-dependent annexin complex mediates plasma membrane repair in muscle. J. Cell Biol. 2016, 213, 705–718. [Google Scholar] [CrossRef] [PubMed]
- McNeil, A.K.; Rescher, U.; Gerke, V.; McNeil, P.L. Requirement for annexin A1 in plasma membrane repair. J. Biol. Chem. 2006, 281, 35202–35207. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rezvanpour, A.; Santamaria-Kisiel, L.; Shaw, G.S. The S100A10-annexin A2 complex provides a novel asymmetric platform for membrane repair. J. Biol. Chem. 2011, 286, 40174–40183. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Swaggart, K.A.; Demonbreun, A.R.; Vo, A.H.; Swanson, K.E.; Kim, E.Y.; Fahrenbach, J.P.; Holley-Cuthrell, J.; Eskin, A.; Chen, Z.; Squire, K.; et al. Annexin A6 modifies muscular dystrophy by mediating sarcolemmal repair. Proc. Natl. Acad. Sci. USA 2014, 111, 6004–6009. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Corrotte, M.; Almeida, P.E.; Tam, C.; Castro-Gomes, T.; Fernandes, M.C.; Millis, B.A.; Cortez, M.; Miller, H.; Song, W.; Maugel, T.K.; et al. Caveolae internalization repairs wounded cells and muscle fibers. elife 2013, 2, e00926. [Google Scholar] [CrossRef]
- Hernández-Deviez, D.J.; Howes, M.T.; Laval, S.H.; Bushby, K.; Hancock, J.F.; Parton, R.G. Caveolin regulates endocytosis of the muscle repair protein, dysferlin. J. Biol. Chem. 2008, 283, 6476–6488. [Google Scholar] [CrossRef] [Green Version]
- Tagawa, K.; Ogawa, M.; Kawabe, K.; Yamanaka, G.; Matsumura, T.; Goto, K.; Nonaka, I.; Nishino, I.; Hayashi, Y.K. Protein and gene analyses of dysferlinopathy in a large group of Japanese muscular dystrophy patients. J. Neurol. Sci. 2003, 211, 23–28. [Google Scholar] [CrossRef]
- Fanin, M.; Angelini, C. Muscle pathology in dysferlin deficiency. Neuropathol. Appl. NeuroBiol. 2002, 28, 461–470. [Google Scholar] [CrossRef]
- Mahjneh, I.; Marconi, G.; Bushby, K.; Anderson, L.V.; Tolvanen-Mahjneh, H.; Somer, H. Dysferlinopathy (LGMD2B): A 23-year follow-up study of 10 patients homozygous for the same frameshifting dysferlin mutations. Neuromuscul. Disord. 2001, 11, 20–26. [Google Scholar] [CrossRef]
- Dincer, P.; Akcoren, Z.; Demir, E.; Richard, I.; Sancak, O.; Kale, G.; Ozme, S.; Karaduman, A.; Tan, E.; Urtizberea, J.A.; et al. A cross section of autosomal recessive limb-girdle muscular dystrophies in 38 families. J. Med. Genet. 2000, 37, 361–367. [Google Scholar] [CrossRef] [Green Version]
- Galbiati, F.; Volonte, D.; Minetti, C.; Chu, J.B.; Lisanti, M.P. Phenotypic behavior of caveolin-3 mutations that cause autosomal dominant limb girdle muscular dystrophy (LGMD-1C). Retention of LGMD-1C caveolin-3 mutants within the golgi complex. J. Biol. Chem. 1999, 274, 25632–25641. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Minetti, C.; Sotgia, F.; Bruno, C.; Scartezzini, P.; Broda, P.; Bado, M.; Masetti, E.; Mazzocco, M.; Egeo, A.; Donati, M.A.; et al. Mutations in the caveolin-3 gene cause autosomal dominant limb-girdle muscular dystrophy. Nat. Genet. 1998, 18, 365–368. [Google Scholar] [CrossRef] [PubMed]
