
Glatiramer Acetate Immunomodulation: Evidence of Neuroprotection and Cognitive Preservation

 ,
,
Abstract
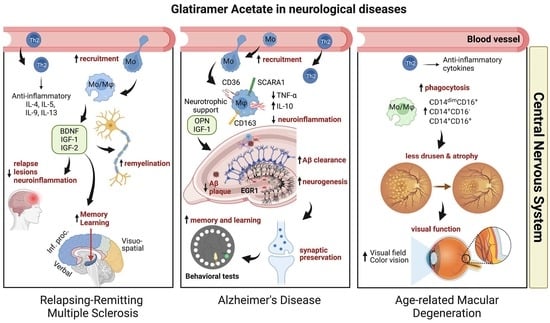
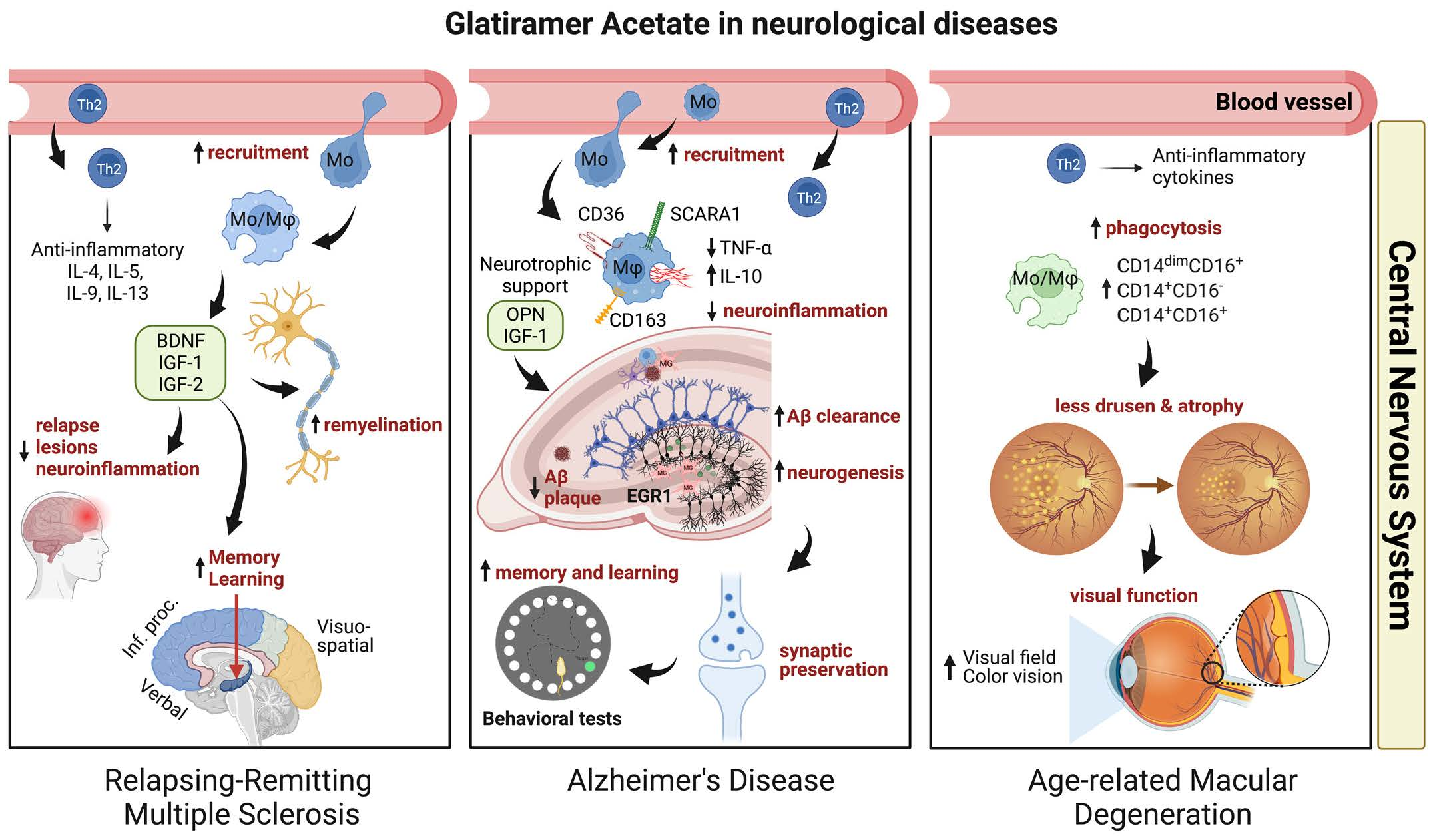
:
1. Introduction
1.1. Mechanism of Action
1.2. Current and Potential Uses of Glatiramer Acetate
2. GA in Clinical Trials
2.1. Role of GA in Preventing Cognitive Decline in Multiple Sclerosis
2.2. Therapeutic Roles of GA in Ophthalmic Disorders
2.3. GA Immunization in Amyotrophic Lateral Sclerosis (ALS)
3. Preclinical Studies Using GA in Neurodegenerative Disease Models
3.1. Effects of GA Immunization in EAE Murine Models of MS
3.2. Effects of GA in Animal Models of ALS
3.3. Role of GA in Repair, Regeneration, and Cognitive Preservation in AD-Model Mice
3.4. GA Immunization in Animal Models of Parkinson’s Disease (PD)
3.5. GA Immunization in Murine Models of Huntington’s Disease (HD)
3.6. Role of GA in Neuropsychology
3.7. Role of GA in Central Ischemia and Vascular Dementia
4. Conclusions and Future Directions
5. Review Methods
6. Side Effects and Safety
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Keith, A.B.; Arnon, R.; Teitelbaum, D.; Caspary, E.A.; Wisniewski, H.M. The effect of Cop 1, a synthetic polypeptide, on chronic relapsing experimental allergic encephalomyelitis in guinea pigs. J. Neurol. Sci. 1979, 42, 267–274. [Google Scholar] [CrossRef]
- Wagner, C.A.; Roqué, P.J.; Mileur, T.R.; Liggitt, D.; Goverman, J.M. Myelin-specific CD8+ T cells exacerbate brain inflammation in CNS autoimmunity. J. Clin. Investig. 2020, 130, 203–213. [Google Scholar] [CrossRef]
- Teitelbaum, D.; Meshorer, A.; Hirshfeld, T.; Arnon, R.; Sela, M. Suppression of experimental allergic encephalomyelitis by a synthetic polypeptide. Eur. J. Immunol. 1971, 1, 242–248. [Google Scholar] [CrossRef] [PubMed]
- Campos-García, V.R.; Herrera-Fernández, D.; Espinosa-de La Garza, C.E.; González, G.; Vallejo-Castillo, L.; Avila, S.; Muñoz-García, L.; Medina-Rivero, E.; Pérez, N.O.; Gracia-Mora, I.; et al. Process signatures in glatiramer acetate synthesis: Structural and functional relationships. Sci. Rep. 2017, 7, 12125. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rommer, P.S.; Milo, R.; Han, M.H.; Satyanarayan, S.; Sellner, J.; Hauer, L.; Illes, Z.; Warnke, C.; Laurent, S.; Weber, M.S.; et al. Immunological Aspects of Approved MS Therapeutics. Front. Immunol. 2019, 10, 1564. [Google Scholar] [CrossRef] [PubMed]
- Bell, C.; Anderson, J.; Ganguly, T.; Prescott, J.; Capila, I.; Lansing, J.C.; Sachleben, R.; Iyer, M.; Fier, I.; Roach, J.; et al. Development of Glatopa® (Glatiramer Acetate): The First FDA-Approved Generic Disease-Modifying Therapy for Relapsing Forms of Multiple Sclerosis. J. Pharm. Pract. 2018, 31, 481–488. [Google Scholar] [CrossRef] [PubMed]
- Leray, E.; Moreau, T.; Fromont, A.; Edan, G. Epidemiology of multiple sclerosis. Rev. Neurol. 2016, 172, 3–13. [Google Scholar] [CrossRef] [PubMed]
- Kamińska, J.; Koper, O.M.; Piechal, K.; Kemona, H. Multiple sclerosis-etiology and diagnostic potential. Postepy Hig. Med. Dosw. 2017, 71, 551–563. [Google Scholar] [CrossRef]
- Yong, V.W. Differential mechanisms of action of interferon-β and glatiramer acetate in MS. Neurology 2002, 59, 802–808. [Google Scholar] [CrossRef] [Green Version]
- Caragnano, M.; Tortorella, P.; Bergami, A.; Ruggieri, M.; Livrea, P.; Specchio, L.M.; Martino, G.; Trojano, M.; Furlan, R.; Avolio, C. Monocytes P2X7 purinergic receptor is modulated by glatiramer acetate in multiple sclerosis. J. Neuroimmunol. 2012, 245, 93–97. [Google Scholar] [CrossRef]
- Aharoni, R. The mechanism of action of glatiramer acetate in multiple sclerosis and beyond. Autoimmun. Rev. 2013, 12, 543–553. [Google Scholar] [CrossRef] [PubMed]
- Aharoni, R. Immunomodulation neuroprotection and remyelination—The fundamental therapeutic effects of glatiramer acetate: A critical review. J. Autoimmun. 2014, 54, 81–92. [Google Scholar] [CrossRef] [PubMed]
- Arnon, R.; Aharoni, R. Glatiramer Acetate: From Bench to Bed and Back. Isr. Med Assoc. J. 2019, 21, 151–157. [Google Scholar] [PubMed]
- Rostami, A.; Ciric, B. Role of Th17 cells in the pathogenesis of CNS inflammatory demyelination. J. Neurol. Sci. 2013, 333, 76–87. [Google Scholar] [CrossRef] [Green Version]
- Romagnani, S. Th1/Th2 Cells. Inflamm. Bowel Dis. 1999, 5, 285–294. [Google Scholar] [CrossRef]
- Aharoni, R.; Eilam, R.; Schottlender, N.; Radomir, L.; Leistner-Segal, S.; Feferman, T.; Hirsch, D.; Sela, M.; Arnon, R. Glatiramer acetate increases T- and B -regulatory cells and decreases granulocyte-macrophage colony-stimulating factor (GM-CSF) in an animal model of multiple sclerosis. J. Neuroimmunol. 2020, 345, 577281. [Google Scholar] [CrossRef]
- Melnikov, M.; Sharanova, S.; Sviridova, A.; Rogovskii, V.; Murugina, N.; Nikolaeva, A.; Dagil, Y.; Murugin, V.; Ospelnikova, T.; Boyko, A.; et al. The influence of glatiramer acetate on Th17-immune response in multiple sclerosis. PLoS ONE 2020, 15, e0240305. [Google Scholar] [CrossRef]
- Butovsky, O.; Koronyo-Hamaoui, M.; Kunis, G.; Ophir, E.; Landa, G.; Cohen, H.; Schwartz, M. Glatiramer acetate fights against Alzheimer’s disease by inducing dendritic-like microglia expressing insulin-like growth factor 1. Proc. Natl. Acad. Sci. USA 2006, 103, 11784–11789. [Google Scholar] [CrossRef] [Green Version]
- Kala, M.; Miravalle, A.; Vollmer, T. Recent insights into the mechanism of action of glatiramer acetate. J. Neuroimmunol. 2011, 235, 9–17. [Google Scholar] [CrossRef]
- Ziemssen, T.; Kümpfel, T.; Schneider, H.; Klinkert, W.E.; Neuhaus, O.; Hohlfeld, R. Secretion of brain-derived neurotrophic factor by glatiramer acetate-reactive T-helper cell lines: Implications for multiple sclerosis therapy. J. Neurol. Sci. 2005, 233, 109–112. [Google Scholar] [CrossRef]
- Lalive, P.H.; Neuhaus, O.; Benkhoucha, M.; Burger, D.; Hohlfeld, R.; Zamvil, S.S.; Weber, M.S. Glatiramer Acetate in the Treatment of Multiple Sclerosis: Emerging concepts regarding its mechanism of action. CNS Drugs 2011, 25, 401–414. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pul, R.; Morbiducci, F.; Škuljec, J.; Skripuletz, T.; Singh, V.; Diederichs, U.; Garde, N.; Voss, E.V.; Trebst, C.; Stangel, M. Glatiramer Acetate Increases Phagocytic Activity of Human Monocytes In Vitro and in Multiple Sclerosis Patients. PLoS ONE 2012, 7, e51867. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pul, R.; Moharregh-Khiabani, D.; Škuljec, J.; Skripuletz, T.; Garde, N.; Voß, E.V.; Stangel, M. Glatiramer Acetate Modulates TNF-α and IL-10 Secretion in Microglia and Promotes Their Phagocytic Activity. J. Neuroimmune Pharmacol. 2011, 6, 381–388. [Google Scholar] [CrossRef] [PubMed]
