
It’s All in the PAN: Crosstalk, Plasticity, Redundancies, Switches, and Interconnectedness Encompassed by PANoptosis Underlying the Totality of Cell Death-Associated Biological Effects



Abstract
:1. Introduction
Key Components in Inflammatory Programmed Cell Death Pathways
2. Evidence of Crosstalk at the Molecular Level
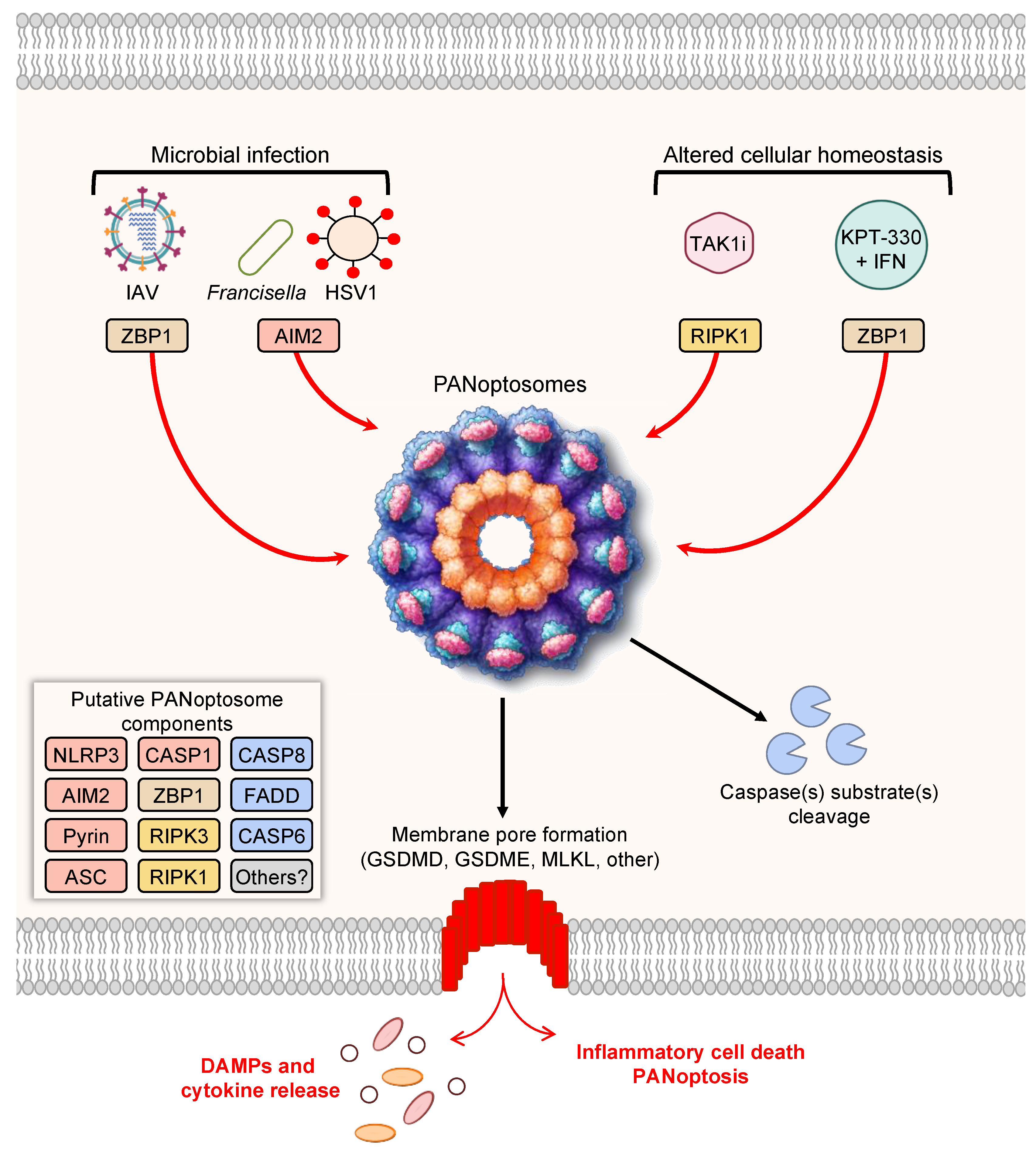
3. Prototypical Examples of PANoptosis
4. PANoptosis Regulation via IRF1
5. A Rose by Any Other Name
6. Discussion and Future Directions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Malireddi, R.K.S.; Kesavardhana, S.; Kanneganti, T.D. ZBP1 and TAK1: Master Regulators of NLRP3 Inflammasome/Pyroptosis, Apoptosis, and Necroptosis (PAN-optosis). Front. Cell. Infect. Microbiol. 2019, 9, 406. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karki, R.; Sharma, B.R.; Tuladhar, S.; Williams, E.P.; Zalduondo, L.; Samir, P.; Zheng, M.; Sundaram, B.; Banoth, B.; Malireddi, R.K.S.; et al. Synergism of TNF-alpha and IFN-gamma Triggers Inflammatory Cell Death, Tissue Damage, and Mortality in SARS-CoV-2 Infection and Cytokine Shock Syndromes. Cell 2021, 184, 149–168.e117. [Google Scholar] [CrossRef] [PubMed]
- Malireddi, R.K.S.; Kesavardhana, S.; Karki, R.; Kancharana, B.; Burton, A.R.; Kanneganti, T.D. RIPK1 Distinctly Regulates Yersinia-Induced Inflammatory Cell Death, PANoptosis. Immunohorizons 2020, 4, 789–796. [Google Scholar] [CrossRef] [PubMed]
- Malireddi, R.K.S.; Karki, R.; Sundaram, B.; Kancharana, B.; Lee, S.; Samir, P.; Kanneganti, T.D. Inflammatory Cell Death, PANoptosis, Mediated by Cytokines in Diverse Cancer Lineages Inhibits Tumor Growth. Immunohorizons 2021, 5, 568–580. [Google Scholar] [CrossRef] [PubMed]
- Malireddi, R.K.S.; Gurung, P.; Mavuluri, J.; Dasari, T.K.; Klco, J.M.; Chi, H.; Kanneganti, T.D. TAK1 restricts spontaneous NLRP3 activation and cell death to control myeloid proliferation. J. Exp. Med. 2018, 215, 1023–1034. [Google Scholar] [CrossRef] [Green Version]
- Malireddi, R.K.; Ippagunta, S.; Lamkanfi, M.; Kanneganti, T.D. Cutting edge: Proteolytic inactivation of poly(ADP-ribose) polymerase 1 by the Nlrp3 and Nlrc4 inflammasomes. J. Immunol. 2010, 185, 3127–3130. [Google Scholar] [CrossRef] [Green Version]
- Karki, R.; Sundaram, B.; Sharma, B.R.; Lee, S.; Malireddi, R.K.S.; Nguyen, L.N.; Christgen, S.; Zheng, M.; Wang, Y.; Samir, P.; et al. ADAR1 restricts ZBP1-mediated immune response and PANoptosis to promote tumorigenesis. Cell Rep. 2021, 37, 109858. [Google Scholar] [CrossRef]
- Karki, R.; Sharma, B.R.; Lee, E.; Banoth, B.; Malireddi, R.K.S.; Samir, P.; Tuladhar, S.; Mummareddy, H.; Burton, A.R.; Vogel, P.; et al. Interferon regulatory factor 1 regulates PANoptosis to prevent colorectal cancer. JCI Insight 2020, 5, e136720. [Google Scholar] [CrossRef]
- Kuriakose, T.; Man, S.M.; Subbarao Malireddi, R.K.; Karki, R.; Kesavardhana, S.; Place, D.E.; Neale, G.; Vogel, P.; Kanneganti, T.D. ZBP1/DAI is an innate sensor of influenza virus triggering the NLRP3 inflammasome and programmed cell death pathways. Sci. Immunol. 2016, 1, aag2045. [Google Scholar] [CrossRef] [Green Version]
- Christgen, S.; Zheng, M.; Kesavardhana, S.; Karki, R.; Malireddi, R.K.S.; Banoth, B.; Place, D.E.; Briard, B.; Sharma, B.R.; Tuladhar, S.; et al. Identification of the PANoptosome: A Molecular Platform Triggering Pyroptosis, Apoptosis, and Necroptosis (PANoptosis). Front. Cell. Infect. Microbiol. 2020, 10, 237. [Google Scholar] [CrossRef]
- Zheng, M.; Karki, R.; Vogel, P.; Kanneganti, T.D. Caspase-6 is a key regulator of innate immunity, inflammasome activation and host defense. Cell 2020, 181, 674–687.e13. [Google Scholar] [CrossRef]
- Lee, S.; Karki, R.; Wang, Y.; Nguyen, L.N.; Kalathur, R.C.; Kanneganti, T.D. AIM2 forms a complex with pyrin and ZBP1 to drive PANoptosis and host defence. Nature 2021, 597, 415–419. [Google Scholar] [CrossRef]
- Kesavardhana, S.; Malireddi, R.K.S.; Burton, A.R.; Porter, S.N.; Vogel, P.; Pruett-Miller, S.M.; Kanneganti, T.D. The Zα2 domain of ZBP1 is a molecular switch regulating influenza-induced PANoptosis and perinatal lethality during development. J. Biol. Chem. 2020, 295, 8325–8330. [Google Scholar] [CrossRef] [PubMed]
- Banoth, B.; Tuladhar, S.; Karki, R.; Sharma, B.R.; Briard, B.; Kesavardhana, S.; Burton, A.; Kanneganti, T.D. ZBP1 promotes fungi-induced inflammasome activation and pyroptosis, apoptosis, and necroptosis (PANoptosis). J. Biol. Chem. 2020, 295, 18276–18283. [Google Scholar] [CrossRef] [PubMed]
- Zheng, M.; Williams, E.P.; Malireddi, R.K.S.; Karki, R.; Banoth, B.; Burton, A.; Webby, R.; Channappanavar, R.; Jonsson, C.B.; Kanneganti, T.D. Impaired NLRP3 inflammasome activation/pyroptosis leads to robust inflammatory cell death via caspase-8/RIPK3 during coronavirus infection. J. Biol. Chem. 2020, 295, 14040–14052. [Google Scholar] [CrossRef] [PubMed]
- Lukens, J.R.; Gurung, P.; Vogel, P.; Johnson, G.R.; Carter, R.A.; McGoldrick, D.J.; Bandi, S.R.; Calabrese, C.R.; Walle, L.V.; Lamkanfi, M.; et al. Dietary modulation of the microbiome affects autoinflammatory disease. Nature 2014, 516, 246–249. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lamkanfi, M.; Kanneganti, T.D.; Van Damme, P.; Vanden Berghe, T.; Vanoverberghe, I.; Vandekerckhove, J.; Vandenabeele, P.; Gevaert, K.; Nunez, G. Targeted peptidecentric proteomics reveals caspase-7 as a substrate of the caspase-1 inflammasomes. Mol. Cell. Proteom. 2008, 7, 2350–2363. [Google Scholar] [CrossRef] [Green Version]
- Gurung, P.; Burton, A.; Kanneganti, T.D. NLRP3 inflammasome plays a redundant role with caspase 8 to promote IL-1β–mediated osteomyelitis. Proc. Natl. Acad. Sci. USA 2016, 113, 4452–4457. [Google Scholar] [CrossRef] [Green Version]
- Gurung, P.; Anand, P.K.; Malireddi, R.K.; Vande Walle, L.; Van Opdenbosch, N.; Dillon, C.P.; Weinlich, R.; Green, D.R.; Lamkanfi, M.; Kanneganti, T.D. FADD and caspase-8 mediate priming and activation of the canonical and noncanonical Nlrp3 inflammasomes. J. Immunol. 2014, 192, 1835–1846. [Google Scholar] [CrossRef] [Green Version]
- Malireddi, R.K.S.; Gurung, P.; Kesavardhana, S.; Samir, P.; Burton, A.; Mummareddy, H.; Vogel, P.; Pelletier, S.; Burgula, S.; Kanneganti, T.D. Innate immune priming in the absence of TAK1 drives RIPK1 kinase activity-independent pyroptosis, apoptosis, necroptosis, and inflammatory disease. J. Exp. Med. 2020, 217, e20191644. [Google Scholar] [CrossRef]
