
Regulation of Mitochondrial Hydrogen Peroxide Availability by Protein S-glutathionylation



{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
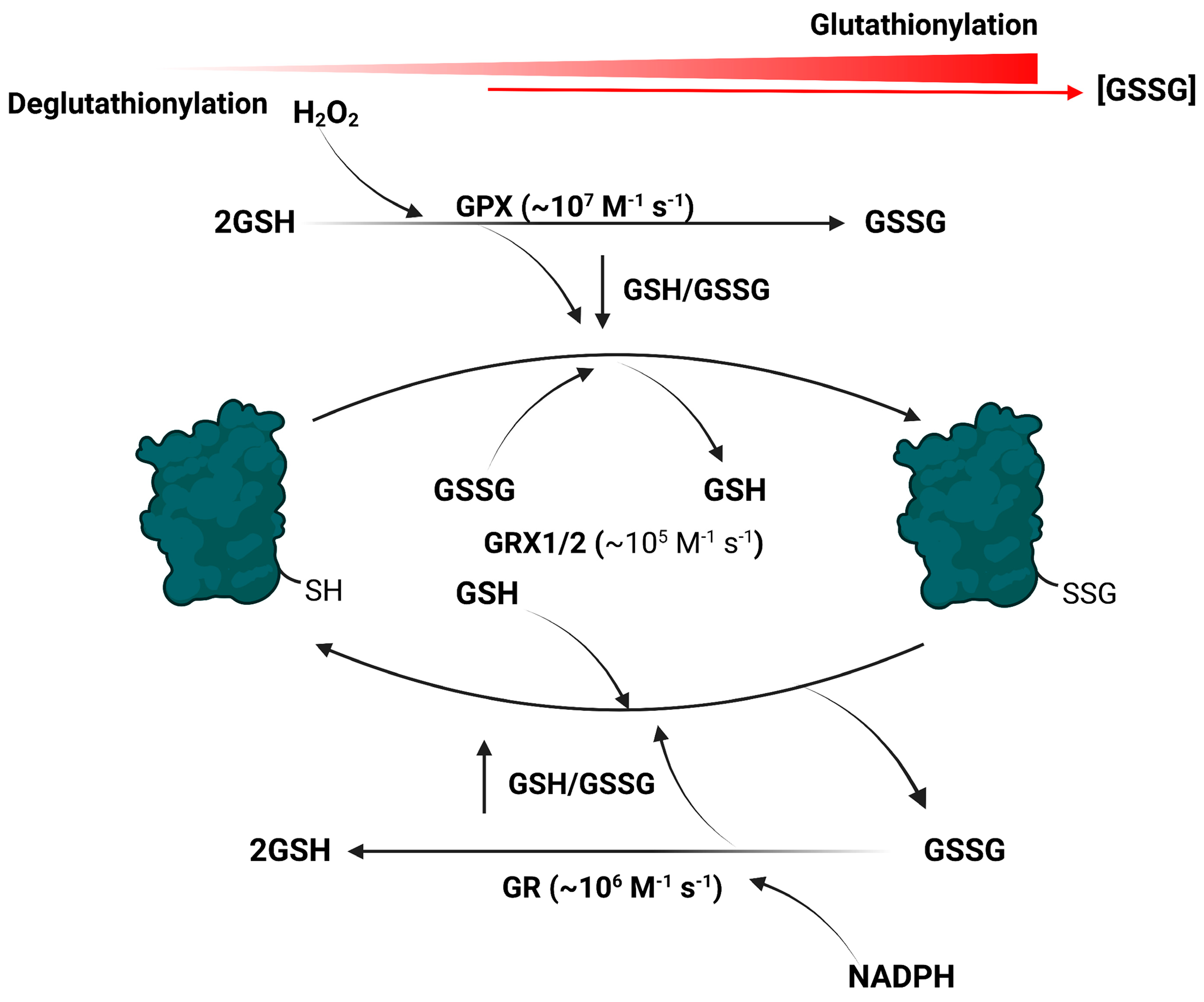
2. Protein S-Glutathionylation
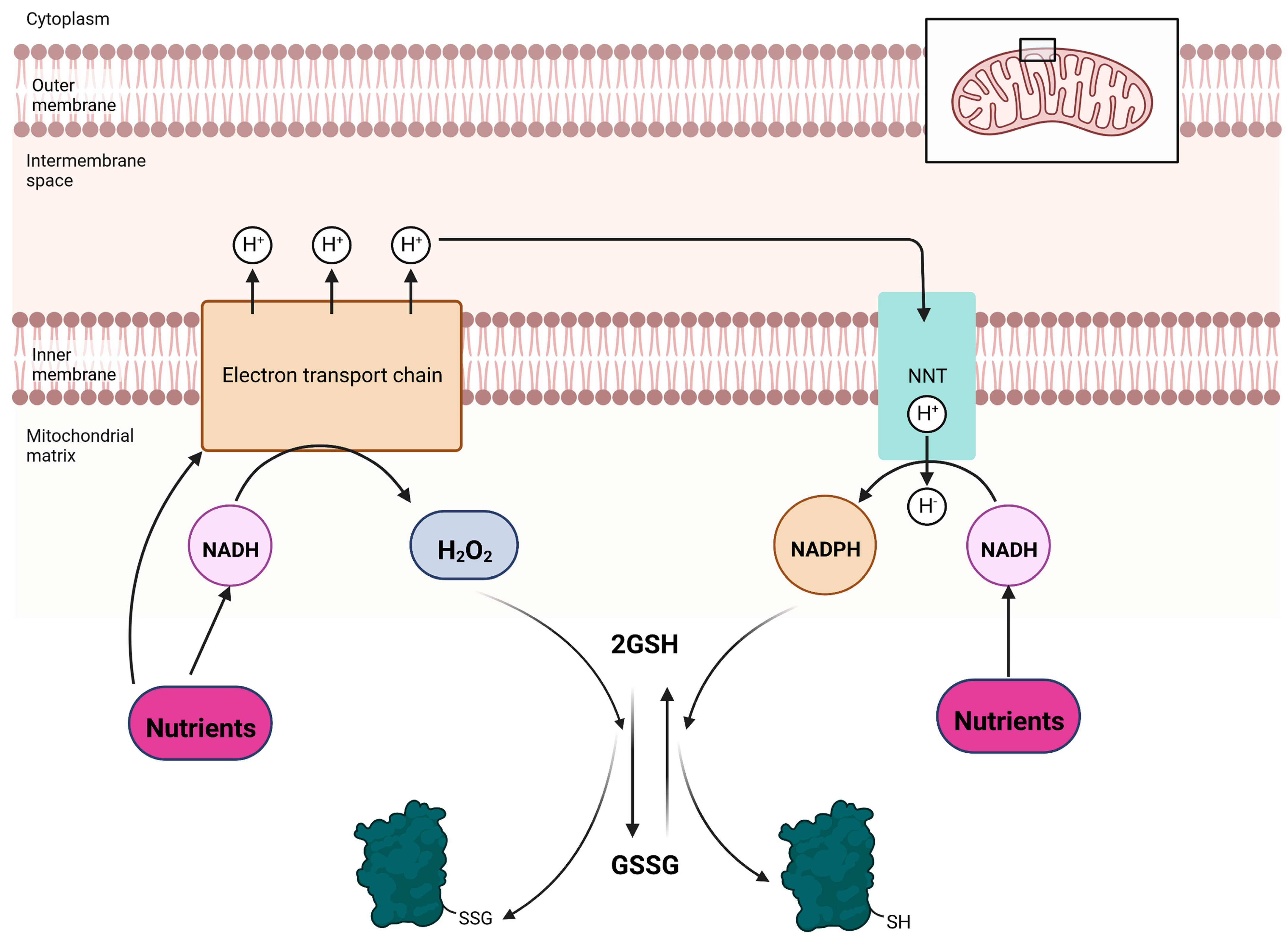
3. Mitochondrial Glutathionylation Reactions
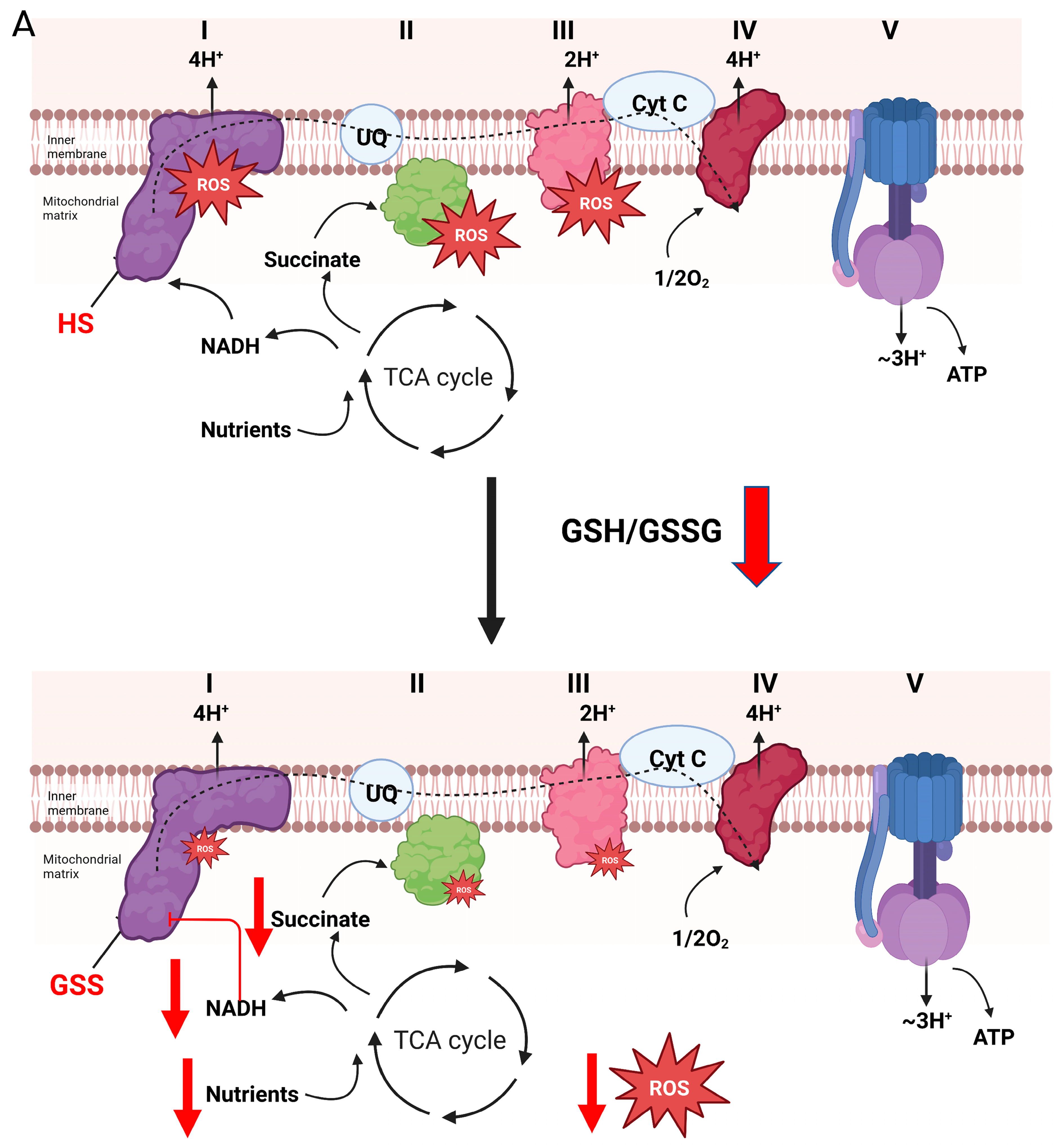
4. Glutathionylation and Complex I-Mediated H2O2 Production
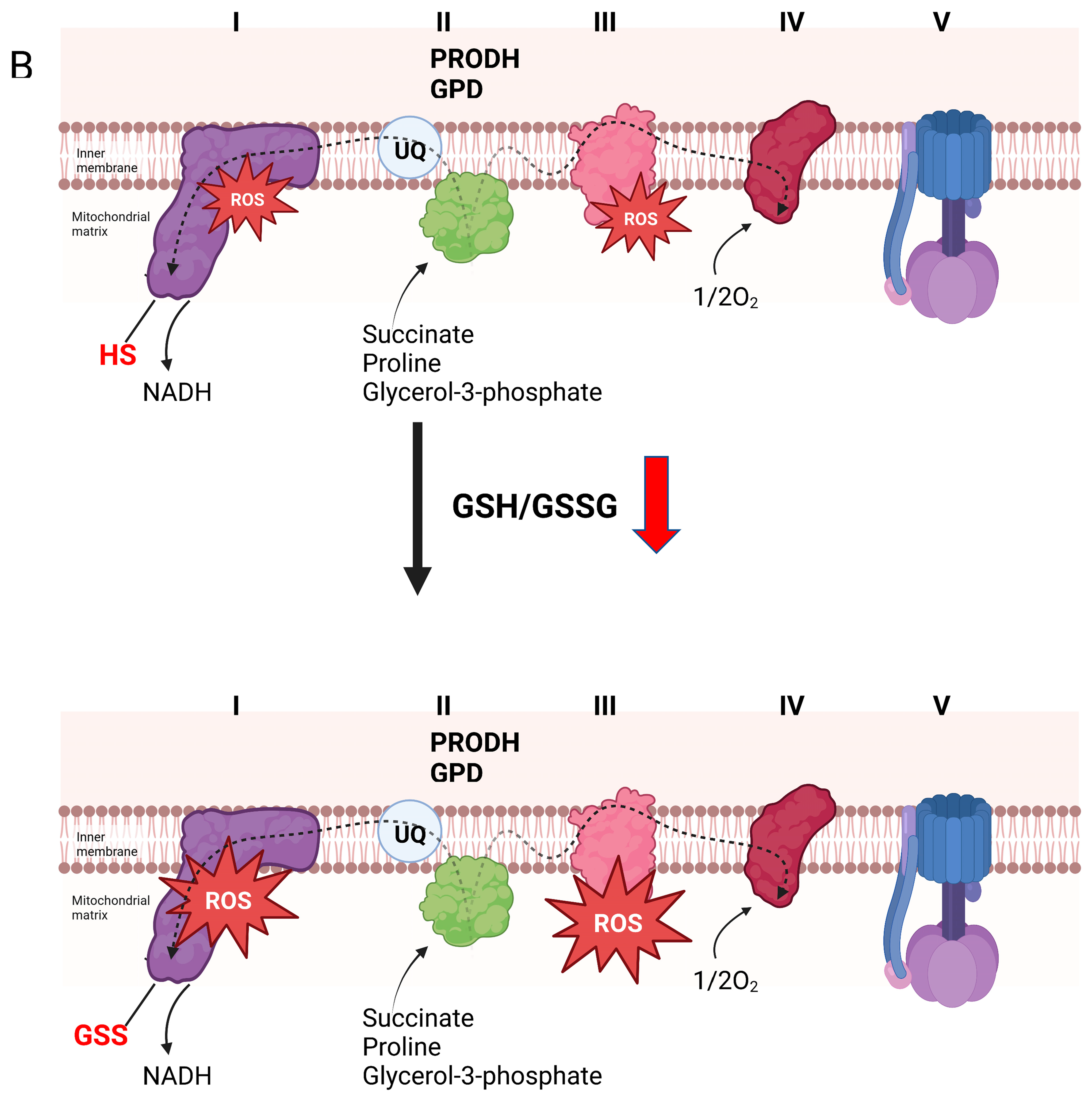
5. Glutathionylation and H2O2 Production by Other Flavoproteins
6. Conclusions
7. General Significance
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Sies, H.; Cadenas, E. Oxidative stress: Damage to intact cells and organs. Philos. Trans. R. Soc. Lond. B Biol. Sci. 1985, 311, 617–631. [Google Scholar] [PubMed]
- Cadenas, E.; Sies, H. Oxidative stress: Excited oxygen species and enzyme activity. Adv. Enzym. Regul. 1985, 23, 217–237. [Google Scholar] [CrossRef] [PubMed]
- Sies, H.; Belousov, V.V.; Chandel, N.S.; Davies, M.J.; Jones, D.P.; Mann, G.E.; Murphy, M.P.; Yamamoto, M.; Winterbourn, C. Defining roles of specific reactive oxygen species (ROS) in cell biology and physiology. Nat. Rev. Mol. Cell Biol. 2022, 23, 499–515. [Google Scholar] [CrossRef] [PubMed]
- Murphy, M.P.; Bayir, H.; Belousov, V.; Chang, C.J.; Davies, K.J.A.; Davies, M.J.; Dick, T.P.; Finkel, T.; Forman, H.J.; Janssen-Heininger, Y.; et al. Guidelines for measuring reactive oxygen species and oxidative damage in cells and in vivo. Nat. Metab. 2022, 4, 651–662. [Google Scholar] [CrossRef] [PubMed]
- Brunelle, J.K.; Bell, E.L.; Quesada, N.M.; Vercauteren, K.; Tiranti, V.; Zeviani, M.; Scarpulla, R.C.; Chandel, N.S. Oxygen sensing requires mitochondrial ROS but not oxidative phosphorylation. Cell Metab. 2005, 1, 409–414. [Google Scholar] [CrossRef] [Green Version]
- Sies, H.; Jones, D.P. Reactive oxygen species (ROS) as pleiotropic physiological signalling agents. Nat. Rev. Mol. Cell Biol. 2020, 21, 363–383. [Google Scholar] [CrossRef]
- Jones, D.; Sies, H. The Redox Code. Antioxid. Redox Signal. 2015, 23, 734–746. [Google Scholar] [CrossRef] [Green Version]
- Yun, J.; Finkel, T. Mitohormesis. Cell Metab. 2014, 19, 757–766. [Google Scholar] [CrossRef] [Green Version]
- Tormos, K.V.; Anso, E.; Hamanaka, R.B.; Eisenbart, J.; Joseph, J.; Kalyanaraman, B.; Chandel, N.S. Mitochondrial Complex III ROS Regulate Adipocyte Differentiation. Cell Metab. 2011, 14, 537–544. [Google Scholar] [CrossRef] [Green Version]
- Mailloux, R.J. An update on methods and approaches for interrogating mitochondrial reactive oxygen species production. Redox Biol. 2021, 45, 102044. [Google Scholar] [CrossRef]
- Horváth, G.; Sváb, G.; Komlódi, T.; Ravasz, D.; Kacsó, G.; Doczi, J.; Chinopoulos, C.; Ambrus, A.; Tretter, L. Reverse and Forward Electron Flow-Induced H2O2 Formation Is Decreased in α-Ketoglutarate Dehydrogenase (α-KGDH) Subunit (E2 or E3) Heterozygote Knock Out Animals. Antioxidants 2022, 11, 1487. [Google Scholar] [CrossRef] [PubMed]
- Fisher-Wellman, K.H.; Gilliam, L.A.; Lin, C.-T.; Cathey, B.L.; Lark, D.S.; Neufer, P.D. Mitochondrial glutathione depletion reveals a novel role for the pyruvate dehydrogenase complex as a key H2O2-emitting source under conditions of nutrient overload. Free Radic. Biol. Med. 2013, 65, 1201–1208. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, W.; Hu, L.; Zhang, M.; Liu, S.; Xu, S.; Chow, V.; Chan, J.; Wong, T. Mitochondrial DHODH regulates hypoxia-inducible factor 1 expression in OTSCC. Am. J. Cancer Res. 2022, 12, 48–67. [Google Scholar] [PubMed]