- Gu, B.J.; Huang, X.; Avula, P.K.; Caruso, E.; Drysdale, C.; Vessey, K.A.; Ou, A.; Fowler, C.; Liu, T.-H.; Lin, Y.; et al. Deficits in Monocyte Function in Age Related Macular Degeneration: A Novel Systemic Change Associated With the Disease. Front. Med. 2021, 8, 634177. [Google Scholar] [CrossRef]
- Kalincik, T.; Diouf, I.; Sharmin, S.; Malpas, C.; Spelman, T.; Horakova, D.; Havrdova, E.K.; Trojano, M.; Izquierdo, G.; Lugaresi, A.; et al. Effect of Disease-Modifying Therapy on Disability in Relapsing-Remitting Multiple Sclerosis Over 15 Years. Neurology 2021, 96, e783–e797. [Google Scholar] [CrossRef]
- Burger, D.; Molnarfi, N.; Weber, M.S.; Brandt, K.J.; Benkhoucha, M.; Gruaz, L.; Chofflon, M.; Zamvil, S.S.; Lalive, P.H. Glatiramer acetate increases IL-1 receptor antagonist but decreases T cell-induced IL-1β in human monocytes and multiple sclerosis. Proc. Natl. Acad. Sci. USA 2009, 106, 4355–4359. [Google Scholar] [CrossRef] [Green Version]
- Nicoletti, F.; Patti, F.; DiMarco, R.; Zaccone, P.; Nicoletti, A.; Meroni, P.; Reggio, A. irculating serum levels of IL-1ra in patients with relapsing remitting multiple sclerosis are normal during remission phases but significantly increased either during exacerbations or in response to IFN-β treatment. Cytokine 1996, 8, 395–400. [Google Scholar] [CrossRef]
- Colonna, M.; Butovsky, O. Microglia Function in the Central Nervous System During Health and Neurodegeneration. Annu. Rev. Immunol. 2017, 35, 441–468. [Google Scholar] [CrossRef]
- Chen, Z.; Trapp, B.D. Microglia and neuroprotection. J. Neurochem. 2016, 136 (Suppl. 1), 10–17. [Google Scholar] [CrossRef]
- Prod’Homme, T.; Zamvil, S.S. The Evolving Mechanisms of Action of Glatiramer Acetate. Cold Spring Harb. Perspect. Med. 2019, 9, a029249. [Google Scholar] [CrossRef]
- Blanchette, F.; Neuhaus, O. Glatiramer Acetate: Evidence for a dual mechanism of action. J. Neurol. 2008, 255 (Suppl. 1), 26–36. [Google Scholar] [CrossRef] [PubMed]
- Koronyo, Y.; Salumbides, B.C.; Sheyn, J.; Pelissier, L.; Li, S.; Ljubimov, V.; Moyseyev, M.; Daley, D.; Fuchs, D.-T.; Pham, M.; et al. Therapeutic effects of glatiramer acetate and grafted CD115+ monocytes in a mouse model of Alzheimer’s disease. Brain 2015, 138, 2399–2422. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scorisa, J.M.; Zanon, R.G.; Freria, C.M.; de Oliveira, A.L.R. Glatiramer acetate positively influences spinal motoneuron survival and synaptic plasticity after ventral root avulsion. Neurosci. Lett. 2009, 451, 34–39. [Google Scholar] [CrossRef] [PubMed]
- Leal, G.; Bramham, C.R.; Duarte, C.B. BDNF and Hippocampal Synaptic Plasticity. Vitam. Horm. 2017, 104, 153–195. [Google Scholar] [CrossRef] [PubMed]
- Vacaras, V.; Major, Z.Z.; Muresanu, D.F.; Krausz, T.L.; Marginean, I.; Buzoianu, D.A. Effect of Glatiramer Acetate on Peripheral Blood Brain-Derived Neurotrophic Factor and Phosphorylated TrkB Levels in Relapsing- Remitting Multiple Sclerosis. CNS Neurol. Disord.-Drug Targets 2014, 13, 647–651. [Google Scholar] [CrossRef]
- Noseworthy, J.H.; Vandervoort, M.K.; Wong, C.J.; Ebers, G.C. Interrater variability with the Expanded Disability Status Scale (EDSS) and Functional Systems (FS) in a multiple sclerosis clinical trial. Neurology 1990, 40, 971–975. [Google Scholar] [CrossRef] [PubMed]
- De Vries, H.; Van Houte, L.R.; Lindeboom, J.; Van Eijk, J.T.; De Haan, M. Paced addition. A neuropsychological test for assessment of divided attention. Tijdschr. Gerontol. Geriatr. 1992, 23, 147–156. [Google Scholar]
- Weinstein, A.; Schwid, S.I.L.; Schiffer, R.B.; McDermott, M.P.; Giang, D.W.; Goodman, A.D. Neuropsychologic Status in Multiple Sclerosis After Treatment With Glatiramer. Arch. Neurol. 1999, 56, 319–324. [Google Scholar] [CrossRef] [Green Version]
- Boringa, J.B.; Lazeron, R.H.; Reuling, I.E.; Adèr, H.J.; Pfennings, L.E.; Lindeboom, J.; De Sonneville, L.M.; Kalkers, N.F.; Polman, C.H. The Brief Repeatable Battery of Neuropsychological Tests: Normative values allow application in multiple sclerosis clinical practice. Mult. Scler. 2001, 7, 263–267. [Google Scholar] [CrossRef]
- Schwid, S.R.; Goodman, A.D.; Weinstein, A.; McDermott, M.P.; Johnson, K.P.; Copaxone Study, G. Cognitive function in relapsing multiple sclerosis: Minimal changes in a 10-year clinical trial. J. Neurol. Sci. 2007, 255, 57–63. [Google Scholar] [CrossRef]
- Suppa, A.; Marsili, L.; Di Stasio, F.; Latorre, A.; Parvez, A.K.; Colosimo, C.; Berardelli, A. Primary motor cortex long-term plasticity in multiple system atrophy. Mov. Disord. 2014, 29, 97–104. [Google Scholar] [CrossRef] [PubMed]
- Mori, F.; Kusayanagi, H.; Buttari, F.; Nicoletti, C.G.; Bernardi, G.; Centonze, D. Glatiramer Acetate Reverses Plasticity and Cognitive Deficits Associated with Acute Inflammation in MS (P04.118). Neurology 2012, 78, P04.118–P104.118. [Google Scholar] [CrossRef]
- Jongen, P.J.; Lehnick, D.; Koeman, J.; Frequin, S.; Heersema, D.; Kornips, B.; Schyns-Soeterboek, A.; Visser, L.H.; Schiphof, P.; Valkenburg, A.; et al. Fatigue and health-related quality of life in relapsing-remitting multiple sclerosis after 2 years glatiramer acetate treatment are predicted by changes at 6 months: An observational multi-center study. J. Neurol. 2014, 261, 1469–1476. [Google Scholar] [CrossRef] [Green Version]
- Wingerchuk, D.M.; Carter, J.L. Multiple Sclerosis: Current and Emerging Disease-Modifying Therapies and Treatment Strategies. Mayo Clin. Proc. 2014, 89, 225–240. [Google Scholar] [CrossRef] [Green Version]
- Vacaras, V.; Major, Z.Z.; Seewooram, R.; Major, K.A.; Muresanu, D.F.; Buzoianu, A.D. Disease activity and disability evolution under glatiramer acetate: A clinical approach. Neuropsychopharmacol. Hung. 2014, 16, 11–18. [Google Scholar]
- Fricska-Nagy, Z.; Füvesi, J.; Rózsa, C.; Komoly, S.; Jakab, G.; Csépány, T.; Jobbágy, Z.; Lencsés, G.; Vécsei, L.; Bencsik, K. The effects of fatigue, depression and the level of disability on the health-related quality of life of glatiramer acetate-treated relapsing-remitting patients with multiple sclerosis in Hungary. Mult. Scler. Relat. Disord. 2016, 7, 26–32. [Google Scholar] [CrossRef]
- Meca-Lallana, J.; Hernández, L.; Caminero, A.B.; Girón, J.M.; Cano-Orgaz, A.; Carcelén-Gadea, M.; Muñoz, D.; Durán-Ferreras, E.; Martín-Hernández, J.; Sanchez-de La Rosa, R.; et al. Fatigue Improvement after Switching Multiple Sclerosis Treatment from Interferon-β to Glatiramer Acetate in Clinical Practice. Eur. Neurol. 2016, 76, 40–47. [Google Scholar] [CrossRef]