- Kesavardhana, S.; Malireddi, R.K.S.; Kanneganti, T.D. Caspases in Cell Death, Inflammation, and Pyroptosis. Annu. Rev. Immunol. 2020, 38, 567–595. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Galluzzi, L.; Vitale, I.; Aaronson, S.A.; Abrams, J.M.; Adam, D.; Agostinis, P.; Alnemri, E.S.; Altucci, L.; Amelio, I.; Andrews, D.W.; et al. Molecular mechanisms of cell death: Recommendations of the Nomenclature Committee on Cell Death 2018. Cell Death Differ. 2018, 25, 486–541. [Google Scholar] [CrossRef] [PubMed]
- Cookson, B.T.; Brennan, M.A. Pro-inflammatory programmed cell death. Trends Microbiol. 2001, 9, 113–114. [Google Scholar] [CrossRef]
- Martinon, F.; Burns, K.; Tschopp, J. The inflammasome: A molecular platform triggering activation of inflammatory caspases and processing of proIL-beta. Mol. Cell 2002, 10, 417–426. [Google Scholar] [CrossRef]
- Kanneganti, T.D.; Ozoren, N.; Body-Malapel, M.; Amer, A.; Park, J.H.; Franchi, L.; Whitfield, J.; Barchet, W.; Colonna, M.; Vandenabeele, P.; et al. Bacterial RNA and small antiviral compounds activate caspase-1 through cryopyrin/Nalp3. Nature 2006, 440, 233–236. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mariathasan, S.; Weiss, D.S.; Newton, K.; McBride, J.; O’Rourke, K.; Roose-Girma, M.; Lee, W.P.; Weinrauch, Y.; Monack, D.M.; Dixit, V.M. Cryopyrin activates the inflammasome in response to toxins and ATP. Nature 2006, 440, 228–232. [Google Scholar] [CrossRef]
- Martinon, F.; Petrilli, V.; Mayor, A.; Tardivel, A.; Tschopp, J. Gout-associated uric acid crystals activate the NALP3 inflammasome. Nature 2006, 440, 237–241. [Google Scholar] [CrossRef] [Green Version]
- Franchi, L.; Amer, A.; Body-Malapel, M.; Kanneganti, T.D.; Ozoren, N.; Jagirdar, R.; Inohara, N.; Vandenabeele, P.; Bertin, J.; Coyle, A.; et al. Cytosolic flagellin requires Ipaf for activation of caspase-1 and interleukin 1beta in salmonella-infected macrophages. Nat. Immunol. 2006, 7, 576–582. [Google Scholar] [CrossRef]
- Miao, E.A.; Alpuche-Aranda, C.M.; Dors, M.; Clark, A.E.; Bader, M.W.; Miller, S.I.; Aderem, A. Cytoplasmic flagellin activates caspase-1 and secretion of interleukin 1beta via Ipaf. Nat. Immunol. 2006, 7, 569–575. [Google Scholar] [CrossRef]
- Mariathasan, S.; Newton, K.; Monack, D.M.; Vucic, D.; French, D.M.; Lee, W.P.; Roose-Girma, M.; Erickson, S.; Dixit, V.M. Differential activation of the inflammasome by caspase-1 adaptors ASC and Ipaf. Nature 2004, 430, 213–218. [Google Scholar] [CrossRef]
- Xu, H.; Yang, J.; Gao, W.; Li, L.; Li, P.; Zhang, L.; Gong, Y.N.; Peng, X.; Xi, J.J.; Chen, S.; et al. Innate immune sensing of bacterial modifications of Rho GTPases by the Pyrin inflammasome. Nature 2014, 513, 237–241. [Google Scholar] [CrossRef] [PubMed]
- Fernandes-Alnemri, T.; Yu, J.W.; Datta, P.; Wu, J.; Alnemri, E.S. AIM2 activates the inflammasome and cell death in response to cytoplasmic DNA. Nature 2009, 458, 509–513. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hornung, V.; Ablasser, A.; Charrel-Dennis, M.; Bauernfeind, F.; Horvath, G.; Caffrey, D.R.; Latz, E.; Fitzgerald, K.A. AIM2 recognizes cytosolic dsDNA and forms a caspase-1-activating inflammasome with ASC. Nature 2009, 458, 514–518. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cai, X.; Chen, J.; Xu, H.; Liu, S.; Jiang, Q.X.; Halfmann, R.; Chen, Z.J. Prion-like polymerization underlies signal transduction in antiviral immune defense and inflammasome activation. Cell 2014, 156, 1207–1222. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, A.; Magupalli, V.G.; Ruan, J.; Yin, Q.; Atianand, M.K.; Vos, M.R.; Schröder, G.F.; Fitzgerald, K.A.; Wu, H.; Egelman, E.H. Unified polymerization mechanism for the assembly of ASC-dependent inflammasomes. Cell 2014, 156, 1193–1206. [Google Scholar] [CrossRef] [Green Version]
- Masumoto, J.; Taniguchi, S.; Ayukawa, K.; Sarvotham, H.; Kishino, T.; Niikawa, N.; Hidaka, E.; Katsuyama, T.; Higuchi, T.; Sagara, J. ASC, a novel 22-kDa protein, aggregates during apoptosis of human promyelocytic leukemia HL-60 cells. J. Biol. Chem. 1999, 274, 33835–33838. [Google Scholar] [CrossRef] [Green Version]
- Sborgi, L.; Ravotti, F.; Dandey, V.P.; Dick, M.S.; Mazur, A.; Reckel, S.; Chami, M.; Scherer, S.; Huber, M.; Böckmann, A.; et al. Structure and assembly of the mouse ASC inflammasome by combined NMR spectroscopy and cryo-electron microscopy. Proc. Natl. Acad. Sci. USA 2015, 112, 13237–13242. [Google Scholar] [CrossRef] [Green Version]
- Dinarello, C.A. Immunological and inflammatory functions of the interleukin-1 family. Annu. Rev. Immunol. 2009, 27, 519–550. [Google Scholar] [CrossRef]
- Shi, J.; Zhao, Y.; Wang, K.; Shi, X.; Wang, Y.; Huang, H.; Zhuang, Y.; Cai, T.; Wang, F.; Shao, F. Cleavage of GSDMD by inflammatory caspases determines pyroptotic cell death. Nature 2015, 526, 660–665. [Google Scholar] [CrossRef]
- He, W.T.; Wan, H.; Hu, L.; Chen, P.; Wang, X.; Huang, Z.; Yang, Z.H.; Zhong, C.Q.; Han, J. Gasdermin D is an executor of pyroptosis and required for interleukin-1beta secretion. Cell Res. 2015, 25, 1285–1298. [Google Scholar] [CrossRef]
- Aglietti, R.A.; Estevez, A.; Gupta, A.; Ramirez, M.G.; Liu, P.S.; Kayagaki, N.; Ciferri, C.; Dixit, V.M.; Dueber, E.C. GsdmD p30 elicited by caspase-11 during pyroptosis forms pores in membranes. Proc. Natl. Acad. Sci. USA 2016, 113, 7858–7863. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ding, J.; Wang, K.; Liu, W.; She, Y.; Sun, Q.; Shi, J.; Sun, H.; Wang, D.C.; Shao, F. Pore-forming activity and structural autoinhibition of the gasdermin family. Nature 2016, 535, 111–116. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Zhang, Z.; Ruan, J.; Pan, Y.; Magupalli, V.G.; Wu, H.; Lieberman, J. Inflammasome-activated gasdermin D causes pyroptosis by forming membrane pores. Nature 2016, 535, 153–158. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sborgi, L.; Ruhl, S.; Mulvihill, E.; Pipercevic, J.; Heilig, R.; Stahlberg, H.; Farady, C.J.; Muller, D.J.; Broz, P.; Hiller, S. GSDMD membrane pore formation constitutes the mechanism of pyroptotic cell death. EMBO J. 2016, 35, 1766–1778. [Google Scholar] [CrossRef]