- Flohé, L. The impact of thiol peroxidases on redox regulation. Free Radic. Res. 2016, 50, 126–142. [Google Scholar] [CrossRef]
- Toppo, S.; Flohé, L.; Ursini, F.; Vanin, S.; Maiorino, M. Catalytic mechanisms and specificities of glutathione peroxidases: Variations of a basic scheme. Biochim. Biophys. Acta (BBA)—Gen. Subj. 2009, 1790, 1486–1500. [Google Scholar] [CrossRef]
- Schafer, F.Q.; Buettner, G.R. Redox environment of the cell as viewed through the redox state of the glutathione disul-fide/glutathione couple. Free Radic. Biol. Med. 2001, 30, 1191–1212. [Google Scholar] [CrossRef]
- Axelsson, K.; Mannervik, B. An essential role of cytosolic thioltransferase in protection of pyruvate kinase from rabbit liver against oxidative inactivation. FEBS Lett. 1983, 152, 114–118. [Google Scholar] [CrossRef] [Green Version]
- Giustarini, D.; Galvagni, F.; Tesei, A.; Farolfi, A.; Zanoni, M.; Pignatta, S.; Milzani, A.; Marone, I.M.; Dalle-Donne, I.; Nassini, R.; et al. Glutathione, glutathione disulfide, and S-glutathionylated proteins in cell cultures. Free Radic. Biol. Med. 2015, 89, 972–981. [Google Scholar] [CrossRef]
- McGarry, D.J.; Chen, W.; Chakravarty, P.; Lamont, D.L.; Wolf, C.R.; Henderson, C.J. Proteome-wide identification and quantification of S-glutathionylation targets in mouse liver. Biochem. J. 2015, 469, 25–32. [Google Scholar] [CrossRef]
- Kramer, P.; Duan, J.; Gaffrey, M.J.; Shukla, A.K.; Wang, L.; Bammler, T.K.; Qian, W.-J.; Marcinek, D.J. Fatiguing contractions increase protein S-glutathionylation occupancy in mouse skeletal muscle. Redox Biol. 2018, 17, 367–376. [Google Scholar] [CrossRef]
- Fratelli, M.; Demol, H.; Puype, M.; Casagrande, S.; Eberini, I.; Salmona, M.; Bonetto, V.; Mengozzi, M.; Duffieux, F.; Miclet, E.; et al. Identification by redox proteomics of glutathionylated proteins in oxidatively stressed human T lymphocytes. Proc. Natl. Acad. Sci. USA 2002, 99, 3505–3510. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiong, Y.; Uys, J.; Tew, K.; Townsend, D. S-glutathionylation: From molecular mechanisms to health outcomes. Antioxid. Redox Signal. 2011, 15, 233–270. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, J.; Ye, Z.-W.; Singh, S.; Townsend, D.M.; Tew, K.D. An evolving understanding of the S-glutathionylation cycle in pathways of redox regulation. Free Radic. Biol. Med. 2018, 120, 204–216. [Google Scholar] [CrossRef]
- Pastore, A.; Piemonte, F. S-Glutathionylation signaling in cell biology: Progress and prospects. Eur. J. Pharm. Sci. 2012, 46, 279–292. [Google Scholar] [CrossRef] [PubMed]
- Mieyal, J.J.; Gallogly, M.M.; Qanungo, S.; Sabens, E.A.; Shelton, M.D. Molecular Mechanisms and Clinical Implications of Reversible ProteinS-Glutathionylation. Antioxid. Redox Signal. 2008, 10, 1941–1988. [Google Scholar] [CrossRef] [PubMed]
- Petrini, S.; Passarelli, C.; Pastore, A.; Tozzi, G.; Coccetti, M.; Colucci, M.; Bianchi, M.; Carrozzo, R.; Bertini, E.; Piemonte, F. Protein glutathionylation in cellular compartments: A constitutive redox signal. Redox Rep. 2012, 17, 63–71. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pastore, A.; Tozzi, G.; Gaeta, L.; Bertini, E.; Serafini, V.; Di Cesare, S.; Bonetto, V.; Casoni, F.; Carrozzo, R.; Federici, G.; et al. Actin glutathionylation increases in fibroblasts of patients with Friedreich’s ataxia: A potential role in the pathogenesis of the disease. J. Biol. Chem. 2003, 278, 42588–42595. [Google Scholar] [CrossRef] [Green Version]