- Penner, I.K.; Raselli, C.; Stöcklin, M.; Opwis, K.; Kappos, L.; Calabrese, P. The Fatigue Scale for Motor and Cognitive Functions (FSMC): Validation of a new instrument to assess multiple sclerosis-related fatigue. Mult. Scler. 2009, 15, 1509–1517. [Google Scholar] [CrossRef]
- Ziemssen, T.; Calabrese, P.; Penner, I.-K.; Apfel, R. QualiCOP: Real-world effectiveness, tolerability, and quality of life in patients with relapsing-remitting multiple sclerosis treated with glatiramer acetate, treatment-naïve patients, and previously treated patients. J. Neurol. 2016, 263, 784–791. [Google Scholar] [CrossRef]
- Cinar, B.P.; Kösehasanoğulları, G.; Yigit, P.; Ozakbas, S. Cognitive dysfunction in patients with multiple sclerosis treated with first-line disease-modifying therapy: A multi-center, controlled study using the BICAMS battery. Neurol. Sci. 2017, 38, 337–342. [Google Scholar] [CrossRef]
- Sazonov, D.V.; Babenko, L.A.; Yarmoschuk, A.V.; Didrikh, E.M. An impact of glatiramer acetate (timexon) on the signs of neurodegeneration process in the neuronal layer of the retina in patients with relapsing-remitting multiple sclerosis. Zhurnal Nevrol. Psikhiatrii Im. SS Korsakova 2018, 118, 25–30. [Google Scholar] [CrossRef] [PubMed]
- Shorobura, M.S. Effect of preventive treatment on cognitive performance in patients with multiple sclerosis. Wiad. Lek. 2018, 71, 648–652. [Google Scholar] [PubMed]
- Zivadinov, R.; Tavazzi, E.; Hagemeier, J.; Carl, E.; Hojnacki, D.; Kolb, C.; Weinstock-Guttman, B. The Effect of Glatiramer Acetate on Retinal Nerve Fiber Layer Thickness in Patients with Relapsing–Remitting Multiple Sclerosis: A Longitudinal Optical Coherence Tomography Study. CNS Drugs 2018, 32, 763–770. [Google Scholar] [CrossRef] [PubMed]
- Johnson, K.P.; Brooks, B.R.; Cohen, J.A.; Ford, C.C.; Goldstein, J.; Lisak, R.P.; Myers, L.W.; Panitch, H.S.; Rose, J.W.; Schiffer, R.B. Copolymer 1 reduces relapse rate and improves disability in relapsing-remitting multiple sclerosis: Results of a phase III multicenter, double-blind, placebo-controlled trial. Neurology 1995, 45, 1268–1276. [Google Scholar] [CrossRef] [PubMed]
- Feinstein, A.; Magalhaes, S.; Richard, J.-F.; Audet, B.; Moore, C.S. The link between multiple sclerosis and depression. Nat. Rev. Neurol. 2014, 10, 507–517. [Google Scholar] [CrossRef] [PubMed]
- Fragoso, Y.D.; Adoni, T.; Anacleto, A.; Da Gama, P.D.; Goncalves, M.V.M.; Matta, A.P.D.C.; Parolin, M.F.K. Recommendations on diagnosis and treatment of depression in patients with multiple sclerosis. Pract. Neurol. 2014, 14, 206–209. [Google Scholar] [CrossRef]
- Beck, A.T.; Ward, C.H.; Mendelson, M.; Mock, J.; Erbaugh, J. An Inventory for Measuring Depression. Arch. Gen. Psychiatry 1961, 4, 561–571. [Google Scholar] [CrossRef] [Green Version]
- Nasreddine, Z.S.; Phillips, N.A.; Bédirian, V.; Charbonneau, S.; Whitehead, V.; Collin, I.; Cummings, J.L.; Chertkow, H. The Montreal Cognitive Assessment, MoCA: A Brief Screening Tool For Mild Cognitive Impairment. J. Am. Geriatr. Soc. 2005, 53, 695–699. [Google Scholar] [CrossRef]
- Weissman, M.M.; Sholomskas, D.; Pottenger, M.; Prusoff, B.A.; Locke, B.Z. Assessing depressive symptoms in five psychiatric populations: A validation study. Am. J. Epidemiol. 1977, 106, 203–214. [Google Scholar] [CrossRef]
- Acaster, S.; Swinburn, P.; Wang, C.; Stemper, B.; Beckmann, K.; Knappertz, V.; Pohl, C.; Sandbrink, R.; Gondek, K.; Edan, G.; et al. Can the functional assessment of multiple sclerosis adapt to changing needs? A psychometric validation in patients with clinically isolated syndrome and early relapsing–remitting multiple sclerosis. Mult. Scler. 2011, 17, 1504–1513. [Google Scholar] [CrossRef]
- Calabrese, P.; Kalbe, E.; Kessler, J. Ein neuropsychologisches Screening zur Erfassung kognitiver Störungen bei MS-Patienten-Das Multiple Sklerose Inventarium Cognition (MUSIC). Psychoneuro 2004, 30, 384–388. [Google Scholar] [CrossRef] [Green Version]
- Hobart, J.; Lamping, D.; Fitzpatrick, R.; Riazi, A.; Thompson, A. The Multiple Sclerosis Impact Scale (MSIS-29): A new patient-based outcome measure. Brain 2001, 124, 962–973. [Google Scholar] [CrossRef] [PubMed]
- Langdon, D.W.; Amato, M.P.; Boringa, J.; Brochet, B.; Foley, F.; Fredrikson, S.; Hämäläinen, P.; Hartung, H.-P.; Krupp, L.; Penner, I.-K.; et al. Recommendations for a Brief International Cognitive Assessment for Multiple Sclerosis (BICAMS). Mult. Scler. 2012, 18, 891–898. [Google Scholar] [CrossRef] [Green Version]
- Riegler, K.E.; Cadden, M.; Guty, E.T.; Bruce, J.M.; Arnett, P.A. Perceived Fatigue Impact and Cognitive Variability in Multiple Sclerosis. J. Int. Neuropsychol. Soc. 2022, 28, 281–291. [Google Scholar] [CrossRef] [PubMed]
- Tiftikcioglu, B.I. Multiple Sclerosis Functional Composite (MSFC): Scoring Instructions. Arch. Neuropsychiatry 2018, 55, S46–S48. [Google Scholar] [CrossRef]
- Tur, C. Fatigue Management in Multiple Sclerosis. Curr. Treat. Options Neurol. 2016, 18, 26. [Google Scholar] [CrossRef] [Green Version]
- Vickrey, B.G.; Hays, R.D.; Harooni, R.; Myers, L.W.; Ellison, G.W. A health-related quality of life measure for multiple sclerosis. Qual. Life Res. 1995, 4, 187–206. [Google Scholar] [CrossRef]
- McGraw, C.A.; Lublin, F.D. Interferon Beta and Glatiramer Acetate Therapy. Neurotherapeutics 2013, 10, 2–18. [Google Scholar] [CrossRef] [Green Version]
- Reilly, M.C.; Zbrozek, A.S.; Dukes, E.M. The Validity and Reproducibility of a Work Productivity and Activity Impairment Instrument. PharmacoEconomics 1993, 4, 353–365. [Google Scholar] [CrossRef]
- Liu, Y.; Vollmer, T.; Havrdova, E.K.; Riester, K.; Lee, A.; Phillips, G.; Wang, P.; Sabatella, G. Impact of daclizumab versus interferon beta-1a on patient-reported outcomes in relapsing-remitting multiple sclerosis. Mult. Scler. Relat. Disord. 2017, 11, 18–24. [Google Scholar] [CrossRef] [Green Version]
- Maurino, J.; Martínez-Ginés, M.L.; García-Domínguez, J.M.; Solar, M.D.; Carcelén-Gadea, M.; Ares-Luque, A.; Ballabriga, J.; Navarro-Cantó, L.; Medrano, N.; Honan, C.A. Workplace difficulties, health-related quality of life, and perception of stigma from the perspective of patients with Multiple Sclerosis. Mult. Scler. Relat. Disord. 2020, 41, 102046. [Google Scholar] [CrossRef] [PubMed]
- Bohlouli, J.; Namjoo, I.; Borzoo-Isfahani, M.; Poorbaferani, F.; Moravejolahkami, A.R.; Clark, C.C.T.; Kermani, M.A.H. Modified Mediterranean diet v. traditional Iranian diet: Efficacy of dietary interventions on dietary inflammatory index score, fatigue severity and disability in multiple sclerosis patients. Br. J. Nutr. 2021, 11, 111. [Google Scholar] [CrossRef] [PubMed]