- Kayagaki, N.; Stowe, I.B.; Lee, B.L.; O’Rourke, K.; Anderson, K.; Warming, S.; Cuellar, T.; Haley, B.; Roose-Girma, M.; Phung, Q.T.; et al. Caspase-11 cleaves gasdermin D for non-canonical inflammasome signalling. Nature 2015, 526, 666–671. [Google Scholar] [CrossRef]
- Kayagaki, N.; Lee, B.L.; Stowe, I.B.; Kornfeld, O.S.; O’Rourke, K.; Mirrashidi, K.M.; Haley, B.; Watanabe, C.; Roose-Girma, M.; Modrusan, Z.; et al. IRF2 transcriptionally induces GSDMD expression for pyroptosis. Sci. Signal. 2019, 12, eaax4917. [Google Scholar] [CrossRef]
- Benaoudia, S.; Martin, A.; Puig Gamez, M.; Gay, G.; Lagrange, B.; Cornut, M.; Krasnykov, K.; Claude, J.B.; Bourgeois, C.F.; Hughes, S.; et al. A genome-wide screen identifies IRF2 as a key regulator of caspase-4 in human cells. EMBO Rep. 2019, 20, e48235. [Google Scholar] [CrossRef]
- Kerr, J.F.; Wyllie, A.H.; Currie, A.R. Apoptosis: A basic biological phenomenon with wide-ranging implications in tissue kinetics. Br. J. Cancer 1972, 26, 239–257. [Google Scholar] [CrossRef] [Green Version]
- Zou, H.; Henzel, W.J.; Liu, X.; Lutschg, A.; Wang, X. Apaf-1, a human protein homologous to C. elegans CED-4, participates in cytochrome c-dependent activation of caspase-3. Cell 1997, 90, 405–413. [Google Scholar] [CrossRef] [Green Version]
- Kim, H.E.; Du, F.; Fang, M.; Wang, X. Formation of apoptosome is initiated by cytochrome c-induced dATP hydrolysis and subsequent nucleotide exchange on Apaf-1. Proc. Natl. Acad. Sci. USA 2005, 102, 17545–17550. [Google Scholar] [CrossRef] [Green Version]
- Li, P.; Nijhawan, D.; Budihardjo, I.; Srinivasula, S.M.; Ahmad, M.; Alnemri, E.S.; Wang, X. Cytochrome c and dATP-dependent formation of Apaf-1/caspase-9 complex initiates an apoptotic protease cascade. Cell 1997, 91, 479–489. [Google Scholar] [CrossRef] [Green Version]
- Boldin, M.P.; Goncharov, T.M.; Goltsev, Y.V.; Wallach, D. Involvement of MACH, a novel MORT1/FADD-interacting protease, in Fas/APO-1- and TNF receptor-induced cell death. Cell 1996, 85, 803–815. [Google Scholar] [CrossRef] [Green Version]
- Muzio, M.; Chinnaiyan, A.M.; Kischkel, F.C.; O’Rourke, K.; Shevchenko, A.; Ni, J.; Scaffidi, C.; Bretz, J.D.; Zhang, M.; Gentz, R.; et al. FLICE, a novel FADD-homologous ICE/CED-3-like protease, is recruited to the CD95 (Fas/APO-1) death--inducing signaling complex. Cell 1996, 85, 817–827. [Google Scholar] [CrossRef] [Green Version]
- Gross, A.; Yin, X.M.; Wang, K.; Wei, M.C.; Jockel, J.; Milliman, C.; Erdjument-Bromage, H.; Tempst, P.; Korsmeyer, S.J. Caspase cleaved BID targets mitochondria and is required for cytochrome c release, while BCL-XL prevents this release but not tumor necrosis factor-R1/Fas death. J. Biol. Chem. 1999, 274, 1156–1163. [Google Scholar] [CrossRef] [Green Version]
- Luo, X.; Budihardjo, I.; Zou, H.; Slaughter, C.; Wang, X. Bid, a Bcl2 interacting protein, mediates cytochrome c release from mitochondria in response to activation of cell surface death receptors. Cell 1998, 94, 481–490. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Zhu, H.; Xu, C.J.; Yuan, J. Cleavage of BID by caspase 8 mediates the mitochondrial damage in the Fas pathway of apoptosis. Cell 1998, 94, 491–501. [Google Scholar] [CrossRef] [Green Version]
- Wei, M.C.; Lindsten, T.; Mootha, V.K.; Weiler, S.; Gross, A.; Ashiya, M.; Thompson, C.B.; Korsmeyer, S.J. tBID, a membrane-targeted death ligand, oligomerizes BAK to release cytochrome c. Genes Dev. 2000, 14, 2060–2071. [Google Scholar] [CrossRef]
- Kuwana, T.; Mackey, M.R.; Perkins, G.; Ellisman, M.H.; Latterich, M.; Schneiter, R.; Green, D.R.; Newmeyer, D.D. Bid, Bax, and lipids cooperate to form supramolecular openings in the outer mitochondrial membrane. Cell 2002, 111, 331–342. [Google Scholar] [CrossRef] [Green Version]
- Gong, Y.; Fan, Z.; Luo, G.; Yang, C.; Huang, Q.; Fan, K.; Cheng, H.; Jin, K.; Ni, Q.; Yu, X.; et al. The role of necroptosis in cancer biology and therapy. Mol Cancer 2019, 18, 100. [Google Scholar] [CrossRef] [Green Version]
- Nailwal, H.; Chan, F.K. Necroptosis in anti-viral inflammation. Cell Death Differ. 2019, 26, 4–13. [Google Scholar] [CrossRef] [Green Version]
- Kang, T.B.; Ben-Moshe, T.; Varfolomeev, E.E.; Pewzner-Jung, Y.; Yogev, N.; Jurewicz, A.; Waisman, A.; Brenner, O.; Haffner, R.; Gustafsson, E.; et al. Caspase-8 serves both apoptotic and nonapoptotic roles. J. Immunol. 2004, 173, 2976–2984. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Newton, K.; Wickliffe, K.E.; Dugger, D.L.; Maltzman, A.; Roose-Girma, M.; Dohse, M.; Kőműves, L.; Webster, J.D.; Dixit, V.M. Cleavage of RIPK1 by caspase-8 is crucial for limiting apoptosis and necroptosis. Nature 2019, 574, 428–431. [Google Scholar] [CrossRef] [PubMed]
- O’Donnell, M.A.; Perez-Jimenez, E.; Oberst, A.; Ng, A.; Massoumi, R.; Xavier, R.; Green, D.R.; Ting, A.T. Caspase 8 inhibits programmed necrosis by processing CYLD. Nat. Cell Biol. 2011, 13, 1437–1442. [Google Scholar] [CrossRef] [PubMed]
- Feng, S.; Yang, Y.; Mei, Y.; Ma, L.; Zhu, D.E.; Hoti, N.; Castanares, M.; Wu, M. Cleavage of RIP3 inactivates its caspase-independent apoptosis pathway by removal of kinase domain. Cell. Signal. 2007, 19, 2056–2067. [Google Scholar] [CrossRef] [PubMed]
- Grootjans, S.; Vanden Berghe, T.; Vandenabeele, P. Initiation and execution mechanisms of necroptosis: An overview. Cell Death Differ. 2017, 24, 1184–1195. [Google Scholar] [CrossRef] [PubMed]
- Vandenabeele, P.; Galluzzi, L.; Vanden Berghe, T.; Kroemer, G. Molecular mechanisms of necroptosis: An ordered cellular explosion. Nat. Rev. Mol. Cell Biol. 2010, 11, 700–714. [Google Scholar] [CrossRef]
- Sun, L.; Wang, H.; Wang, Z.; He, S.; Chen, S.; Liao, D.; Wang, L.; Yan, J.; Liu, W.; Lei, X.; et al. Mixed lineage kinase domain-like protein mediates necrosis signaling downstream of RIP3 kinase. Cell 2012, 148, 213–227. [Google Scholar] [CrossRef] [Green Version]
- Zhao, J.; Jitkaew, S.; Cai, Z.; Choksi, S.; Li, Q.; Luo, J.; Liu, Z.G. Mixed lineage kinase domain-like is a key receptor interacting protein 3 downstream component of TNF-induced necrosis. Proc. Natl. Acad. Sci. USA 2012, 109, 5322–5327. [Google Scholar] [CrossRef] [Green Version]