- Waddell, L.B.; Lemckert, F.A.; Zheng, X.F.; Tran, J.; Evesson, F.J.; Hawkes, J.M.; Lek, A.; Street, N.E.; Lin, P.; Clarke, N.F.; et al. Dysferlin, annexin A1, and mitsugumin 53 are upregulated in muscular dystrophy and localize to longitudinal tubules of the T-system with stretch. J. Neuropathol. Exp. Neurol. 2011, 70, 302–313. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Corrotte, M.; Fernandes, M.C.; Tam, C.; Andrews, N.W. Toxin pores endocytosed during plasma membrane repair traffic into the lumen of MVBs for degradation. Traffic 2012, 13, 483–494. [Google Scholar] [CrossRef] [Green Version]
- Gu, J.H.; Ge, J.B.; Li, M.; Xu, H.D.; Wu, F.; Qin, Z.H. Poloxamer 188 protects neurons against ischemia/reperfusion injury through preserving integrity of cell membranes and blood brain barrier. PLoS ONE 2013, 8, e61641. [Google Scholar] [CrossRef]
- Yao, Y.; Zhang, B.; Zhu, H.; Li, H.; Han, Y.; Chen, K.; Wang, Z.; Zeng, J.; Liu, Y.; Wang, X.; et al. MG53 permeates through blood-brain barrier to protect ischemic brain injury. Oncotarget 2016, 7, 22474–22485. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.; Zhu, H.; Zheng, Y.; Xu, Z.; Li, L.; Tan, T.; Park, K.H.; Hou, J.; Zhang, C.; Li, D.; et al. Cardioprotection of recombinant human MG53 protein in a porcine model of ischemia and reperfusion injury. J. Mol. Cell. Cardiol. 2015, 80, 10–19. [Google Scholar] [CrossRef] [Green Version]
- Zhu, H.; Hou, J.; Roe, J.L.; Park, K.H.; Tan, T.; Zheng, Y.; Li, L.; Zhang, C.; Liu, J.; Liu, Z.; et al. Amelioration of ischemia-reperfusion-induced muscle injury by the recombinant human MG53 protein. Muscle Nerve 2015, 52, 852–858. [Google Scholar] [CrossRef]
- Liu, J.; Aoki, M.; Illa, I.; Wu, C.; Fardeau, M.; Angelini, C.; Serrano, C.; Urtizberea, J.A.; Hentati, F.; Hamida, M.B.; et al. Dysferlin, a novel skeletal muscle gene, is mutated in Miyoshi myopathy and limb girdle muscular dystrophy. Nat. Genet. 1998, 20, 31–36. [Google Scholar] [CrossRef]
- Ryder, S.; Leadley, R.M.; Armstrong, N.; Westwood, M.; de Kock, S.; Butt, T.; Jain, M.; Kleijnen, J. The burden, epidemiology, costs and treatment for Duchenne muscular dystrophy: An evidence review. Orphanet J. Rare Dis. 2017, 12, 79. [Google Scholar] [CrossRef] [Green Version]
- Mendell, J.R.; Lloyd-Puryear, M. Report of MDA muscle disease symposium on newborn screening for Duchenne muscular dystrophy. Muscle Nerve 2013, 48, 21–26. [Google Scholar] [CrossRef] [PubMed]
- Weisleder, N.; Takizawa, N.; Lin, P.; Wang, X.; Cao, C.; Zhang, Y.; Tan, T.; Ferrante, C.; Zhu, H.; Chen, P.J.; et al. Recombinant MG53 protein modulates therapeutic cell membrane repair in treatment of muscular dystrophy. Sci. Transl. Med. 2012, 4, 139ra185. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Markham, B.E.; Kernodle, S.; Nemzek, J.; Wilkinson, J.E.; Sigler, R. Chronic Dosing with Membrane Sealant Poloxamer 188 NF Improves Respiratory Dysfunction in Dystrophic Mdx and Mdx/Utrophin-/- Mice. PLoS ONE 2015, 10, e0134832. [Google Scholar] [CrossRef] [PubMed]
- Houang, E.M.; Haman, K.J.; Filareto, A.; Perlingeiro, R.C.; Bates, F.S.; Lowe, D.A.; Metzger, J.M. Membrane-stabilizing copolymers confer marked protection to dystrophic skeletal muscle in vivo. Mol. Ther. Methods Clin. Dev. 2015, 2, 15042. [Google Scholar] [CrossRef]
- Renzini, A.; Marroncelli, N.; Cavioli, G.; Di Francescantonio, S.; Forcina, L.; Lambridis, A.; Di Giorgio, E.; Valente, S.; Mai, A.; Brancolini, C.; et al. Cytoplasmic HDAC4 regulates the membrane repair mechanism in Duchenne muscular dystrophy. J. Cachexia Sarcopenia Muscle 2022, 13, 1339–1359. [Google Scholar] [CrossRef]