- Melanson, M.; Grossberndt, A.; Klowak, M.; Leong, C.; Frost, E.E.; Prout, M.; Le Dorze, J.-A.; Gramlich, C.; Doupe, M.; Wong, L.; et al. Fatigue and Cognition in Patients with Relapsing Multiple Sclerosis Treated with Interferon Beta. Int. J. Neurosci. 2010, 120, 631–640. [Google Scholar] [CrossRef] [PubMed]
- Pavan, K.; Schmidt, K.; Marangoni, B.; Mendes, M.F.; Tilbery, C.P.; Lianza, S. Multiple sclerosis: Cross-cultural adaptation and validation of the modified fatigue impact scale. Arq. Neuro-Psiquiatr. 2007, 65, 669–673. [Google Scholar] [CrossRef]
- Losonczi, E.; Bencsik, K.; Rajda, C.; Lencsés, G.; Török, M.; Vécsei, L. Validation of the Fatigue Impact Scale in Hungarian patients with multiple sclerosis. Qual. Life Res. 2011, 20, 301–306. [Google Scholar] [CrossRef]
- Melamud, L.; Golan, D.; Luboshitzky, R.; Lavi, I.; Miller, A. Melatonin dysregulation, sleep disturbances and fatigue in multiple sclerosis. J. Neurol. Sci. 2012, 314, 37–40. [Google Scholar] [CrossRef]
- Ozkul, C.; Guclu-Gunduz, A.; Eldemir, K.; Apaydin, Y.; Yazici, G.; Irkec, C. Combined exercise training improves cognitive functions in multiple sclerosis patients with cognitive impairment: A single-blinded randomized controlled trial. Mult. Scler. Relat. Disord. 2020, 45, 102419. [Google Scholar] [CrossRef]
- Capra, R.; Morra, V.B.; Mirabella, M.; Gasperini, C.; Scandellari, C.; Totaro, R.; De Rossi, N.; Masera, S.; Zipoli, V.; Patti, F.; et al. Natalizumab is associated with early improvement of working ability in relapsing-remitting multiple sclerosis patients: WANT observational study results. Neurol. Sci. 2021, 42, 2837–2845. [Google Scholar] [CrossRef]
- Nohara, C.; Hase, M.; Liebert, R.; Wu, N. The burden of multiple sclerosis in Japan. J. Med Econ. 2017, 20, 1290–1298. [Google Scholar] [CrossRef] [PubMed]
- Khoy, K.; Mariotte, D.; Defer, G.; Petit, G.; Toutirais, O.; Le Mauff, B. Natalizumab in Multiple Sclerosis Treatment: From Biological Effects to Immune Monitoring. Front. Immunol. 2020, 11, 549842. [Google Scholar] [CrossRef]
- Skorve, E.; Lundervold, A.J.; Torkildsen, Ø.; Myhr, K.-M. A two-year longitudinal follow-up of cognitive performance assessed by BICAMS in newly diagnosed patients with MS. Mult. Scler. Relat. Disord. 2020, 46, 102577. [Google Scholar] [CrossRef] [PubMed]
- Woods, S.P.; Delis, D.C.; Scott, J.C.; Kramer, J.H.; Holdnack, J.A. The California Verbal Learning Test–second edition: Test-retest reliability, practice effects, and reliable change indices for the standard and alternate forms. Arch. Clin. Neuropsychol. 2006, 21, 413–420. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fellows, R.P.; Schmitter-Edgecombe, M. Symbol Digit Modalities Test: Regression-Based Normative Data and Clinical Utility. Arch. Clin. Neuropsychol. 2019, 35, 105–115. [Google Scholar] [CrossRef]
- Tam, J.W.; Schmitter-Edgecombe, M. The Role of Processing Speed in the Brief Visuospatial Memory Test–Revised. Clin. Neuropsychol. 2013, 27, 962–972. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amato, M.P.; Langdon, D.; Montalban, X.; Benedict, R.H.B.; DeLuca, J.; Krupp, L.B.; Thompson, A.J.; Comi, G. Treatment of cognitive impairment in multiple sclerosis: Position paper. J. Neurol. 2013, 260, 1452–1468. [Google Scholar] [CrossRef]
- Landmeyer, N.C.; Bürkner, P.-C.; Wiendl, H.; Ruck, T.; Hartung, H.-P.; Holling, H.; Meuth, S.G.; Johnen, A. Disease-modifying treatments and cognition in relapsing-remitting multiple sclerosis: A meta-analysis. Neurology 2020, 94, e2373–e2383. [Google Scholar] [CrossRef]
- Von Bismarck, O.; Dankowski, T.; Ambrosius, B.; Hessler, N.; Antony, G.; Ziegler, A.; Hoshi, M.-M.; Aly, L.; Luessi, F.; Groppa, S.; et al. Treatment choices and neuropsychological symptoms of a large cohort of early MS. Neurol.-Neuroimmunol. Neuroinflammation 2018, 5, e446. [Google Scholar] [CrossRef] [Green Version]
- Lublin, F.D.; Cofield, S.S.; Cutter, G.R.; Gustafson, T.; Krieger, S.; Narayana, P.A.; Nelson, F.; Salter, A.R.; Wolinsky, J.S. Long-term follow-up of a randomized study of combination interferon and glatiramer acetate in multiple sclerosis: Efficacy and safety results up to 7 years. Mult. Scler. Relat. Disord. 2017, 18, 95–102. [Google Scholar] [CrossRef]
- Bagert, B.; Camplair, P.; Bourdette, D. Cognitive Dysfunction in Multiple Sclerosis: Natural history, pathophysiology and management. CNS Drugs 2002, 16, 445–455. [Google Scholar] [CrossRef]
- Matsui, Y.; Kondo, M.; Uchiyama, E.; Mityata, R.; Matsubara, H. New clinical ultrahigh-resolution SD-OCT using A-scan matching algorithm. Graefe’s Arch. Clin. Exp. Ophthalmol. 2019, 257, 255–263. [Google Scholar] [CrossRef]
- Schrempf, W.; Ziemssen, T. Glatiramer acetate: Mechanisms of action in multiple sclerosis. Autoimmun. Rev. 2007, 6, 469–475. [Google Scholar] [CrossRef] [PubMed]
- Ambati, J.; Anand, A.; Fernandez, S.; Sakurai, E.; Lynn, B.C.; Kuziel, W.A.; Rollins, B.J.; Ambati, B.K. An animal model of age-related macular degeneration in senescent Ccl-2- or Ccr-2-deficient mice. Nat. Med. 2003, 9, 1390–1397. [Google Scholar] [CrossRef]
- Landa, G.; Butovsky, O.; Shoshani, J.; Schwartz, M.; Pollack, A. Weekly Vaccination with Copaxone (Glatiramer Acetate) as a Potential Therapy for Dry Age-Related Macular Degeneration. Curr. Eye Res. 2008, 33, 1011–1013. [Google Scholar] [CrossRef] [PubMed]
- Landa, G.; Rosen, R.B.; Patel, A.; Lima, V.C.; Tai, K.W.; Perez, V.R.; Aizman, A.; Garcia, P.M. Qualitative Spectral OCT/SLO Analysis of Drusen Change in Dry Age-Related Macular Degeneration Patients Treated with Copaxone. J. Ocul. Pharmacol. Ther. 2011, 27, 77–82. [Google Scholar] [CrossRef]
- Bakalash, S.; Pham, M.; Koronyo, Y.; Salumbides, B.C.; Kramerov, A.; Seidenberg, H.; Berel, D.; Black, K.L.; Koronyo-Hamaoui, M. Egr1 Expression Is Induced Following Glatiramer Acetate Immunotherapy in Rodent Models of Glaucoma and Alzheimer’s Disease. Investig. Opthalmology Vis. Sci. 2011, 52, 9033–9046. [Google Scholar] [CrossRef]
- Fan, K.R.; Baskaran, M.; Nongpiur, M.E.; Htoon, H.M.; De Leon, J.M.S.; Perera, S.A.; Belkin, M.; Aung, T. Investigating the neuroprotective effect of Copolymer-1 in acute primary angle closure—Interim report of a randomized placebo-controlled double-masked clinical trial. Acta Ophthalmol. 2019, 97, e827–e832. [Google Scholar] [CrossRef]
- Gordon, P.H.; Doorish, C.; Montes, J.; Mosley, R.L.; Diamond, B.; MacArthur, R.B.; Weimer, L.H.; Kaufmann, P.; Hays, A.P.; Rowland, L.P.; et al. Randomized controlled phase II trial of glatiramer acetate in ALS. Neurology 2006, 66, 1117–1119. [Google Scholar] [CrossRef] [PubMed]