- Garcia, L.R.; Tenev, T.; Newman, R.; Haich, R.O.; Liccardi, G.; John, S.W.; Annibaldi, A.; Yu, L.; Pardo, M.; Young, S.N.; et al. Ubiquitylation of MLKL at lysine 219 positively regulates necroptosis-induced tissue injury and pathogen clearance. Nat. Commun. 2021, 12, 3364. [Google Scholar] [CrossRef]
- Sharif, H.; Wang, L.; Wang, W.L.; Magupalli, V.G.; Andreeva, L.; Qiao, Q.; Hauenstein, A.V.; Wu, Z.; Núñez, G.; Mao, Y.; et al. Structural mechanism for NEK7-licensed activation of NLRP3 inflammasome. Nature 2019, 570, 338–343. [Google Scholar] [CrossRef]
- Shi, H.; Wang, Y.; Li, X.; Zhan, X.; Tang, M.; Fina, M.; Su, L.; Pratt, D.; Bu, C.H.; Hildebrand, S.; et al. NLRP3 activation and mitosis are mutually exclusive events coordinated by NEK7, a new inflammasome component. Nat. Immunol. 2016, 17, 250–258. [Google Scholar] [CrossRef] [PubMed]
- Schmid-Burgk, J.L.; Chauhan, D.; Schmidt, T.; Ebert, T.S.; Reinhardt, J.; Endl, E.; Hornung, V. A Genome-wide CRISPR (Clustered Regularly Interspaced Short Palindromic Repeats) Screen Identifies NEK7 as an Essential Component of NLRP3 Inflammasome Activation. J. Biol. Chem. 2016, 291, 103–109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, Y.; Zeng, M.Y.; Yang, D.; Motro, B.; Nunez, G. NEK7 is an essential mediator of NLRP3 activation downstream of potassium efflux. Nature 2016, 530, 354–357. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Samir, P.; Kesavardhana, S.; Patmore, D.M.; Gingras, S.; Malireddi, R.K.S.; Karki, R.; Guy, C.S.; Briard, B.; Place, D.E.; Bhattacharya, A.; et al. DDX3X acts as a live-or-die checkpoint in stressed cells by regulating NLRP3 inflammasome. Nature 2019, 573, 590–594. [Google Scholar] [CrossRef] [PubMed]
- Kuriakose, T.; Zheng, M.; Neale, G.; Kanneganti, T.D. IRF1 Is a Transcriptional Regulator of ZBP1 Promoting NLRP3 Inflammasome Activation and Cell Death during Influenza Virus Infection. J. Immunol. 2018, 200, 1489–1495. [Google Scholar] [CrossRef] [Green Version]
- Karki, R.; Lee, E.; Place, D.; Samir, P.; Mavuluri, J.; Sharma, B.R.; Balakrishnan, A.; Malireddi, R.K.S.; Geiger, R.; Zhu, Q.; et al. IRF8 Regulates Transcription of Naips for NLRC4 Inflammasome Activation. Cell 2018, 173, 920–933.e13. [Google Scholar] [CrossRef] [Green Version]
- Kayagaki, N.; Kornfeld, O.S.; Lee, B.L.; Stowe, I.B.; O’Rourke, K.; Li, Q.; Sandoval, W.; Yan, D.; Kang, J.; Xu, M.; et al. NINJ1 mediates plasma membrane rupture during lytic cell death. Nature 2021, 591, 131–136. [Google Scholar] [CrossRef]
- Christgen, S.; Place, D.E.; Kanneganti, T.D. Toward targeting inflammasomes: Insights into their regulation and activation. Cell Res. 2020, 30, 315–327. [Google Scholar] [CrossRef] [Green Version]
- Bertheloot, D.; Latz, E.; Franklin, B.S. Necroptosis, pyroptosis and apoptosis: An intricate game of cell death. Cell Mol. Immunol. 2021, 18, 1106–1121. [Google Scholar] [CrossRef]
- Holler, N.; Zaru, R.; Micheau, O.; Thome, M.; Attinger, A.; Valitutti, S.; Bodmer, J.L.; Schneider, P.; Seed, B.; Tschopp, J. Fas triggers an alternative, caspase-8-independent cell death pathway using the kinase RIP as effector molecule. Nat. Immunol. 2000, 1, 489–495. [Google Scholar] [CrossRef]
- Oberst, A.; Dillon, C.P.; Weinlich, R.; McCormick, L.L.; Fitzgerald, P.; Pop, C.; Hakem, R.; Salvesen, G.S.; Green, D.R. Catalytic activity of the caspase-8-FLIP(L) complex inhibits RIPK3-dependent necrosis. Nature 2011, 471, 363–367. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaiser, W.J.; Upton, J.W.; Long, A.B.; Livingston-Rosanoff, D.; Daley-Bauer, L.P.; Hakem, R.; Caspary, T.; Mocarski, E.S. RIP3 mediates the embryonic lethality of caspase-8-deficient mice. Nature 2011, 471, 368–372. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alvarez-Diaz, S.; Dillon, C.P.; Lalaoui, N.; Tanzer, M.C.; Rodriguez, D.A.; Lin, A.; Lebois, M.; Hakem, R.; Josefsson, E.C.; O’Reilly, L.A.; et al. The Pseudokinase MLKL and the Kinase RIPK3 Have Distinct Roles in Autoimmune Disease Caused by Loss of Death-Receptor-Induced Apoptosis. Immunity 2016, 45, 513–526. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Varfolomeev, E.E.; Schuchmann, M.; Luria, V.; Chiannilkulchai, N.; Beckmann, J.S.; Mett, I.L.; Rebrikov, D.; Brodianski, V.M.; Kemper, O.C.; Kollet, O.; et al. Targeted disruption of the mouse Caspase 8 gene ablates cell death induction by the TNF receptors, Fas/Apo1, and DR3 and is lethal prenatally. Immunity 1998, 9, 267–276. [Google Scholar] [CrossRef] [Green Version]
- Lalaoui, N.; Boyden, S.E.; Oda, H.; Wood, G.M.; Stone, D.L.; Chau, D.; Liu, L.; Stoffels, M.; Kratina, T.; Lawlor, K.E.; et al. Mutations that prevent caspase cleavage of RIPK1 cause autoinflammatory disease. Nature 2020, 577, 103–108. [Google Scholar] [CrossRef]
- Chun, H.J.; Zheng, L.; Ahmad, M.; Wang, J.; Speirs, C.K.; Siegel, R.M.; Dale, J.K.; Puck, J.; Davis, J.; Hall, C.G.; et al. Pleiotropic defects in lymphocyte activation caused by caspase-8 mutations lead to human immunodeficiency. Nature 2002, 419, 395–399. [Google Scholar] [CrossRef] [Green Version]
- Puri, A.W.; Broz, P.; Shen, A.; Monack, D.M.; Bogyo, M. Caspase-1 activity is required to bypass macrophage apoptosis upon Salmonella infection. Nat. Chem. Biol. 2012, 8, 745–747. [Google Scholar] [CrossRef] [Green Version]
- Yu, J.; Nagasu, H.; Murakami, T.; Hoang, H.; Broderick, L.; Hoffman, H.M.; Horng, T. Inflammasome activation leads to Caspase-1-dependent mitochondrial damage and block of mitophagy. Proc. Natl. Acad. Sci. USA 2014, 111, 15514–15519. [Google Scholar] [CrossRef] [Green Version]
- Pierini, R.; Juruj, C.; Perret, M.; Jones, C.L.; Mangeot, P.; Weiss, D.S.; Henry, T. AIM2/ASC triggers caspase-8-dependent apoptosis in Francisella-infected caspase-1-deficient macrophages. Cell Death Differ. 2012, 19, 1709–1721. [Google Scholar] [CrossRef] [Green Version]
- Sagulenko, V.; Thygesen, S.J.; Sester, D.P.; Idris, A.; Cridland, J.A.; Vajjhala, P.R.; Roberts, T.L.; Schroder, K.; Vince, J.E.; Hill, J.M.; et al. AIM2 and NLRP3 inflammasomes activate both apoptotic and pyroptotic death pathways via ASC. Cell Death Differ. 2013, 20, 1149–1160. [Google Scholar] [CrossRef] [Green Version]