- Cooper, A.J.; Pinto, J.T.; Callery, P.S. Reversible and irreversible protein glutathionylation: Biological and clinical aspects. Expert Opin. Drug Metab. Toxicol. 2011, 7, 891–910. [Google Scholar] [CrossRef] [Green Version]
- O’Brien, M.; Chalker, J.; Slade, L.; Gardiner, D.; Mailloux, R.J. Protein S-glutathionylation alters superoxide/hydrogen peroxide emission from pyruvate dehydrogenase complex. Free Radic. Biol. Med. 2017, 106, 302–314. [Google Scholar] [CrossRef]
- Chalker, J.; Gardiner, D.; Kuksal, N.; Mailloux, R.J. Characterization of the impact of glutaredoxin-2 (GRX2) deficiency on superoxide/hydrogen peroxide release from cardiac and liver mitochondria. Redox Biol. 2018, 15, 216–227. [Google Scholar] [CrossRef]
- Gill, R.M.; O’Brien, M.; Young, A.; Gardiner, D.; Mailloux, R.J. Protein S-glutathionylation lowers superoxide/hydrogen peroxide release from skeletal muscle mitochondria through modification of complex I and inhibition of pyruvate uptake. PLoS ONE 2018, 13, e0192801. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Letourneau, M.; Wang, K.; Mailloux, R.J. Protein S-glutathionylation decreases superoxide/hydrogen peroxide production xanthine oxidoreductase. Free Radic. Biol. Med. 2021, 175, 184–192. [Google Scholar] [CrossRef] [PubMed]
- Beer, S.; Taylor, E.; Brown, S.; Dahm, C.; Costa, N.; Runswick, M.; Murphy, M. Glutaredoxin 2 catalyzes the reversible oxidation and glutathionylation of mitochondrial membrane thiol proteins: Implications for mitochondrial redox regulation and an-tioxidant DEFENSE. J. Biol. Chem. 2004, 279, 47939–47951. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gallogly, M.M.; Starke, D.W.; Mieyal, J.J. Mechanistic and Kinetic Details of Catalysis of Thiol-Disulfide Exchange by Glu-taredoxins and Potential Mechanisms of Regulation. Antioxid. Redox Signal. 2009, 11, 1059–1081. [Google Scholar] [CrossRef] [Green Version]
- Bechtel, T.J.; Weerapana, E. From structure to redox: The diverse functional roles of disulfides and implications in disease. Proteomics 2017, 17, 1600391. [Google Scholar] [CrossRef] [Green Version]
- Hatahet, F.; Ruddock, L.W. Protein Disulfide Isomerase: A Critical Evaluation of Its Function in Disulfide Bond Formation. Antioxid. Redox Signal. 2009, 11, 2807–2850. [Google Scholar] [CrossRef]
- Mailloux, R.; Xuan, J.Y.; McBride, S.; Maharsy, W.; Thorn, S.; Holterman, C.; Kennedy, C.R.; Rippstein, P.; Dekemp, R.; da Silva, J.; et al. Glutaredoxin-2 Is Required to Control Oxidative Phosphorylation in Cardiac Muscle by Mediating Deglutathi-onylation Reactions. J. Biol. Chem. 2014, 289, 14812–14828. [Google Scholar] [CrossRef] [Green Version]
- Van’t Erve, T.J.; Wagner, B.; Ryckman, K.; Raife, T.; Buettner, G. The concentration of glutathione in human erythrocytes is a heritable trait. Free Radic. Biol. Med. 2013, 65, 742–749. [Google Scholar] [CrossRef] [Green Version]
- Enns, G.M.; Cowan, T.M. Glutathione as a Redox Biomarker in Mitochondrial Disease—Implications for Therapy. J. Clin. Med. 2017, 6, 50. [Google Scholar] [CrossRef] [Green Version]