- Mâncio, R.; Hermes, T.; Macedo, A.; Mizobuti, D.; Valduga, A.; Rupcic, I.; Minatel, E. Vitamin E treatment decreases muscle injury in mdx mice. Nutrition 2017, 43–44, 39–46. [Google Scholar] [CrossRef]
- Quattrocelli, M.; Salamone, I.; Page, P.; Warner, J.; Demonbreun, A.; McNally, E. Intermittent Glucocorticoid Dosing Improves Muscle Repair and Function in Mice with Limb-Girdle Muscular Dystrophy. Am. J. Pathol. 2017, 187, 2520–2535. [Google Scholar] [CrossRef] [Green Version]
- Vila, M.; Rayavarapu, S.; Hogarth, M.; Van der Meulen, J.; Horn, A.; Defour, A.; Takeda, S.; Brown, K.; Hathout, Y.; Nagaraju, K.; et al. Mitochondria mediate cell membrane repair and contribute to Duchenne muscular dystrophy. Cell Death Differ. 2017, 24, 330–342. [Google Scholar] [CrossRef] [Green Version]
- Wright, P.; McKinney, E.; Berry, S.; Evers, A.; Kent, C. A functional membrane repair system in Duchenne muscular dystrophy fibroblasts. J. Neurol. Sci. 1984, 64, 259–264. [Google Scholar] [CrossRef]
- Han, R.; Rader, E.P.; Levy, J.R.; Bansal, D.; Campbell, K.P. Dystrophin deficiency exacerbates skeletal muscle pathology in dysferlin-null mice. Skelet Muscle 2011, 1, 35. [Google Scholar] [CrossRef] [Green Version]
- Matsuda, C.; Miyake, K.; Kameyama, K.; Keduka, E.; Takeshima, H.; Imamura, T.; Araki, N.; Nishino, I.; Hayashi, Y. The C2A domain in dysferlin is important for association with MG53 (TRIM72). PLoS Curr. 2012, 4, e5035add8caff4. [Google Scholar] [CrossRef] [PubMed]
- Radley-Crabb, H.G.; Marini, J.C.; Sosa, H.A.; Castillo, L.I.; Grounds, M.D.; Fiorotto, M.L. Dystropathology increases energy expenditure and protein turnover in the mdx mouse model of duchenne muscular dystrophy. PLoS ONE 2014, 9, e89277. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dupont-Versteegden, E.E.; McCarter, R.J. Differential expression of muscular dystrophy in diaphragm versus hindlimb muscles of mdx mice. Muscle Nerve 1992, 15, 1105–1110. [Google Scholar] [CrossRef] [PubMed]
- Motohashi, N.; Alexander, M.S.; Shimizu-Motohashi, Y.; Myers, J.A.; Kawahara, G.; Kunkel, L.M. Regulation of IRS1/Akt insulin signaling by microRNA-128a during myogenesis. J. Cell Sci. 2013, 126, 2678–2691. [Google Scholar] [CrossRef] [Green Version]
- Yi, J.-S.; Park, J.S.; Ham, Y.-M.; Nguyen, N.; Lee, N.-R.; Hong, J.; Kim, B.-W.; Lee, H.; Lee, C.-S.; Jeong, B.-C.; et al. MG53-induced IRS-1 ubiquitination negatively regulates skeletal myogenesis and insulin signalling. Nat. Commun. 2013, 4, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.; Li, X.; Ong, H.; Tan, T.; Park, K.; Bian, Z.; Zou, X.; Haggard, E.; Janssen, P.; Merritt, R.; et al. MG53 suppresses NF-κB activation to mitigate age-related heart failure. JCI Insight 2021, 6, e148375. [Google Scholar] [CrossRef]
- Jiang, P.; Ren, L.; Zhi, L.; Yu, Z.; Lv, F.; Xu, F.; Peng, W.; Bai, X.; Cheng, K.; Quan, L.; et al. Negative regulation of AMPK signaling by high glucose via E3 ubiquitin ligase MG53. Mol. Cell 2020, 81, 629–637. [Google Scholar] [CrossRef]
- Wynn, T.A. Cellular and molecular mechanisms of fibrosis. J. Pathol. 2008, 214, 199–210. [Google Scholar] [CrossRef] [Green Version]
- Tidball, J.G.; Villalta, S.A. Regulatory interactions between muscle and the immune system during muscle regeneration. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2010, 298, R1173–R1187. [Google Scholar] [CrossRef] [Green Version]