- Man, S.M.; Tourlomousis, P.; Hopkins, L.; Monie, T.P.; Fitzgerald, K.A.; Bryant, C.E. Salmonella infection induces recruitment of Caspase-8 to the inflammasome to modulate IL-1β production. J. Immunol. 2013, 191, 5239–5246. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mascarenhas, D.P.A.; Cerqueira, D.M.; Pereira, M.S.F.; Castanheira, F.V.S.; Fernandes, T.D.; Manin, G.Z.; Cunha, L.D.; Zamboni, D.S. Inhibition of caspase-1 or gasdermin-D enable caspase-8 activation in the Naip5/NLRC4/ASC inflammasome. PLoS Pathog. 2017, 13, e1006502. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Opdenbosch, N.; Van Gorp, H.; Verdonckt, M.; Saavedra, P.H.V.; de Vasconcelos, N.M.; Goncalves, A.; Vande Walle, L.; Demon, D.; Matusiak, M.; Van Hauwermeiren, F.; et al. Caspase-1 Engagement and TLR-Induced c-FLIP Expression Suppress ASC/Caspase-8-Dependent Apoptosis by Inflammasome Sensors NLRP1b and NLRC4. Cell Rep. 2017, 21, 3427–3444. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Man, S.M.; Hopkins, L.J.; Nugent, E.; Cox, S.; Glück, I.M.; Tourlomousis, P.; Wright, J.A.; Cicuta, P.; Monie, T.P.; Bryant, C.E. Inflammasome activation causes dual recruitment of NLRC4 and NLRP3 to the same macromolecular complex. Proc. Natl. Acad. Sci. USA 2014, 111, 7403–7408. [Google Scholar] [CrossRef] [Green Version]
- Kang, S.; Fernandes-Alnemri, T.; Rogers, C.; Mayes, L.; Wang, Y.; Dillon, C.; Roback, L.; Kaiser, W.; Oberst, A.; Sagara, J.; et al. Caspase-8 scaffolding function and MLKL regulate NLRP3 inflammasome activation downstream of TLR3. Nat. Commun. 2015, 6, 7515. [Google Scholar] [CrossRef]
- Conos, S.A.; Chen, K.W.; De Nardo, D.; Hara, H.; Whitehead, L.; Nunez, G.; Masters, S.L.; Murphy, J.M.; Schroder, K.; Vaux, D.L.; et al. Active MLKL triggers the NLRP3 inflammasome in a cell-intrinsic manner. Proc. Natl. Acad. Sci. USA 2017, 114, E961–E969. [Google Scholar] [CrossRef] [Green Version]
- Gutierrez, K.D.; Davis, M.A.; Daniels, B.P.; Olsen, T.M.; Ralli-Jain, P.; Tait, S.W.; Gale, M., Jr.; Oberst, A. MLKL Activation Triggers NLRP3-Mediated Processing and Release of IL-1β Independently of Gasdermin-D. J. Immunol. 2017, 198, 2156–2164. [Google Scholar] [CrossRef] [Green Version]
- Sarhan, J.; Liu, B.C.; Muendlein, H.I.; Li, P.; Nilson, R.; Tang, A.Y.; Rongvaux, A.; Bunnell, S.C.; Shao, F.; Green, D.R.; et al. Caspase-8 induces cleavage of gasdermin D to elicit pyroptosis during Yersinia infection. Proc. Natl. Acad. Sci. USA 2018, 115, E10888–E10897. [Google Scholar] [CrossRef] [Green Version]
- Demarco, B.; Grayczyk, J.P.; Bjanes, E.; Le Roy, D.; Tonnus, W.; Assenmacher, C.A.; Radaelli, E.; Fettrelet, T.; Mack, V.; Linkermann, A.; et al. Caspase-8-dependent gasdermin D cleavage promotes antimicrobial defense but confers susceptibility to TNF-induced lethality. Sci. Adv. 2020, 6, eabc3465. [Google Scholar] [CrossRef]
- Orning, P.; Weng, D.; Starheim, K.; Ratner, D.; Best, Z.; Lee, B.; Brooks, A.; Xia, S.; Wu, H.; Kelliher, M.A.; et al. Pathogen blockade of TAK1 triggers caspase-8-dependent cleavage of gasdermin D and cell death. Science 2018, 362, 1064–1069. [Google Scholar] [CrossRef] [Green Version]
- Taabazuing, C.Y.; Okondo, M.C.; Bachovchin, D.A. Pyroptosis and apoptosis pathways engage in bidirectional crosstalk in monocytes and macrophages. Cell Chem. Biol. 2017, 24, 507–514. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Platnich, J.M.; Chung, H.; Lau, A.; Sandall, C.F.; Bondzi-Simpson, A.; Chen, H.M.; Komada, T.; Trotman-Grant, A.C.; Brandelli, J.R.; Chun, J.; et al. Shiga Toxin/Lipopolysaccharide Activates Caspase-4 and Gasdermin D to Trigger Mitochondrial Reactive Oxygen Species Upstream of the NLRP3 Inflammasome. Cell Rep. 2018, 25, 1525–1536.e1527. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rogers, C.; Erkes, D.A.; Nardone, A.; Aplin, A.E.; Fernandes-Alnemri, T.; Alnemri, E.S. Gasdermin pores permeabilize mitochondria to augment caspase-3 activation during apoptosis and inflammasome activation. Nat. Commun. 2019, 10, 1689. [Google Scholar] [CrossRef] [PubMed]
- de Vasconcelos, N.M.; Van Opdenbosch, N.; Van Gorp, H.; Parthoens, E.; Lamkanfi, M. Single-cell analysis of pyroptosis dynamics reveals conserved GSDMD-mediated subcellular events that precede plasma membrane rupture. Cell Death Differ. 2019, 26, 146–161. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Op de Beeck, K.; Van Camp, G.; Thys, S.; Cools, N.; Callebaut, I.; Vrijens, K.; Van Nassauw, L.; Van Tendeloo, V.F.; Timmermans, J.P.; Van Laer, L. The DFNA5 gene, responsible for hearing loss and involved in cancer, encodes a novel apoptosis-inducing protein. Eur. J. Hum. Genet. 2011, 19, 965–973. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Gao, W.; Shi, X.; Ding, J.; Liu, W.; He, H.; Wang, K.; Shao, F. Chemotherapy drugs induce pyroptosis through caspase-3 cleavage of a gasdermin. Nature 2017, 547, 99–103. [Google Scholar] [CrossRef]
- Rogers, C.; Fernandes-Alnemri, T.; Mayes, L.; Alnemri, D.; Cingolani, G.; Alnemri, E.S. Cleavage of DFNA5 by caspase-3 during apoptosis mediates progression to secondary necrotic/pyroptotic cell death. Nat. Commun. 2017, 8, 14128. [Google Scholar] [CrossRef] [Green Version]
- Zhou, B.; Abbott, D.W. Gasdermin E permits interleukin-1 beta release in distinct sublytic and pyroptotic phases. Cell Rep. 2021, 35, 108998. [Google Scholar] [CrossRef]
- Wang, C.; Yang, T.; Xiao, J.; Xu, C.; Alippe, Y.; Sun, K.; Kanneganti, T.D.; Monahan, J.B.; Abu-Amer, Y.; Lieberman, J.; et al. NLRP3 inflammasome activation triggers gasdermin D-independent inflammation. Sci. Immunol. 2021, 6, eabj3859. [Google Scholar] [CrossRef]
- Tsuchiya, K.; Nakajima, S.; Hosojima, S.; Thi Nguyen, D.; Hattori, T.; Manh Le, T.; Hori, O.; Mahib, M.R.; Yamaguchi, Y.; Miura, M.; et al. Caspase-1 initiates apoptosis in the absence of gasdermin D. Nat. Commun. 2019, 10, 2091. [Google Scholar] [CrossRef] [Green Version]
- Heilig, R.; Dilucca, M.; Boucher, D.; Chen, K.W.; Hancz, D.; Demarco, B.; Shkarina, K.; Broz, P. Caspase-1 cleaves Bid to release mitochondrial SMAC and drive secondary necrosis in the absence of GSDMD. Life Sci. Alliance 2020, 3, e202000735. [Google Scholar] [CrossRef]
- Xu, W.; Che, Y.; Zhang, Q.; Huang, H.; Ding, C.; Wang, Y.; Wang, G.; Cao, L.; Hao, H. Apaf-1 Pyroptosome Senses Mitochondrial Permeability Transition. Cell Metab. 2021, 33, 424–436.e410. [Google Scholar] [CrossRef] [PubMed]