- Mandal, P.K.; Saharan, S.; Tripathi, M.; Murari, G. Brain Glutathione Levels—A Novel Biomarker for Mild Cognitive Im-pairment and Alzheimer’s Disease. Biol. Psychiatry 2015, 78, 702–710. [Google Scholar] [CrossRef]
- Campbell, M.D.; Duan, J.; Samuelson, A.T.; Gaffrey, M.J.; Merrihew, G.E.; Egertson, J.D.; Wang, L.; Bammler, T.K.; Moore, R.J.; White, C.C.; et al. Improving mitochondrial function with SS-31 reverses age-related redox stress and improves exercise tolerance in aged mice. Free Radic. Biol. Med. 2019, 134, 268–281. [Google Scholar] [CrossRef] [PubMed]
- Whitson, J.A.; Martín-Pérez, M.; Zhang, T.; Gaffrey, M.J.; Merrihew, G.E.; Huang, E.; White, C.C.; Kavanagh, T.J.; Qian, W.-J.; Campbell, M.D.; et al. Elamipretide (SS-31) treatment attenuates age-associated post-translational modifications of heart proteins. GeroScience 2021, 43, 2395–2412. [Google Scholar] [CrossRef] [PubMed]
- Lou, M.F. Glutathione and Glutaredoxin in Redox Regulation and Cell Signaling of the Lens. Antioxidants 2022, 11, 1973. [Google Scholar] [CrossRef] [PubMed]
- Young, A.; Gardiner, D.; Kuksal, N.; Gill, R.; O’Brien, M.; Mailloux, R.J. Deletion of the Glutaredoxin-2 Gene Protects Mice from Diet-Induced Weight Gain, Which Correlates with Increased Mitochondrial Respiration and Proton Leaks in Skeletal Muscle. Antioxid. Redox Signal. 2019, 31, 1272–1288. [Google Scholar] [CrossRef] [Green Version]
- Dong, K.; Wu, M.; Liu, X.; Huang, Y.; Zhang, D.; Wang, Y.; Yan, L.-J.; Shi, D. Glutaredoxins concomitant with optimal ROS activate AMPK through S-glutathionylation to improve glucose metabolism in type 2 diabetes. Free Radic. Biol. Med. 2016, 101, 334–347. [Google Scholar] [CrossRef]
- Zamora, D.; Downs, K.; Ullevig, S.; Tavakoli, S.; Kim, H.; Qiao, M.; Greaves, D.; Asmis, R. Glutaredoxin 2a overexpression in macrophages promotes mitochondrial dysfunction but has little or no effect on atherogenesis in LDL-receptor null mice. Atherosclerosis 2015, 241, 69–78. [Google Scholar] [CrossRef] [Green Version]
- Wohua, Z.; Weiming, X. Glutaredoxin 2 (GRX2) deficiency exacerbates high fat diet (HFD)-induced insulin resistance, in-flammation and mitochondrial dysfunction in brain injury: A mechanism involving GSK-3β. Biomed. Pharmacother. 2019, 118, 108940. [Google Scholar] [CrossRef]
- Almutairi, M.; Gopal, K.; Greenwell, A.; Young, A.; Gill, R.; Aburasayn, H.; Al Batran, R.; Chahade, J.J.; Gandhi, M.; Eaton, F.; et al. The GLP-1 Receptor Agonist Liraglutide Increases Myocardial Glucose Oxidation Rates via Indirect Mechanisms and Mitigates Experimental Diabetic Cardiomyopathy. Can. J. Cardiol. 2021, 37, 140–150. [Google Scholar] [CrossRef]
- Wu, H.; Yu, Y.; David, L.; Ho, Y.-S.; Lou, M.F. Glutaredoxin 2 (Grx2) Gene Deletion Induces Early Onset of Age-dependent Cataracts in Mice. J. Biol. Chem. 2014, 289, 36125–36139. [Google Scholar] [CrossRef] [Green Version]
- Gallogly, M.M.; Starke, D.W.; Leonberg, A.K.; Ospina, S.M.E.; Mieyal, J.J. Kinetic and Mechanistic Characterization and Versatile Catalytic Properties of Mammalian Glutaredoxin 2: Implications for Intracellular Roles. Biochemistry 2008, 47, 11144–11157. [Google Scholar] [CrossRef]
- Gallogly, M.M.; Mieyal, J.J. Mechanisms of reversible protein glutathionylation in redox signaling and oxidative stress. Curr. Opin. Pharmacol. 2007, 7, 381–391. [Google Scholar] [CrossRef] [PubMed]