- Mosley, R.L.; Gordon, P.H.; Hasiak, C.M.; Van Wetering, F.J.; Mitsumoto, H.; Gendelman, H.E. Glatiramer acetate immunization induces specific antibody and cytokine responses in ALS patients. Amyotroph. Lateral Scler. 2007, 8, 235–242. [Google Scholar] [CrossRef] [PubMed]
- Angelov, D.N.; Waibel, S.; Guntinas-Lichius, O.; Lenzen, M.; Neiss, W.F.; Tomov, T.L.; Yoles, E.; Kipnis, J.; Schori, H.; Reuter, A.; et al. Therapeutic vaccine for acute and chronic motor neuron diseases: Implications for amyotrophic lateral sclerosis. Proc. Natl. Acad. Sci. USA 2003, 100, 4790–4795. [Google Scholar] [CrossRef] [Green Version]
- Meininger, V.; Drory, V.E.; Leigh, P.N.; Ludolph, A.; Robberecht, W.; Silani, V. Glatiramer acetate has no impact on disease progression in ALS at 40 mg/day: A double- blind, randomized, multicentre, placebo-controlled trial. Amyotroph. Lateral Scler. 2009, 10, 378–383. [Google Scholar] [CrossRef]
- Butovsky, O.; Landa, G.; Kunis, G.; Ziv, Y.; Avidan, H.; Greenberg, N.; Schwartz, A.; Smirnov, I.; Pollack, A.; Jung, S.; et al. Induction and blockage of oligodendrogenesis by differently activated microglia in an animal model of multiple sclerosis. J. Clin. Investig. 2006, 116, 905–915. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Herges, K.; Millward, J.M.; Hentschel, N.; Infante-Duarte, C.; Aktas, O.; Zipp, F. Neuroprotective Effect of Combination Therapy of Glatiramer Acetate and Epigallocatechin-3-Gallate in Neuroinflammation. PLoS ONE 2011, 6, e25456. [Google Scholar] [CrossRef] [PubMed]
- Kipnis, J.; Cohen, H.; Cardon, M.; Ziv, Y.; Schwartz, M. T cell deficiency leads to cognitive dysfunction: Implications for therapeutic vaccination for schizophrenia and other psychiatric conditions. Proc. Natl. Acad. Sci. USA 2004, 101, 8180–8185. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kiyota, T.; Okuyama, S.; Swan, R.J.; Jacobsen, M.T.; Gendelman, H.E.; Ikezu, T. CNS expression of anti-inflammatory cytokine interleukin-4 attenuates Alzheimer’s disease-like pathogenesis in APP+PS1 bigenic mice. FASEB J. 2010, 24, 3093–3102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Doustar, J.; Rentsendorj, A.; Torbati, T.; Regis, G.C.; Fuchs, D.T.; Sheyn, J.; Mirzaei, N.; Graham, S.L.; Shah, P.K.; Mastali, M.; et al. Parallels between retinal and brain pathology and response to immunotherapy in old, late-stage Alzheimer’s disease mouse models. Aging Cell 2020, 19, e13246. [Google Scholar] [CrossRef] [PubMed]
- Constantinescu, C.S.; Farooqi, N.; O’Brien, K.; Gran, B. Experimental autoimmune encephalomyelitis (EAE) as a model for multiple sclerosis (MS). Br. J. Pharmacol. 2011, 164, 1079–1106. [Google Scholar] [CrossRef]
- Lopresti, P. Glatiramer Acetate Guards Against Rapid Memory Decline During Relapsing-Remitting Experimental Autoimmune Encephalomyelitis. Neurochem. Res. 2015, 40, 473–479. [Google Scholar] [CrossRef]
- Hamilton, J.A.; Anderson, G.P. GM-CSF Biology. Growth Factors 2004, 22, 225–231. [Google Scholar] [CrossRef]
- Eilam, R.; Segal, M.; Malach, R.; Sela, M.; Arnon, R.; Aharoni, R. Astrocyte disruption of neurovascular communication is linked to cortical damage in an animal model of multiple sclerosis. Glia 2018, 66, 1098–1117. [Google Scholar] [CrossRef]
- Aharoni, R.; Schottlender, N.; Bar-Lev, D.D.; Eilam, R.; Sela, M.; Tsoory, M.; Arnon, R. Cognitive impairment in an animal model of multiple sclerosis and its amelioration by glatiramer acetate. Sci. Rep. 2019, 9, 4140. [Google Scholar] [CrossRef]
- Li, A.; Wu, Y.; Pulli, B.; Wojtkiewicz, G.R.; Iwamoto, Y.; Wang, C.; Li, J.-H.; Ali, M.; Feng, X.; Yao, Z.; et al. Myeloperoxidase Molecular MRI Reveals Synergistic Combination Therapy in Murine Experimental Autoimmune Neuroinflammation. Radiology 2019, 293, 158–165. [Google Scholar] [CrossRef] [PubMed]
- Lawson, R.; Cross, H.A.; Tambe, J.T. Effects of large and small rewards on maze performance after different prior experiences with reward amounts. J. Comp. Physiol. Psychol. 1959, 52, 717–720. [Google Scholar] [CrossRef] [PubMed]
- Longa, E.Z.; Weinstein, P.R.; Carlson, S.; Cummins, R. Reversible middle cerebral artery occlusion without craniectomy in rats. Stroke 1989, 20, 84–91. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Markowska, A.L.; Olton, D.S.; Murray, E.A.; Gaffan, D. A comparative analysis of the role of fornix and cingulate cortex in memory: Rats. Exp. Brain Res. 1989, 74, 187–201. [Google Scholar] [CrossRef] [PubMed]
- Shibata, N. Transgenic mouse model for familial amyotrophic lateral sclerosis with superoxide dismutase-1 mutation. Neuropathology 2001, 21, 82–92. [Google Scholar] [CrossRef] [PubMed]
- Habisch, H.-J.; Schwalenstöcker, B.; Danzeisen, R.; Neuhaus, O.; Hartung, H.-P.; Ludolph, A. Limited effects of glatiramer acetate in the high-copy number hSOD1-G93A mouse model of ALS. Exp. Neurol. 2007, 206, 288–295. [Google Scholar] [CrossRef]
- Haenggeli, C.; Julien, J.-P.; Mosley, R.L.; Perez, N.; Dhar, A.; Gendelman, H.E.; Rothstein, J.D. Therapeutic immunization with a glatiramer acetate derivative does not alter survival in G93A and G37R SOD1 mouse models of familial ALS. Neurobiol. Dis. 2007, 26, 146–152. [Google Scholar] [CrossRef]
- Zuroff, L.; Daley, D.; Black, K.L.; Koronyo-Hamaoui, M. Clearance of cerebral Aβ in Alzheimer’s disease: Reassessing the role of microglia and monocytes. Cell. Mol. Life Sci. 2017, 74, 2167–2201. [Google Scholar] [CrossRef] [Green Version]
- Koronyo-Hamaoui, M.; Ko, M.K.; Koronyo, Y.; Azoulay, D.; Seksenyan, A.; Kunis, G.; Pham, M.; Bakhsheshian, J.; Rogeri, P.; Black, K.L.; et al. Attenuation of AD-like neuropathology by harnessing peripheral immune cells: Local elevation of IL-10 and MMP-9. J. Neurochem. 2009, 111, 1409–1424. [Google Scholar] [CrossRef]
- Lassmann, H. Mechanisms of neurodegeneration shared between multiple sclerosis and Alzheimer’s disease. J. Neural Transm. 2011, 118, 747–752. [Google Scholar] [CrossRef]
- Dal Bianco, A.; Bradl, M.; Frischer, J.; Kutzelnigg, A.; Jellinger, K.; Lassmann, H. Multiple sclerosis and Alzheimer’s disease. Ann. Neurol. 2008, 63, 174–183. [Google Scholar] [CrossRef] [PubMed]
- Pons, V.; Rivest, S. Targeting Systemic Innate Immune Cells as a Therapeutic Avenue for Alzheimer Disease. Pharmacol. Rev. 2022, 74, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Myers, A.; Mcgonigle, P. Overview of Transgenic Mouse Models for Alzheimer’s Disease. Curr. Protoc. Neurosci. 2019, 89, e81. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Hayden, E.Y.; Garcia, V.J.; Fuchs, D.-T.; Sheyn, J.; Daley, D.A.; Rentsendorj, A.; Torbati, T.; Black, K.L.; Rutishauser, U.; et al. Activated Bone Marrow-Derived Macrophages Eradicate Alzheimer’s-Related Aβ42 Oligomers and Protect Synapses. Front. Immunol. 2020, 11, 49. [Google Scholar] [CrossRef]