- Hamrick, M.; Ding, K.; Pennington, C.; Chao, Y.; Wu, Y.; Howard, B.; Immel, D.; Borlongan, C.; McNeil, P.; Bollag, W.; et al. Age-related loss of muscle mass and bone strength in mice is associated with a decline in physical activity and serum leptin. Bone 2006, 39, 845–853. [Google Scholar] [CrossRef]
- Cohen, T.V.; Cohen, J.E.; Partridge, T.A. Myogenesis in dysferlin-deficient myoblasts is inhibited by an intrinsic inflammatory response. Neuromuscul. Disord. 2012, 22, 648–658. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moens, P.; Baatsen, P.H.; Marechal, G. Increased susceptibility of EDL muscles from mdx mice to damage induced by contractions with stretch. J. Muscle Res. Cell Motil. 1993, 14, 446–451. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.K.; Zhang, Y.; Cao, C.M.; Hu, X.; Fang, M.; Yao, Y.; Jin, L.; Chen, G.; Jiang, P.; Zhang, S.; et al. Glucose-Sensitive Myokine/Cardiokine MG53 Regulates Systemic Insulin Response and Metabolic Homeostasis. Circulation 2019, 139, 901–914. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; de Morrée, A.; van Remoortere, A.; Bushby, K.; Frants, R.R.; den Dunnen, J.T.; van der Maarel, S.M. Calpain 3 is a modulator of the dysferlin protein complex in skeletal muscle. Hum. Mol. Genet. 2008, 17, 1855–1866. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duguez, S.; Bartoli, M.; Richard, I. Calpain 3: A key regulator of the sarcomere? FEBS J. 2006, 273, 3427–3436. [Google Scholar] [CrossRef] [PubMed]
- Mellgren, R.L.; Miyake, K.; Kramerova, I.; Spencer, M.J.; Bourg, N.; Bartoli, M.; Richard, I.; Greer, P.A.; McNeil, P.L. Calcium-dependent plasma membrane repair requires m- or mu-calpain, but not calpain-3, the proteasome, or caspases. Biochim. Biophys. Acta 2009, 1793, 1886–1893. [Google Scholar] [CrossRef] [Green Version]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Paleo, B.J.; McElhanon, K.E.; Bulgart, H.R.; Banford, K.K.; Beck, E.X.; Sattler, K.M.; Goines, B.N.; Ratcliff, S.L.; Crowe, K.E.; Weisleder, N. Reduced Sarcolemmal Membrane Repair Exacerbates Striated Muscle Pathology in a Mouse Model of Duchenne Muscular Dystrophy. Cells 2022, 11, 1417. https://doi.org/10.3390/cells11091417
Paleo BJ, McElhanon KE, Bulgart HR, Banford KK, Beck EX, Sattler KM, Goines BN, Ratcliff SL, Crowe KE, Weisleder N. Reduced Sarcolemmal Membrane Repair Exacerbates Striated Muscle Pathology in a Mouse Model of Duchenne Muscular Dystrophy. Cells. 2022; 11(9):1417. https://doi.org/10.3390/cells11091417
Chicago/Turabian StylePaleo, Brian J., Kevin E. McElhanon, Hannah R. Bulgart, Kassidy K. Banford, Eric X Beck, Kristina M. Sattler, Briana N. Goines, Shelby L. Ratcliff, Kelly E. Crowe, and Noah Weisleder. 2022. "Reduced Sarcolemmal Membrane Repair Exacerbates Striated Muscle Pathology in a Mouse Model of Duchenne Muscular Dystrophy" Cells 11, no. 9: 1417. https://doi.org/10.3390/cells11091417
APA StylePaleo, B. J., McElhanon, K. E., Bulgart, H. R., Banford, K. K., Beck, E. X., Sattler, K. M., Goines, B. N., Ratcliff, S. L., Crowe, K. E., & Weisleder, N. (2022). Reduced Sarcolemmal Membrane Repair Exacerbates Striated Muscle Pathology in a Mouse Model of Duchenne Muscular Dystrophy. Cells, 11(9), 1417. https://doi.org/10.3390/cells11091417