- Alegre-Cebollada, J.; Kosuri, P.; Giganti, D.; Eckels, E.; Rivas-Pardo, J.A.; Hamdani, N.; Warren, C.M.; Solaro, R.J.; Linke, W.A.; Fernández, J.M. S-Glutathionylation of Cryptic Cysteines Enhances Titin Elasticity by Blocking Protein Folding. Cell 2014, 156, 1235–1246. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kukulage, D.S.; Don, N.N.M.; Ahn, Y.-H. Emerging chemistry and biology in protein glutathionylation. Curr. Opin. Chem. Biol. 2022, 71, 102221. [Google Scholar] [CrossRef] [PubMed]
- Winterbourn, C.C. The biological chemistry of hydrogen peroxide. Methods Enzymol. 2013, 528, 3–25. [Google Scholar]
- Holmgren, A. Hydrogen donor system for Escherichia coli ribonucleoside-diphosphate reductase dependent upon gluta-thione. Proc. Natl. Acad. Sci. USA 1976, 73, 2275–2279. [Google Scholar] [CrossRef] [Green Version]
- Mannervik, B.; Axelsson, K. Role of cytoplasmic thioltransferase in cellular regulation by thiol-disulphide interchange. Bio-chem. J. 1980, 190, 125–130. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Boja, E.S.; Tan, W.; Tekle, E.; Fales, H.M.; English, S.; Mieyal, J.J.; Chock, P.B. Reversible Glutathionylation Regulates Actin Polymerization in A431 Cells. J. Biol. Chem. 2001, 276, 47763–47766. [Google Scholar] [CrossRef] [Green Version]
- Sakai, J.; Li, J.; Subramanian, K.K.; Mondal, S.; Bajrami, B.; Hattori, H.; Jia, Y.; Dickinson, B.C.; Zhong, J.; Ye, K.; et al. Reactive Oxygen Species-Induced Actin Glutathionylation Controls Actin Dynamics in Neutrophils. Immunity 2012, 37, 1037–1049. [Google Scholar] [CrossRef] [Green Version]
- Stojkov, D.; Amini, P.; Oberson, K.; Sokollik, C.; Duppenthaler, A.; Simon, H.-U.; Yousefi, S. ROS and glutathionylation balance cytoskeletal dynamics in neutrophil extracellular trap formation. J. Cell Biol. 2017, 216, 4073–4090. [Google Scholar] [CrossRef]
- Forman, H.J.; Ursini, F.; Maiorino, M. An overview of mechanisms of redox signaling. J. Mol. Cell. Cardiol. 2014, 73, 2–9. [Google Scholar] [CrossRef] [Green Version]
- Mailloux, R.J.; Jin, X.; Willmore, W.G. Redox regulation of mitochondrial function with emphasis on cysteine oxidation reactions. Redox Biol. 2014, 2, 123–139. [Google Scholar] [CrossRef] [PubMed]
- Montagna, C.; Cirotti, C.; Rizza, S.; Filomeni, G. WhenS-Nitrosylation Gets to Mitochondria: From Signaling to Age-Related Diseases. Antioxid. Redox Signal. 2020, 32, 884–905. [Google Scholar] [CrossRef] [PubMed]
- Hildebrandt, T.; Knuesting, J.; Berndt, C.; Morgan, B.; Scheibe, R. Cytosolic thiol switches regulating basic cellular functions: GAPDH as an information hub? Biol. Chem. 2015, 396, 523–537. [Google Scholar] [CrossRef] [PubMed]
- Miljkovic, J.L.; Burger, N.; Gawel, J.M.; Mulvey, J.F.; Norman, A.A.; Nishimura, T.; Tsujihata, Y.; Logan, A.; Sauchanka, O.; Caldwell, S.T.; et al. Rapid and selective generation of H2S within mitochondria protects against cardiac ischemia-reperfusion injury. Redox Biol. 2022, 55, 102429. [Google Scholar] [CrossRef] [PubMed]
- Mieyal, J.J.; Chock, P.B. Posttranslational Modification of Cysteine in Redox Signaling and Oxidative Stress: Focus on S-Glutathionylation. Antioxid. Redox Signal. 2012, 16, 471–475. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pal, D.; Rai, A.; Checker, R.; Patwardhan, R.; Singh, B.; Sharma, D.; Sandur, S.K. Role of protein S-Glutathionylation in cancer progression and development of resistance to anti-cancer drugs. Arch. Biochem. Biophys. 2021, 704, 108890. [Google Scholar] [CrossRef] [PubMed]