- Frenkel, D.; Maron, R.; Burt, D.S.; Weiner, H.L. Nasal vaccination with a proteosome-based adjuvant and glatiramer acetate clears β-amyloid in a mouse model of Alzheimer disease. J. Clin. Investig. 2005, 115, 2423–2433. [Google Scholar] [CrossRef] [Green Version]
- Dionisio-Santos, D.A.; Karaahmet, B.; Belcher, E.K.; Owlett, L.D.; Trojanczyk, L.A.; Olschowka, J.A.; O’Banion, M.K. Evaluating Effects of Glatiramer Acetate Treatment on Amyloid Deposition and Tau Phosphorylation in the 3xTg Mouse Model of Alzheimer’s Disease. Front. Neurosci. 2021, 15, 758677. [Google Scholar] [CrossRef]
- Rentsendorj, A.; Sheyn, J.; Fuchs, D.-T.; Daley, D.; Salumbides, B.C.; Schubloom, H.E.; Hart, N.J.; Li, S.; Hayden, E.Y.; Teplow, D.B.; et al. A novel role for osteopontin in macrophage-mediated amyloid-β clearance in Alzheimer’s models. Brain Behav. Immun. 2018, 67, 163–180. [Google Scholar] [CrossRef] [Green Version]
- Butovsky, O.; Kunis, G.; Koronyo-Hamaoui, M.; Schwartz, M. Selective ablation of bone marrow-derived dendritic cells increases amyloid plaques in a mouse Alzheimer’s disease model. Eur. J. Neurosci. 2007, 26, 413–416. [Google Scholar] [CrossRef]
- Baruch, K.; Rosenzweig, N.; Kertser, A.; Deczkowska, A.; Sharif, A.M.; Spinrad, A.; Tsitsou-Kampeli, A.; Sarel, A.; Cahalon, L.; Schwartz, M. Breaking immune tolerance by targeting Foxp3+ regulatory T cells mitigates Alzheimer’s disease pathology. Nat. Commun. 2015, 6, 7967. [Google Scholar] [CrossRef]
- Jung, S.; Unutmaz, D.; Wong, P.; Sano, G.-I.; De los Santos, K.; Sparwasser, T.; Wu, S.; Vuthoori, S.; Ko, K.; Zavala, F.; et al. In Vivo Depletion of CD11c+ Dendritic Cells Abrogates Priming of CD8+ T Cells by Exogenous Cell-Associated Antigens. Immunity 2002, 17, 211–220. [Google Scholar] [CrossRef] [Green Version]
- Oddo, S.; Caccamo, A.; Shepherd, J.D.; Murphy, M.P.; Golde, T.E.; Kayed, R.; Metherate, R.; Mattson, M.P.; Akbari, Y.; LaFerla, F.M. Triple-Transgenic Model of Alzheimer’s Disease with Plaques and Tangles: Intracellular Abeta and Synaptic Dysfunction. Neuron 2003, 39, 409–421. [Google Scholar] [CrossRef] [Green Version]
- Barnes, R.H.; Cunnold, S.R.; Zimmermann, R.R.; Simmons, H.; MacLeod, R.B.; Krook, L. Influence of Nutritional Deprivations in Early Life on Learning Behavior of Rats as Measured by Performance in a Water Maze. J. Nutr. 1966, 89, 399–410. [Google Scholar] [CrossRef] [PubMed]
- Burešová, O.; Bureš, J.; Oitzl, M.S.; Zahálka, A. Radial maze in the water tank: An aversively motivated spatial working memory task. Physiol. Behav. 1985, 34, 1003–1005. [Google Scholar] [CrossRef]
- Morris, R. Developments of a water-maze procedure for studying spatial learning in the rat. J. Neurosci. Methods 1984, 11, 47–60. [Google Scholar] [CrossRef]
- Churchill, M.J.; Cantu, M.A.; Kasanga, E.A.; Moore, C.; Salvatore, M.F.; Meshul, C.K. Glatiramer Acetate Reverses Motor Dysfunction and the Decrease in Tyrosine Hydroxylase Levels in a Mouse Model of Parkinson’s Disease. Neuroscience 2019, 414, 8–27. [Google Scholar] [CrossRef]
- Jackson-Lewis, V.; Przedborski, S. Protocol for the MPTP mouse model of Parkinson’s disease. Nat. Protoc. 2007, 2, 141–151. [Google Scholar] [CrossRef]
- Laurie, C.; Reynolds, A.; Coskun, O.; Bowman, E.; Gendelman, H.E.; Mosley, R.L. CD4+ T cells from Copolymer-1 immunized mice protect dopaminergic neurons in the 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine model of Parkinson’s disease. J. Neuroimmunol. 2007, 183, 60–68. [Google Scholar] [CrossRef]
- Pallier, P.N.; Drew, C.J.; Morton, A.J. The detection and measurement of locomotor deficits in a transgenic mouse model of Huntington’s disease are task- and protocol-dependent: Influence of non-motor factors on locomotor function. Brain Res. Bull. 2009, 78, 347–355. [Google Scholar] [CrossRef]
- Corey-Bloom, J.; Aikin, A.M.; Gutierrez, A.M.; Nadhem, J.S.; Howell, T.L.; Thomas, E.A. Beneficial effects of glatiramer acetate in Huntington’s disease mouse models: Evidence for BDNF-elevating and immunomodulatory mechanisms. Brain Res. 2017, 1673, 102–110. [Google Scholar] [CrossRef] [PubMed]
- Reick, C.; Ellrichmann, G.; Tsai, T.; Lee, D.-H.; Wiese, S.; Gold, R.; Saft, C.; Linker, R.A. Expression of brain-derived neurotrophic factor in astrocytes—Beneficial effects of glatiramer acetate in the R6/2 and YAC128 mouse models of Huntington’s disease. Exp. Neurol. 2016, 285, 12–23. [Google Scholar] [CrossRef] [PubMed]
- Ryu, J.K.; Kim, J.; Cho, S.J.; Hatori, K.; Nagai, A.; Choi, H.B.; Lee, M.C.; McLarnon, J.G.; Kim, S.U. Proactive transplantation of human neural stem cells prevents degeneration of striatal neurons in a rat model of Huntington disease. Neurobiol. Dis. 2004, 16, 68–77. [Google Scholar] [CrossRef] [PubMed]
- Corey-Bloom, J.; Jia, H.; Aikin, A.M.; Thomas, E.A. Disease Modifying Potential of Glatiramer Acetate in Huntington’s Disease. J. Huntingt. Dis. 2014, 3, 311–316. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guyenet, S.J.; Furrer, S.A.; Damian, V.M.; Baughan, T.D.; La Spada, A.R.; Garden, G.A. A Simple Composite Phenotype Scoring System for Evaluating Mouse Models of Cerebellar Ataxia. J. Vis. Exp. 2010, 39, e1787. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bouchon, R.; Will, B. Effects of post-weaning environment and apparatus dimension on spontaneous alternation as a function of phenotype in “dwarf” mice. Physiol. Behav. 1983, 30, 213–219. [Google Scholar] [CrossRef]
- Costall, B.; Naylor, R.J.; Nohria, V. Climbing behaviour induced by apomorphine in mice: A potential model for the detection of neuroleptic activity. Eur. J. Pharmacol. 1978, 50, 39–50. [Google Scholar] [CrossRef]
- Meyer, O.A.; Tilson, H.A.; Byrd, W.C.; Riley, M.T. A method for the routine assessment of fore- and hindlimb grip strength of rats and mice. Neurobehav. Toxicol. 1979, 1, 233–236. [Google Scholar]
- Coughenour, L.L.; McLean, J.R.; Parker, R.B. A new device for the rapid measurement of impaired motor function in mice. Pharmacol. Biochem. Behav. 1977, 6, 351–353. [Google Scholar] [CrossRef]
- Markel, A.L.; Khusainov, R.A. Method of complex recording of the behavioral and autonomic reactions of rats during conduction of the “open field” test. Zhurnal Vyss. Nervn. Deiatelnosti Im. IP Pavlov. 1976, 26, 1314–1318. [Google Scholar]
- Nonoyama, S.; Ochs, H.D. Immune Deficiency in SCID Mice. Int. Rev. Immunol. 1996, 13, 289–300. [Google Scholar] [CrossRef]