- Chen, K.W.; Demarco, B.; Heilig, R.; Shkarina, K.; Boettcher, A.; Farady, C.J.; Pelczar, P.; Broz, P. Extrinsic and intrinsic apoptosis activate pannexin-1 to drive NLRP3 inflammasome assembly. EMBO J. 2019, 38, e101638. [Google Scholar] [CrossRef] [PubMed]
- Yan, W.T.; Yang, Y.D.; Hu, W.M.; Ning, W.Y.; Liao, L.S.; Lu, S.; Zhao, W.J.; Zhang, Q.; Xiong, K. Do pyroptosis, apoptosis, and necroptosis (PANoptosis) exist in cerebral ischemia? Evidence from cell and rodent studies. Neural Regen. Res. 2022, 17, 1761–1768. [Google Scholar] [CrossRef] [PubMed]
- Jiang, W.; Deng, Z.; Dai, X.; Zhao, W. PANoptosis: A New Insight Into Oral Infectious Diseases. Front. Immunol. 2021, 12, 789610. [Google Scholar] [CrossRef]
- Chi, D.; Lin, X.; Meng, Q.; Tan, J.; Gong, Q.; Tong, Z. Real-Time Induction of Macrophage Apoptosis, Pyroptosis, and Necroptosis by Enterococcus faecalis OG1RF and Two Root Canal Isolated Strains. Front. Cell. Infect. Microbiol. 2021, 11, 720147. [Google Scholar] [CrossRef]
- Lin, J.F.; Hu, P.S.; Wang, Y.Y.; Tan, Y.T.; Yu, K.; Liao, K.; Wu, Q.N.; Li, T.; Meng, Q.; Lin, J.Z.; et al. Phosphorylated NFS1 weakens oxaliplatin-based chemosensitivity of colorectal cancer by preventing PANoptosis. Signal Transduct. Target. Ther. 2022, 7, 54. [Google Scholar] [CrossRef]
- Song, M.; Xia, W.; Tao, Z.; Zhu, B.; Zhang, W.; Liu, C.; Chen, S. Self-assembled polymeric nanocarrier-mediated co-delivery of metformin and doxorubicin for melanoma therapy. Drug Deliv. 2021, 28, 594–606. [Google Scholar] [CrossRef]
- Upton, J.W.; Kaiser, W.J.; Mocarski, E.S. DAI/ZBP1/DLM-1 complexes with RIP3 to mediate virus-induced programmed necrosis that is targeted by murine cytomegalovirus vIRA. Cell Host Microbe 2012, 11, 290–297. [Google Scholar] [CrossRef] [Green Version]
- Kaiser, W.J.; Upton, J.W.; Mocarski, E.S. Receptor-interacting protein homotypic interaction motif-dependent control of NF-kappa B activation via the DNA-dependent activator of IFN regulatory factors. J. Immunol. 2008, 181, 6427–6434. [Google Scholar] [CrossRef] [Green Version]
- Thapa, R.J.; Ingram, J.P.; Ragan, K.B.; Nogusa, S.; Boyd, D.F.; Benitez, A.A.; Sridharan, H.; Kosoff, R.; Shubina, M.; Landsteiner, V.J.; et al. DAI senses influenza A virus genomic RNA and activates RIPK3-dependent cell death. Cell Host Microbe 2016, 20, 674–681. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Kanneganti, T.D. From pyroptosis, apoptosis and necroptosis to PANoptosis: A mechanistic compendium of programmed cell death pathways. Comput. Struct. Biotechnol. J. 2021, 19, 4641–4657. [Google Scholar] [CrossRef] [PubMed]
- Tamura, T.; Ishihara, M.; Lamphier, M.S.; Tanaka, N.; Oishi, I.; Aizawa, S.; Matsuyama, T.; Mak, T.W.; Taki, S.; Taniguchi, T. An IRF-1-dependent pathway of DNA damage-induced apoptosis in mitogen-activated T lymphocytes. Nature 1995, 376, 596–599. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, N.; Ishihara, M.; Kitagawa, M.; Harada, H.; Kimura, T.; Matsuyama, T.; Lamphier, M.S.; Aizawa, S.; Mak, T.W.; Taniguchi, T. Cellular commitment to oncogene-induced transformation or apoptosis is dependent on the transcription factor IRF-1. Cell 1994, 77, 829–839. [Google Scholar] [CrossRef]
- Karki, R.; Kanneganti, T.D. The ‘cytokine storm’: Molecular mechanisms and therapeutic prospects. Trends Immunol. 2021, 42, 681–705. [Google Scholar] [CrossRef]
- Man, S.M.; Karki, R.; Malireddi, R.K.; Neale, G.; Vogel, P.; Yamamoto, M.; Lamkanfi, M.; Kanneganti, T.D. The transcription factor IRF1 and guanylate-binding proteins target activation of the AIM2 inflammasome by Francisella infection. Nat. Immunol. 2015, 16, 467–475. [Google Scholar] [CrossRef] [Green Version]
- Vanden Berghe, T.; Kaiser, W.J.; Bertrand, M.J.; Vandenabeele, P. Molecular crosstalk between apoptosis, necroptosis, and survival signaling. Mol. Cell. Oncol. 2015, 2, e975093. [Google Scholar] [CrossRef] [Green Version]
- Fritsch, M.; Gunther, S.D.; Schwarzer, R.; Albert, M.C.; Schorn, F.; Werthenbach, J.P.; Schiffmann, L.M.; Stair, N.; Stocks, H.; Seeger, J.M.; et al. Caspase-8 is the molecular switch for apoptosis, necroptosis and pyroptosis. Nature 2019, 575, 683–687. [Google Scholar] [CrossRef]
- Tummers, B.; Mari, L.; Guy, C.S.; Heckmann, B.L.; Rodriguez, D.A.; Rühl, S.; Moretti, J.; Crawford, J.C.; Fitzgerald, P.; Kanneganti, T.D.; et al. Caspase-8-Dependent Inflammatory Responses Are Controlled by Its Adaptor, FADD, and Necroptosis. Immunity 2020, 52, 994–1006.e8. [Google Scholar] [CrossRef]
- Newton, K.; Wickliffe, K.E.; Maltzman, A.; Dugger, D.L.; Reja, R.; Zhang, Y.; Roose-Girma, M.; Modrusan, Z.; Sagolla, M.S.; Webster, J.D.; et al. Activity of caspase-8 determines plasticity between cell death pathways. Nature 2019, 575, 679–682. [Google Scholar] [CrossRef]
- Schwarzer, R.; Jiao, H.; Wachsmuth, L.; Tresch, A.; Pasparakis, M. FADD and Caspase-8 Regulate Gut Homeostasis and Inflammation by Controlling MLKL- and GSDMD-Mediated Death of Intestinal Epithelial Cells. Immunity 2020, 52, 978–993.e6. [Google Scholar] [CrossRef] [PubMed]
- Kang, T.B.; Oh, G.S.; Scandella, E.; Bolinger, B.; Ludewig, B.; Kovalenko, A.; Wallach, D. Mutation of a self-processing site in caspase-8 compromises its apoptotic but not its nonapoptotic functions in bacterial artificial chromosome-transgenic mice. J. Immunol. 2008, 181, 2522–2532. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Karki, R.; Zheng, M.; Kancharana, B.; Lee, S.; Kesavardhana, S.; Hansen, B.S.; Pruett-Miller, S.M.; Kanneganti, T.D. Cutting Edge: Caspase-8 Is a Linchpin in Caspase-3 and Gasdermin D Activation to Control Cell Death, Cytokine Release, and Host Defense during Influenza A Virus Infection. J. Immunol. 2021, 207, 2411–2416. [Google Scholar] [CrossRef] [PubMed]
- Belhocine, K.; Monack, D.M. Francisella infection triggers activation of the AIM2 inflammasome in murine dendritic cells. Cell Microbiol. 2012, 14, 71–80. [Google Scholar] [CrossRef] [Green Version]
- Doerflinger, M.; Deng, Y.; Whitney, P.; Salvamoser, R.; Engel, S.; Kueh, A.J.; Tai, L.; Bachem, A.; Gressier, E.; Geoghegan, N.D.; et al. Flexible Usage and Interconnectivity of Diverse Cell Death Pathways Protect against Intracellular Infection. Immunity 2020, 53, 533–547.e7. [Google Scholar] [CrossRef]