- Ukuwela, A.A.; Bush, A.I.; Wedd, A.G.; Xiao, Z. Reduction potentials of protein disulfides and catalysis of glutathionylation and deglutathionylation by glutaredoxin enzymes. Biochem. J. 2017, 474, 3799–3815. [Google Scholar] [CrossRef]
- Begas, P.; Liedgens, L.; Moseler, A.; Meyer, A.J.; Deponte, M. Glutaredoxin catalysis requires two distinct glutathione inter-action sites. Nat. Commun. 2017, 8, 14835. [Google Scholar] [CrossRef] [Green Version]
- Can, B.; Erkmen, G.K.; Dalmizrak, O.; Ogus, I.H.; Ozer, N. Purification and Characterisation of Rat Kidney Glutathione Re-ductase. J. Protein Chem. 2010, 29, 250–256. [Google Scholar] [CrossRef]
- Mailloux, R.J.; McBride, S.L.; Harper, M.-E. Unearthing the secrets of mitochondrial ROS and glutathione in bioenergetics. Trends Biochem. Sci. 2013, 38, 592–602. [Google Scholar] [CrossRef]
- Lundberg, M.; Johansson, C.; Chandra, J.; Enoksson, M.; Jacobsson, G.; Ljung, J.; Johansson, M.; Holmgren, A. Cloning and Expression of a Novel Human Glutaredoxin (Grx2) with Mitochondrial and Nuclear Isoforms. J. Biol. Chem. 2001, 276, 26269–26275. [Google Scholar] [CrossRef] [PubMed]
- Gladyshev, V.N.; Liu, A.; Novoselov, S.V.; Krysan, K.; Sun, Q.-A.; Kryukov, V.M.; Kryukov, G.V.; Lou, M.F. Identification and Characterization of a New Mammalian Glutaredoxin (Thioltransferase), Grx2. J. Biol. Chem. 2001, 276, 30374–30380. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Piemonte, F.; Passarelli, C.; Tozzi, G.; Pastore, A.; Bertini, E. GSSG-mediated Complex I defect in isolated cardiac mitochondria. Int. J. Mol. Med. 2010, 26, 95–99. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gill, R.; Mallay, S.; Young, A.; Mailloux, R.J. An investigation into the impact of deleting one copy of the glutaredoxin-2 gene on diet-induced weight gain and the bioenergetics of muscle mitochondria in female mice fed a high fat diet. Redox Rep. 2020, 25, 87–94. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.; Lin, L.; Giblin, F.; Ho, Y.-S.; Lou, M.F. Glutaredoxin 2 knockout increases sensitivity to oxidative stress in mouse lens epithelial cells. Free Radic. Biol. Med. 2011, 51, 2108–2117. [Google Scholar] [CrossRef] [Green Version]
- Wang, K.; Hirschenson, J.; Moore, A.; Mailloux, R.J. Conditions Conducive to the Glutathionylation of Complex I Subunit NDUFS1 Augment ROS Production following the Oxidation of Ubiquinone Linked Substrates, Glycerol-3-Phosphate and Proline. Antioxidants 2022, 11, 2043. [Google Scholar] [CrossRef]
- Chen, J.; Chen, C.-L.; Rawale, S.; Chen, C.-A.; Zweier, J.L.; Kaumaya, P.T.; Chen, Y.-R. Peptide-based Antibodies against Glutathione-binding Domains Suppress Superoxide Production Mediated by Mitochondrial Complex I. J. Biol. Chem. 2010, 285, 3168–3180. [Google Scholar] [CrossRef] [Green Version]
- Hurd, T.; Requejo, R.; Filipovska, A.; Brown, S.; Prime, T.; Robinson, A.; Fearnley, I.; Murphy, M. Complex I within oxidatively stressed bovine heart mitochondria is glutathionylated on Cys-531 and Cys-704 of the 75-kDa subunit: Potential role of CYS residues in decreasing oxidative damage. J. Biol. Chem. 2008, 283, 24801–24815. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, V.; Kleffmann, T.; Hampton, M.B.; Cannell, M.B.; Winterbourn, C.C. Redox proteomics of thiol proteins in mouse heart during ischemia/reperfusion using ICAT reagents and mass spectrometry. Free Radic. Biol. Med. 2013, 58, 109–117. [Google Scholar] [CrossRef]