- Avni-Magen, N.; Zafrir, B.; King, R.; Bdolah-Abram, T.; Shilo-Benjamini, Y. Immobilization of captive Persian fallow deer (Dama dama mesopotamica) using medetomidine-ketamine or medetomidine-midazolam. Vet. Anaesth. Analg. 2019, 46, 662–666. [Google Scholar] [CrossRef]
- Kuypers, K.P.C. Psychedelic medicine: The biology underlying the persisting psychedelic effects. Med. Hypotheses 2019, 125, 21–24. [Google Scholar] [CrossRef] [PubMed]
- Louthan, A.; Gray, L.; Gabriele, M.L. Multi-sensory (auditory and somatosensory) pre-pulse inhibition in mice. Physiol. Behav. 2020, 222, 112901. [Google Scholar] [CrossRef] [PubMed]
- Jafer, A.; Sylvius, N.; Adewoye, A.B.; Dubrova, Y.E. The long-term effects of exposure to ionising radiation on gene expression in mice. Mutat. Res. 2020, 821, 111723. [Google Scholar] [CrossRef] [PubMed]
- He, F.; Zou, J.-T.; Zhou, Q.-F.; Niu, D.-L.; Jia, W.-H. Glatiramer acetate reverses cognitive deficits from cranial-irradiated rat by inducing hippocampal neurogenesis. J. Neuroimmunol. 2014, 271, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Willner, P.; Muscat, R.; Papp, M. Chronic mild stress-induced anhedonia: A realistic animal model of depression. Neurosci. Biobehav. Rev. 1992, 16, 525–534. [Google Scholar] [CrossRef]
- Buhot-Averseng, M.-C. Nest-box choice in the laboratory mouse: Preferences for nest-boxes differing in design (size and/or shape) and composition. Behav. Process. 1981, 6, 337–384. [Google Scholar] [CrossRef]
- Pascuan, C.G.; Simon, E.H.; Genaro, A.M.; Palumbo, M.L. Involvement of nitric oxide in improving stress-induced behavioural alteration by glatiramer acetate treatment in female BALB/c mice. Psychopharmacology 2015, 232, 1595–1605. [Google Scholar] [CrossRef]
- Goujon, E.; Parnet, P.; Laye, S.; Combe, C.; Kelley, K.W.; Dantzer, R. Stress Downregulates Lipopolysaccharide-Induced Expression of Proinflammatory Cytokines in the Spleen, Pituitary, and Brain of Mice. Brain Behav. Immun. 1995, 9, 292–303. [Google Scholar] [CrossRef] [Green Version]
- Kraeuter, A.-K.; Guest, P.C.; Sarnyai, Z. The Y-Maze for Assessment of Spatial Working and Reference Memory in Mice. Methods Mol. Biol. 2019, 1916, 105–111. [Google Scholar] [CrossRef]
- Tucker, A.R.; Gibbs, M.E.; Stanes, M.D. Cycloheximide and passive avoidance memory in mice: Time-response, dose-response and short-term memory. Pharmacol. Biochem. Behav. 1976, 4, 441–446. [Google Scholar] [CrossRef]
- Mohammadi, F.; Rahimian, R.; Fakhraei, N.; Rezayat, S.M.; Javadi-Paydar, M.; Dehpour, A.R.; Afshari, K.; Ejtemaei Mehr, S. Effect of glatiramer acetate on short-term memory impairment induced by lipopolysaccharide in male mice. Fundam. Clin. Pharmacol. 2016, 30, 347–356. [Google Scholar] [CrossRef] [PubMed]
- Valko, M.; Leibfritz, D.; Moncol, J.; Cronin, M.T.D.; Mazur, M.; Telser, J. Free radicals and antioxidants in normal physiological functions and human disease. Int. J. Biochem. Cell Biol. 2007, 39, 44–84. [Google Scholar] [CrossRef] [PubMed]
- Jeong, Y.J.; Son, Y.; Han, N.-K.; Choi, H.-D.; Pack, J.-K.; Kim, N.; Lee, Y.-S.; Lee, H.-J. Impact of Long-Term RF-EMF on Oxidative Stress and Neuroinflammation in Aging Brains of C57BL/6 Mice. Int. J. Mol. Sci. 2018, 19, 2103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ibarra, A.; Avendaño, H.; Cruz, Y. Copolymer-1 (Cop-1) improves neurological recovery after middle cerebral artery occlusion in rats. Neurosci. Lett. 2007, 425, 110–113. [Google Scholar] [CrossRef]
- Shekhar, S.; Cunningham, M.W.; Pabbidi, M.R.; Wang, S.; Booz, G.W.; Fan, F. Targeting vascular inflammation in ischemic stroke: Recent developments on novel immunomodulatory approaches. Eur. J. Pharmacol. 2018, 833, 531–544. [Google Scholar] [CrossRef] [PubMed]
- Qi, J.; Rong, Y.; Wang, L.; Xu, J.; Zhao, K. Rab7b Overexpression–Ameliorated Ischemic Brain Damage Following tMCAO Involves Suppression of TLR4 and NF-κB p65. J. Mol. Neurosci. 2019, 68, 163–170. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Yao, Y.; Wei, C.; Sun, Y.; Ma, X.; Zhang, R.; Xu, X.; Hao, J. T cell immunity to glatiramer acetate ameliorates cognitive deficits induced by chronic cerebral hypoperfusion by modulating the microenvironment. Sci. Rep. 2015, 5, 14308. [Google Scholar] [CrossRef] [Green Version]
- Cruz, Y.; Lorea, J.; Mestre, H.; Kim-Lee, J.H.; Herrera, J.; Mellado, R.; Gálvez, V.; Cuellar, L.; Musri, C.; Ibarra, A. Copolymer-1 Promotes Neurogenesis and Improves Functional Recovery after Acute Ischemic Stroke in Rats. PLoS ONE 2015, 10, e0121854. [Google Scholar] [CrossRef] [Green Version]
- Ibarra, A.; Cruz, Y.; García, E.E.; Gálvez, J.V.; Arias-Santiago, S.V.; Carvajal, H.G.; Silva-García, R.; Bonilla-Jaime, H.; Rojas-Castañeda, J.; Ibarra, A. Release of interleukin-10 and neurotrophic factors in the choroid plexus: Possible inductors of neurogenesis following copolymer-1 immunization after cerebral ischemia. Neural Regen. Res. 2018, 13, 1743–1752. [Google Scholar] [CrossRef]
- Chiang, T.; Messing, R.O.; Chou, W.-H. Mouse Model of Middle Cerebral Artery Occlusion. J. Vis. Exp. 2011, 48, e2761. [Google Scholar] [CrossRef] [Green Version]
- Sharp, J.L.; Miller-Cahill, M.E.; Riccio, D.C.; Fountain, S.B. Serial pattern retention in male and female rats. Neurobiol. Learn. Mem. 2018, 155, 578–582. [Google Scholar] [CrossRef] [PubMed]
- Luong, T.N.; Carlisle, H.J.; Southwell, A.; Patterson, P.H. Assessment of Motor Balance and Coordination in Mice using the Balance Beam. J. Vis. Exp. 2011, 49, e2376. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matsuura, K.; Kabuto, H.; Makino, H.; Ogawa, N. Pole test is a useful method for evaluating the mouse movement disorder caused by striatal dopamine depletion. J. Neurosci. Methods 1997, 73, 45–48. [Google Scholar] [CrossRef]
- Mangin, G.; Poittevin, M.; Charriaut-Marlangue, C.; Giannesini, C.; Merkoulova-Rainon, T.; Kubis, N. Glatiramer acetate reduces infarct volume in diabetic mice with cerebral ischemia and prevents long-term memory loss. Brain Behav. Immun. 2019, 80, 315–327. [Google Scholar] [CrossRef]
- Duncombe, J.; Kitamura, A.; Hase, Y.; Ihara, M.; Kalaria, R.N.; Horsburgh, K. Chronic cerebral hypoperfusion: A key mechanism leading to vascular cognitive impairment and dementia. Closing the translational gap between rodent models and human vascular cognitive impairment and dementia. Clin. Sci. 2017, 131, 2451–2468. [Google Scholar] [CrossRef] [Green Version]
- Benedek, G.; Meza-Romero, R.; Jordan, K.; Zhang, Y.; Nguyen, H.; Kent, G.; Li, J.; Siu, E.; Frazer, J.; Piecychna, M.; et al. MIF and D-DT are potential disease severity modifiers in male MS subjects. Proc. Natl. Acad. Sci. USA 2017, 114, E8421–E8429. [Google Scholar] [CrossRef] [Green Version]