- Broz, P.; Ruby, T.; Belhocine, K.; Bouley, D.M.; Kayagaki, N.; Dixit, V.M.; Monack, D.M. Caspase-11 increases susceptibility to Salmonella infection in the absence of caspase-1. Nature 2012, 490, 288–291. [Google Scholar] [CrossRef]
- Sundaram, B.; Karki, R.; Kanneganti, T.D. NLRC4 Deficiency Leads to Enhanced Phosphorylation of MLKL and Necroptosis. Immunohorizons 2022, 6, 243–252. [Google Scholar] [CrossRef]
- Sutterwala, F.S.; Mijares, L.A.; Li, L.; Ogura, Y.; Kazmierczak, B.I.; Flavell, R.A. Immune recognition of Pseudomonas aeruginosa mediated by the IPAF/NLRC4 inflammasome. J. Exp. Med. 2007, 204, 3235–3245. [Google Scholar] [CrossRef] [Green Version]
- Miao, E.A.; Ernst, R.K.; Dors, M.; Mao, D.P.; Aderem, A. Pseudomonas aeruginosa activates caspase 1 through Ipaf. Proc. Natl. Acad. Sci. USA 2008, 105, 2562–2567. [Google Scholar] [CrossRef] [Green Version]
- Ferguson, P.J.; Bing, X.; Vasef, M.A.; Ochoa, L.A.; Mahgoub, A.; Waldschmidt, T.J.; Tygrett, L.T.; Schlueter, A.J.; El-Shanti, H. A missense mutation in pstpip2 is associated with the murine autoinflammatory disorder chronic multifocal osteomyelitis. Bone 2006, 38, 41–47. [Google Scholar] [CrossRef] [Green Version]
- Cassel, S.L.; Janczy, J.R.; Bing, X.; Wilson, S.P.; Olivier, A.K.; Otero, J.E.; Iwakura, Y.; Shayakhmetov, D.M.; Bassuk, A.G.; Abu-Amer, Y.; et al. Inflammasome-independent IL-1β mediates autoinflammatory disease in Pstpip2-deficient mice. Proc. Natl. Acad. Sci. USA 2014, 111, 1072–1077. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Douglas, T.; Champagne, C.; Morizot, A.; Lapointe, J.M.; Saleh, M. The Inflammatory Caspases-1 and -11 Mediate the Pathogenesis of Dermatitis in Sharpin-Deficient Mice. J. Immunol. 2015, 195, 2365–2373. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rickard, J.A.; Anderton, H.; Etemadi, N.; Nachbur, U.; Darding, M.; Peltzer, N.; Lalaoui, N.; Lawlor, K.E.; Vanyai, H.; Hall, C.; et al. TNFR1-dependent cell death drives inflammation in Sharpin-deficient mice. eLife 2014, 3, e03464. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumari, S.; Redouane, Y.; Lopez-Mosqueda, J.; Shiraishi, R.; Romanowska, M.; Lutzmayer, S.; Kuiper, J.; Martinez, C.; Dikic, I.; Pasparakis, M.; et al. Sharpin prevents skin inflammation by inhibiting TNFR1-induced keratinocyte apoptosis. eLife 2014, 3, e03422. [Google Scholar] [CrossRef] [PubMed]
- HogenEsch, H.; Gijbels, M.J.; Offerman, E.; van Hooft, J.; van Bekkum, D.W.; Zurcher, C. A spontaneous mutation characterized by chronic proliferative dermatitis in C57BL mice. Am. J. Pathol. 1993, 143, 972–982. [Google Scholar]
- Berger, S.B.; Kasparcova, V.; Hoffman, S.; Swift, B.; Dare, L.; Schaeffer, M.; Capriotti, C.; Cook, M.; Finger, J.; Hughes-Earle, A.; et al. Cutting Edge: RIP1 kinase activity is dispensable for normal development but is a key regulator of inflammation in SHARPIN-deficient mice. J. Immunol. 2014, 192, 5476–5480. [Google Scholar] [CrossRef]
- Lukens, J.R.; Vogel, P.; Johnson, G.R.; Kelliher, M.A.; Iwakura, Y.; Lamkanfi, M.; Kanneganti, T.D. RIP1-driven autoinflammation targets IL-1alpha independently of inflammasomes and RIP3. Nature 2013, 498, 224–227. [Google Scholar] [CrossRef] [Green Version]
- Gurung, P.; Fan, G.; Lukens, J.R.; Vogel, P.; Tonks, N.K.; Kanneganti, T.D. Tyrosine Kinase SYK Licenses MyD88 Adaptor Protein to Instigate IL-1alpha-Mediated Inflammatory Disease. Immunity 2017, 46, 635–648. [Google Scholar] [CrossRef] [Green Version]
- Peltzer, N.; Darding, M.; Montinaro, A.; Draber, P.; Draberova, H.; Kupka, S.; Rieser, E.; Fisher, A.; Hutchinson, C.; Taraborrelli, L.; et al. LUBAC is essential for embryogenesis by preventing cell death and enabling haematopoiesis. Nature 2018, 557, 112–117. [Google Scholar] [CrossRef]
- Mandal, P.; Feng, Y.; Lyons, J.D.; Berger, S.B.; Otani, S.; DeLaney, A.; Tharp, G.K.; Maner-Smith, K.; Burd, E.M.; Schaeffer, M.; et al. Caspase-8 Collaborates with Caspase-11 to Drive Tissue Damage and Execution of Endotoxic Shock. Immunity 2018, 49, 42–55.e6. [Google Scholar] [CrossRef] [Green Version]
- Rauch, I.; Deets, K.A.; Ji, D.X.; von Moltke, J.; Tenthorey, J.L.; Lee, A.Y.; Philip, N.H.; Ayres, J.S.; Brodsky, I.E.; Gronert, K.; et al. NAIP-NLRC4 Inflammasomes Coordinate Intestinal Epithelial Cell Expulsion with Eicosanoid and IL-18 Release via Activation of Caspase-1 and -8. Immunity 2017, 46, 649–659. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- von Moltke, J.; Trinidad, N.J.; Moayeri, M.; Kintzer, A.F.; Wang, S.B.; van Rooijen, N.; Brown, C.R.; Krantz, B.A.; Leppla, S.H.; Gronert, K.; et al. Rapid induction of inflammatory lipid mediators by the inflammasome in vivo. Nature 2012, 490, 107–111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maccioni, R.B.; Rojo, L.E.; Fernández, J.A.; Kuljis, R.O. The role of neuroimmunomodulation in Alzheimer’s disease. Ann. N. Y. Acad. Sci. 2009, 1153, 240–246. [Google Scholar] [CrossRef] [PubMed]
- Ofengeim, D.; Mazzitelli, S.; Ito, Y.; DeWitt, J.P.; Mifflin, L.; Zou, C.; Das, S.; Adiconis, X.; Chen, H.; Zhu, H.; et al. RIPK1 mediates a disease-associated microglial response in Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 2017, 114, E8788–E8797. [Google Scholar] [CrossRef] [Green Version]
- Nogusa, S.; Thapa, R.J.; Dillon, C.P.; Liedmann, S.; Oguin, T.H., 3rd; Ingram, J.P.; Rodriguez, D.A.; Kosoff, R.; Sharma, S.; Sturm, O.; et al. RIPK3 activates parallel pathways of MLKL-criven necroptosis and FADD-mediated apoptosis to protect against Influenza A virus. Cell Host Microbe 2016, 20, 13–24. [Google Scholar] [CrossRef] [Green Version]
- Wallace, H.L.; Wang, L.; Gardner, C.L.; Corkum, C.P.; Grant, M.D.; Hirasawa, K.; Russell, R.S. Crosstalk Between Pyroptosis and Apoptosis in Hepatitis C Virus-induced Cell Death. Front. Immunol. 2022, 13, 788138. [Google Scholar] [CrossRef]
- Van Hauwermeiren, F.; Van Opdenbosch, N.; Van Gorp, H.; de Vasconcelos, N.; van Loo, G.; Vandenabeele, P.; Kanneganti, T.D.; Lamkanfi, M. Bacillus anthracis induces NLRP3 inflammasome activation and caspase-8-mediated apoptosis of macrophages to promote lethal anthrax. Proc. Natl. Acad. Sci. USA 2022, 119, e2116415119. [Google Scholar] [CrossRef]
- Turner, S.; Kenshole, B.; Ruby, J. Viral modulation of the host response via crmA/SPI-2 expression. Immunol. Cell Biol. 1999, 77, 236–241. [Google Scholar] [CrossRef]