- Diotte, N.M.; Xiong, Y.; Gao, J.; Chua, B.H.; Ho, Y.-S. Attenuation of doxorubicin-induced cardiac injury by mitochondrial glutaredoxin 2. Biochim. Biophys. Acta (BBA)—Mol. Cell Res. 2009, 1793, 427–438. [Google Scholar] [CrossRef] [Green Version]
- Hurd, T.; Costa, N.; Dahm, C.; Beer, S.; Brown, S.; Filipovska, A.; Murphy, M. Glutathionylation of mitochondrial proteins. Antioxid. Redox Signal. 2005, 7, 999–1010. [Google Scholar] [CrossRef] [PubMed]
- Massey, V. Activation of molecular oxygen by flavins and flavoproteins. J. Biol. Chem. 1994, 269, 22459–22462. [Google Scholar] [CrossRef] [PubMed]
- Starkov, A.; Fiskum, G.; Chinopoulos, C.; Lorenzo, B.J.; Browne, S.E.; Patel, M.S.; Beal, M.F. Mitochondrial -Ketoglutarate Dehydrogenase Complex Generates Reactive Oxygen Species. J. Neurosci. 2004, 24, 7779–7788. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mailloux, R.; Gardiner, D.; O’Brien, M. 2-Oxoglutarate dehydrogenase is a more significant source of O2(.-)/H2O2 than py-ruvate dehydrogenase in cardiac and liver tissue. Free Radic. Biol. Med. 2016, 97, 501–512. [Google Scholar]
- Murphy, M.P. How mitochondria produce reactive oxygen species. Biochem. J. 2009, 417, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Francisco, A.; Figueira, T.R.; Castilho, R.F. Mitochondrial NAD(P)+ Transhydrogenase: From Molecular Features to Physiology and Disease. Antioxid. Redox Signal. 2022, 36, 864–884. [Google Scholar] [CrossRef]
- Korshunov, S.S.; Skulachev, V.P.; Starkov, A.A. High protonic potential actuates a mechanism of production of reactive oxygen species in mitochondria. FEBS Lett. 1997, 416, 15–18. [Google Scholar] [CrossRef] [Green Version]
- Young, A.; Gill, R.; Mailloux, R.J. Protein S-glutathionylation: The linchpin for the transmission of regulatory information on redox buffering capacity in mitochondria. Chem. Interact. 2018, 299, 151–162. [Google Scholar] [CrossRef]
- Mailloux, R.J.; Treberg, J.R. Protein S-glutathionlyation links energy metabolism to redox signaling in mitochondria. Redox Biol. 2016, 8, 110–118. [Google Scholar] [CrossRef] [Green Version]
- Doulias, P.-T.; Tenopoulou, M.; Greene, J.L.; Raju, K.; Ischiropoulos, H. Nitric Oxide Regulates Mitochondrial Fatty Acid Metabolism Through Reversible Protein S -Nitrosylation. Sci. Signal. 2013, 6, rs1. [Google Scholar] [CrossRef] [Green Version]
- Randi, E.B.; Zuhra, K.; Pecze, L.; Panagaki, T.; Szabo, C. Physiological concentrations of cyanide stimulate mitochondrial Complex IV and enhance cellular bioenergetics. Proc. Natl. Acad. Sci. USA 2021, 118, e2026245118. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Foster, D.; Rucker, J.; O’Rourke, B.; Kass, D.; Van Eyk, J. Redox regulation of mitochondrial ATP synthase: Impli-cations for cardiac resynchronization therapy. Circ. Res. 2011, 109, 750–757. [Google Scholar] [CrossRef] [PubMed]
- Vanhecke, G.C.; Abeywardana, M.Y.; Ahn, Y.-H. Proteomic Identification of Protein Glutathionylation in Cardiomyocytes. J. Proteome Res. 2019, 18, 1806–1818. [Google Scholar] [CrossRef] [PubMed]