- Cavalli, E.; Mazzon, E.; Basile, M.S.; Mangano, K.; Di Marco, R.; Bramanti, P.; Nicoletti, F.; Fagone, P.; Petralia, M.C. Upregulated Expression of Macrophage Migration Inhibitory Factor, Its Analogue D-Dopachrome Tautomerase, and the CD44 Receptor in Peripheral CD4 T Cells from Clinically Isolated Syndrome Patients with Rapid Conversion to Clinical Defined Multiple Sclerosis. Medicina 2019, 55, 667. [Google Scholar] [CrossRef] [Green Version]
- Günther, S.; Fagone, P.; Jalce, G.; Atanasov, A.G.; Guignabert, C.; Nicoletti, F. Role of MIF and D-DT in immune-inflammatory, autoimmune, and chronic respiratory diseases: From pathogenic factors to therapeutic targets. Drug Discov. Today 2019, 24, 428–439. [Google Scholar] [CrossRef]
- Cavalli, E.; Mazzon, E.; Basile, M.S.; Mammana, S.; Pennisi, M.; Fagone, P.; Kalfin, R.; Martinovic, V.; Ivanovic, J.; Andabaka, M.; et al. In Silico and In Vivo Analysis of IL37 in Multiple Sclerosis Reveals Its Probable Homeostatic Role on the Clinical Activity, Disability, and Treatment with Fingolimod. Molecules 2019, 25, 20. [Google Scholar] [CrossRef] [Green Version]
- Ziemssen, T.; Ashtamker, N.; Rubinchick, S.; Knappertz, V.; Comi, G. Long-term safety and tolerability of glatiramer acetate 20 mg/mL in the treatment of relapsing forms of multiple sclerosis. Expert Opin. Drug Saf. 2017, 16, 247–255. [Google Scholar] [CrossRef]
- Wolinsky, J.S.; Borresen, T.E.; Dietrich, D.W.; Wynn, D.; Sidi, Y.; Steinerman, J.R.; Knappertz, V.; Kolodny, S. GLACIER: An open-label, randomized, multicenter study to assess the safety and tolerability of glatiramer acetate 40 mg three-times weekly versus 20mg daily in patients with relapsing-remitting multiple sclerosis. Mult. Scler. Relat. Disord. 2015, 4, 370–376. [Google Scholar] [CrossRef] [PubMed] [Green Version]



{kind=link}
{kind=link}
{kind=link}
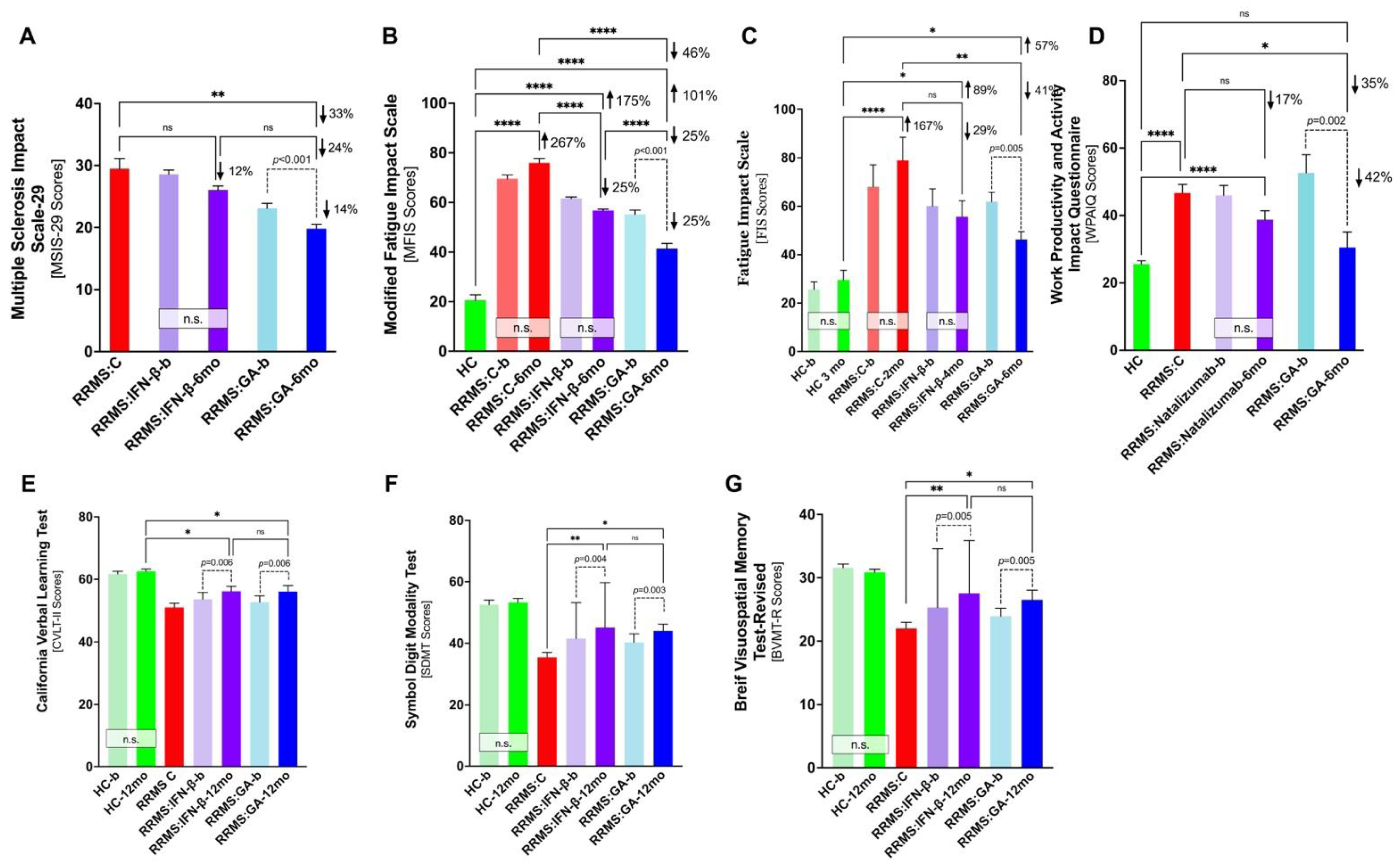
| Disease State | Research Design and Methodology | Findings | Ref. |
|---|---|---|---|
| MS |
|
| Weinstein, A. et al., 1999 [38] |
| MS |
|
| Schwid, R. et al., 2007 [40] |
| MS |
|
| Mori, F. et al., 2012 [42] |
| MS |
|
| Jongen, P. et al., 2014 [43] |
| MS |
| EDSS
| Vacaras, V. et al., 2014 [45] |
| MS |
|
| Fricksa-Nagy, Z. et al., 2016 [46] |
| MS |
| MFIS GA–b vs. GA–6 months [mean ± SD]:
| Meca-Lallana, J. et al., 2016 [47] |
| MS |
| GA—b vs. GA—2 years
| Ziemssen, T. et al., 2016 [49] |
| MS |
| BICAMS GA—b vs. GA—12 months [mean ± SD]:
| Cinar, B. et al., 2017 [50] |
| MS |
|
| Sazonov, D. et al., 2018 [51] |
| MS |
|
| Shorobura, M., 2018 [52] |
| MS |
|
| Zivadinov, R. et al., 2018 [53] |
| Disease State | Research Design and Methodology | Findings | Ref. |
|---|---|---|---|
| AMD |
|
| Landa, G. et al., 2008 [93] |
| Glaucoma (animal model) |
|
| Bakalash et al., 2011 [95] |
| AMD |
|
| Landa et al., 2011 [94] |
| Glaucoma |
|
| Fan et al., 2019 [96] |
| AMD |
|
| Gu, B. et al., 2021 [24] |
| Disease State | Research Design and Methodology | Findings | Ref. |
|---|---|---|---|
| ALS |
|
| Gordon, P. et al., 2006 [97] |
| ALS |
|
| Mosley, R. et al., 2007 [98] |
| Disease Model | Research Model and Methodology | Findings | Ref. |
|---|---|---|---|
| MS |
|
| Herges, K. et al., 2011 [102] |
| MS |
|
| LoPresti, P. 2015 [107] |
| MS |
|
| Eilam, R. et al., 2018 [109] |
| MS |
|
| Aharoni, R. et al., 2019 [110] |
| MS |
| GA and combo treatment vs. placebo
| Li, A. et al., 2019 [111] |
| Disease Model | Research Model and Methodology | Findings | Ref. |
|---|---|---|---|
| ALS |
|
| Angelov, D. et al., 2003 [99] |
| ALS |
|
| Habisch, H. et al., 2007 [116] |
| ALS |
|
Study utilized TV-5010 (synthetic HMW polymer formulation of the same amino acids of GA). | Haenggeli, C. et al., 2007 [117] |
| Disease Model | Research Model and Methodology | GA Effects/Findings | Ref. |
|---|---|---|---|
| AD |
|
| Frenkel, D. et al., 2005 [125] |
| AD |
|
| Butovsky, O. et al., 2006 [18] |
| AD |
|
| Butovsky, O. et al., 2007 [128] |
| AD |
| GA vs. controls, mice and rats
| Bakalash, S. et al., 2011 [95] |
| AD |
| GA vs. controls
| Koronyo, Y. et al., 2015 [32] |
| AD |
|
| Baruch et al., 2015 [129] |
| AD |
| GA vs. controls
| Rentsendorj, A. et al., 2018 [127] |
| AD |
| GA vs. controls
| Doustar, J. et al., 2020 [105] |
| AD |
| GA vs. controls
| Li, S. et al., 2020 [124] |
| AD |
|
| Dionisio-Santos, D. et al., 2021 [126] |
| Disease Model | Research Model and Methodology | Findings | Ref. |
|---|---|---|---|
| PD |
|
| Laurie, C. et al., 2007 [137] |
| PD |
| GA vs. controls
| Churchill, M. et al., 2019 [135] |
| Disease Model | Research Model and Methodology | Findings | Ref. |
|---|---|---|---|
| HD |
| GA vs. controls
| Corey-Bloom, C. et al., 2014 [142] |
| HD |
| GA vs. controls
| Reick, C. et al., 2016 [140] |
| HD |
| GA vs. controls
| Corey-Bloom, J. et al., 2017 [139] |
| Disease Model | Research Model and Methodology | Findings | Ref. |
|---|---|---|---|
| Psych |
|
| Kipnis, J. et al., 2004 [103] |
| Neuro psych |
|
| He, F. et al., 2014 [154] |
| Neuro psych |
| OFBA and OIPT [156] GA vs. control
| Pascuan, G. et al., 2015 [157] |
| Neuro psych |
| GA vs. control
| Mohammadi, F. et al., 2016 [161] |
| Disease Model | Research Model and Methodology | Findings | Ref. |
|---|---|---|---|
| CNSi |
|
| Ibarra, A. et al., 2007 [164] |
| CNSi |
| MWMT, GA vs. control
| Chen, L. et al., 2015 [167] |
| CNSi |
| GA vs. control [mean ± SD]
| Cruz, Y. et al., 2015 [168] |
| CNSi |
| GA vs. control
| Cruz, Y. et al., 2018 [169] |
| CNSi |
| GA vs. control
| Mangin, G. et al., 2019 [174] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kasindi, A.; Fuchs, D.-T.; Koronyo, Y.; Rentsendorj, A.; Black, K.L.; Koronyo-Hamaoui, M. Glatiramer Acetate Immunomodulation: Evidence of Neuroprotection and Cognitive Preservation. Cells 2022, 11, 1578. https://doi.org/10.3390/cells11091578
Kasindi A, Fuchs D-T, Koronyo Y, Rentsendorj A, Black KL, Koronyo-Hamaoui M. Glatiramer Acetate Immunomodulation: Evidence of Neuroprotection and Cognitive Preservation. Cells. 2022; 11(9):1578. https://doi.org/10.3390/cells11091578
Chicago/Turabian StyleKasindi, Arielle, Dieu-Trang Fuchs, Yosef Koronyo, Altan Rentsendorj, Keith L. Black, and Maya Koronyo-Hamaoui. 2022. "Glatiramer Acetate Immunomodulation: Evidence of Neuroprotection and Cognitive Preservation" Cells 11, no. 9: 1578. https://doi.org/10.3390/cells11091578
APA StyleKasindi, A., Fuchs, D. -T., Koronyo, Y., Rentsendorj, A., Black, K. L., & Koronyo-Hamaoui, M. (2022). Glatiramer Acetate Immunomodulation: Evidence of Neuroprotection and Cognitive Preservation. Cells, 11(9), 1578. https://doi.org/10.3390/cells11091578