- He, S.; Han, J. Manipulation of Host Cell Death Pathways by Herpes Simplex Virus. In Current Topics in Microbiology and Immunology; Springer: Berlin, Heidelberg, 2020. [Google Scholar]
- Pauleau, A.L.; Larochette, N.; Giordanetto, F.; Scholz, S.R.; Poncet, D.; Zamzami, N.; Goldmacher, V.S.; Kroemer, G. Structure-function analysis of the interaction between Bax and the cytomegalovirus-encoded protein vMIA. Oncogene 2007, 26, 7067–7080. [Google Scholar] [CrossRef]
- Ashida, H.; Sasakawa, C.; Suzuki, T. A unique bacterial tactic to circumvent the cell death crosstalk induced by blockade of caspase-8. EMBO J. 2020, 39, e104469. [Google Scholar] [CrossRef]


{kind=link}
| Trigger | PANoptosome Sensor | Regulator | Pyroptosis | Apoptosis | Necroptosis | PANoptosis | ||||
|---|---|---|---|---|---|---|---|---|---|---|
| Genotype | Cell Death? | Genotype | Cell Death? | Genotype | Cell Death? | Genotype | Cell Death? | |||
| IAV [9,10,11,133] | ZBP1 | IRF1 | Nlrp3−/− | ✔ | Casp8DA/DA | ✔ | Ripk3−/− | D | Fadd−/−Ripk3−/− | D |
| Casp1/11−/− | ✔ | Casp6−/− | D | Mlkl−/− | ✔ | Casp8−/−Ripk3−/− | X | |||
| Ripk1K45A | ✔ | Casp1/11−/−Casp8−/−Ripk3−/− | X | |||||||
| KPT + IFN [7] | ZBP1 | IRF1 | Nlrp3−/− | ✔ | Casp3−/− | ✔ | Ripk3−/− | D | Casp8−/−Ripk3−/− | X |
| Asc−/− | ✔ | |||||||||
| Casp1−/− | ✔ | Casp7−/− | ✔ | Mlkl−/− | ✔ | |||||
| Casp11−/− | ✔ | |||||||||
| Francisella [12,134] | AIM2 | IRF1 | Aim2−/− | X | Ripk3−/− | D | Casp8−/−Ripk3−/− | X | ||
| Casp1/11−/− | X | |||||||||
| Asc−/− | X | |||||||||
| Mefv−/− | D | Fadd−/−Ripk3−/− | X | |||||||
| Nlrp3−/− | ✔ | |||||||||
| Nlrc4−/− | ✔ | |||||||||
| HSV1 [12] | AIM2 | Aim2−/− | X | Ripk3−/− | D | Casp8−/−Ripk3−/− | X | |||
| Mefv−/− | D | |||||||||
| Nlrp3−/− | ✔ | |||||||||
| Nlrc4−/− | ✔ | |||||||||
| Yersinia [3] | RIPK1 | Casp1/11−/− | ✔ | Casp3−/− | ✔ | Ripk3−/− | ✔ | Casp8−/−Ripk3−/− | D | |
| Ripk1−/− | D | |||||||||
| Gsdmd−/− | ✔ | Casp7−/− | ✔ | Mlkl−/− | ✔ | Casp1/11−/−Casp8−/−Ripk3−/− | X | |||
| TAK1i [5,20] | RIPK1 | Nlrp3−/− | ✔ | Ripk3−/− | ✔ | Casp8−/−Ripk3−/− | X | |||
| Aim2−/− | ✔ | Ripk1−/− | X | |||||||
| Nlrc4−/− | ✔ | Ripk1K45A | X | |||||||
| Asc−/− | ✔ | |||||||||
| TNF + IFN-γ [8] | IRF1 | Casp1/11−/− | ✔ | Casp3−/− | D | Ripk3−/− | ✔ | Casp8−/−Ripk3−/− | X | |
| Casp1−/− | ✔ | |||||||||
| Casp11−/− | ✔ | Casp7−/− | ✔ | Fadd−/− Ripk3−/− | X | |||||
| MHV [15] | Nlrp3−/− | I | Ripk3−/− | D | Casp8−/−Ripk3−/− | X | ||||
| Casp1/11−/− | I | |||||||||
| Aim2−/− | ✔ | |||||||||
| Nlrc4−/− | ✔ | Casp1/11−/−Casp8−/−Ripk3−/− | X | |||||||
| Casp11−/− | ✔ | |||||||||
| Salmonella [10,30,135,136] | Nlrc4−/− | D | Ripk3−/− | ✔ | Casp8−/−Ripk3−/− | ✔ | ||||
| Casp1/11−/− | D | |||||||||
| Casp11−/− | D | Casp1/11−/−Casp8−/−Ripk3−/− | X | |||||||
| Asc−/− | D | |||||||||
| Pseudomonas [137,138,139] | Nlrc4−/− | D | Casp1/11−/−Casp8−/−Ripk3−/− | X | ||||||
| Asc−/− | ✔ | |||||||||
| Casp1−/− | X | |||||||||
| Model | Pathology | Pyroptosis | Apoptosis | Necroptosis | PANoptosis | ||||
|---|---|---|---|---|---|---|---|---|---|
| Genotype | Disease? | Genotype | Disease? | Genotype | Disease? | Genotype | Disease? | ||
| Pstpip2cmo [16,18,140,141] | Osteomyelitis | Nlrp3−/− | ✔ | Casp8−/−Ripk3−/− a | ✔ | Ripk3−/− | ✔ | Nlrp3−/−Casp8−/−Ripk3−/− | X |
| Casp1−/− | ✔ | Casp1−/−Casp8−/−Ripk3−/− | X | ||||||
| Sharpincpdm [142,143,144,145,146] | Dermatitis | Nlrp3−/− | D | Bid−/− | D | Mlkl−/− | D | Ripk3−/−FaddE-KO | X |
| Ripk3−/− | D | Ripk3−/−TraddE-KO | X | ||||||
| Casp1/11−/− | D | Ripk1K45A | X | Casp8−/−Ripk3−/− | X b | ||||
| Ptpn6spin [147,148] | Dermatosis | Nlrp3−/− | ✔ | Casp8−/−Ripk3−/− a | ✔ | Ripk3−/− | ✔ | Casp8−/−Ripk1−/−Ripk3−/− | X |
| Mlkl−/− | ✔ | ||||||||
| Casp1−/− | ✔ | Ripk1K45A | ✔ | ||||||
| Hoildeficiency [149] | Embryonic lethality | Casp8−/− | ✔ | Ripk3−/− | D | Casp8−/−Mlkl−/− | D | ||
| Casp8−/−Ripk3−/− a | D c | Mlkl−/− | D | Casp8−/−Ripk1−/− Ripk3−/− | X | ||||
| Ripk1K45A | D | ||||||||
| Caspase-8 deficiency [62,81,82,83,84,131] | Embryonic lethality | Casp8−/− | ✔ | Ripk1D138N | D | Casp8−/−Ripk3−/− | X | ||
| Casp8−/−Mlkl−/− | X | ||||||||
| Casp8C362A [130] | Embryonic lethality | Nlrp3−/− Mlkl−/− | ✔ | Fadd−/− Mlkl−/− | D | Mlkl−/− | ✔ | Casp1/11−/− Ripk3−/− | X |
| Casp1−/− Mlkl−/− | ✔ | ||||||||
| Casp11−/− Mlkl−/− | D | Ripk3−/− | D | Casp1/11−/−Mlkl−/− | X | ||||
| Asc−/− Mlkl−/− | D | ||||||||
| LPS shock [2,150] | Lethality | Asc−/− | ✔ | Ripk3−/− | ✔ | Casp8−/−Ripk3−/− | X | ||
| Casp8−/−Ripk3K51A | X | ||||||||
| Casp11−/− | D | Casp8−/−Ripk3−/−Ripk1−/− | X | ||||||
| TNF + IFN-γshock [2] | Lethality | Ripk3−/− | ✔ | Casp8−/−Ripk3−/− | X | ||||
| FlaTox injection [151,152] | Hypothermia | Nlc4−/− | X | Casp8−/−Ripk3−/− a | ✔ | Ripk3−/− | ✔ | Casp1−/−Ripk3−/− | D |
| Asc−/− | ✔ | Casp1−/−Casp8−/−Ripk3−/− | X | ||||||
| Casp1−/− | D | Asc−/−Casp8−/−Ripk3−/− | X | ||||||
| Casp1/11−/− | D | ||||||||
| Gsdmd−/− | D | ||||||||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gullett, J.M.; Tweedell, R.E.; Kanneganti, T.-D. It’s All in the PAN: Crosstalk, Plasticity, Redundancies, Switches, and Interconnectedness Encompassed by PANoptosis Underlying the Totality of Cell Death-Associated Biological Effects. Cells 2022, 11, 1495. https://doi.org/10.3390/cells11091495
Gullett JM, Tweedell RE, Kanneganti T-D. It’s All in the PAN: Crosstalk, Plasticity, Redundancies, Switches, and Interconnectedness Encompassed by PANoptosis Underlying the Totality of Cell Death-Associated Biological Effects. Cells. 2022; 11(9):1495. https://doi.org/10.3390/cells11091495
Chicago/Turabian StyleGullett, Jessica M., Rebecca E. Tweedell, and Thirumala-Devi Kanneganti. 2022. "It’s All in the PAN: Crosstalk, Plasticity, Redundancies, Switches, and Interconnectedness Encompassed by PANoptosis Underlying the Totality of Cell Death-Associated Biological Effects" Cells 11, no. 9: 1495. https://doi.org/10.3390/cells11091495
APA StyleGullett, J. M., Tweedell, R. E., & Kanneganti, T. -D. (2022). It’s All in the PAN: Crosstalk, Plasticity, Redundancies, Switches, and Interconnectedness Encompassed by PANoptosis Underlying the Totality of Cell Death-Associated Biological Effects. Cells, 11(9), 1495. https://doi.org/10.3390/cells11091495