- Bleier, L.; Wittig, I.; Heide, H.; Steger, M.; Brandt, U.; Dröse, S. Generator-specific targets of mitochondrial reactive oxygen species. Free Radic. Biol. Med. 2015, 78, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Chouchani, E.T.; Kazak, L.; Jedrychowski, M.P.; Lu, G.Z.; Erickson, B.K.; Szpyt, J.; Pierce, K.A.; Laznik-Bogoslavski, D.; Vetrivelan, R.; Clish, C.B.; et al. Mitochondrial ROS regulate thermogenic energy expenditure and sulfenylation of UCP1. Nature 2016, 532, 112–116. [Google Scholar] [CrossRef] [Green Version]
- Scialo’, F.; Sriram, A.; Fernández-Ayala, D.J.M.; Gubina, N.; Lõhmus, M.; Nelson, G.; Logan, A.; Cooper, H.M.; Navas, P.; Enríquez, J.A.; et al. Mitochondrial ROS Produced via Reverse Electron Transport Extend Animal Lifespan. Cell Metab. 2016, 23, 725–734. [Google Scholar] [CrossRef] [Green Version]
- Scialò, F.; Sriram, A.; Stefanatos, R.; Spriggs, R.V.; Loh, S.H.; Martins, L.M.; Sanz, A. Mitochondrial complex I derived ROS regulate stress adaptation in Drosophila melanogaster. Redox Biol. 2020, 32, 101450. [Google Scholar] [CrossRef]
- Jackson, M.J.; Stretton, C.; McArdle, A. Hydrogen peroxide as a signal for skeletal muscle adaptations to exercise: What do concentrations tell us about potential mechanisms? Redox Biol. 2020, 35, 101484. [Google Scholar] [CrossRef]
- Taylor, E.R.; Hurrell, F.; Shannon, R.J.; Lin, T.-K.; Hirst, J.; Murphy, M.P. Reversible Glutathionylation of Complex I Increases Mitochondrial Superoxide Formation. J. Biol. Chem. 2003, 278, 19603–19610. [Google Scholar] [CrossRef] [Green Version]
- Mailloux, R.J.; Willmore, W.G. S-glutathionylation reactions in mitochondrial function and disease. Front. Cell Dev. Biol. 2014, 2, 68. [Google Scholar] [CrossRef] [Green Version]
- Mimaki, M.; Wang, X.; McKenzie, M.; Thorburn, D.R.; Ryan, M.T. Understanding mitochondrial complex I assembly in health and disease. Biochim. Biophys. Acta (BBA)-Bioenerg. 2012, 1817, 851–862. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hirschenson, J.; Mailloux, R.J. The glutathionylation agent disulfiram augments superoxide/hydrogen peroxide production when liver mitochondria are oxidizing ubiquinone pool-linked and branched chain amino acid substrates. Free Radic. Biol. Med. 2021, 172, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Chouchani, E.; Pell, V.; Gaude, E.; Aksentijevic, D.; Sundier, S.; Robb, E.; Logan, A.; Nadtochiy, S.; Ord, E.; Smith, A.; et al. Ischaemic accumulation of succinate controls reperfusion injury through mitochondrial ROS. Nature 2014, 515, 431–435. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kanaan, G.N.; Ichim, B.; Gharibeh, L.; Maharsy, W.; Patten, D.A.; Xuan, J.Y.; Reunov, A.; Marshall, P.; Veinot, J.; Menzies, K.; et al. Glutaredoxin-2 controls cardiac mitochondrial dynamics and energetics in mice, and protects against human cardiac pa-thologies. Redox Biol. 2018, 14, 509–521. [Google Scholar] [CrossRef]
- Tretter, L.; Adam-Vizi, V. Generation of reactive oxygen species in the reaction catalyzed by alpha-ketoglutarate dehydro-genase. J. Neurosci. 2004, 24, 7771–7778. [Google Scholar] [CrossRef] [Green Version]
- Quinlan, C.L.; Goncalves, R.L.; Hey-Mogensen, M.; Yadava, N.; Bunik, V.I.; Brand, M.D. The 2-Oxoacid Dehydrogenase Complexes in Mitochondria Can Produce Superoxide/Hydrogen Peroxide at Much Higher Rates Than Complex I. J. Biol. Chem. 2014, 289, 8312–8325. [Google Scholar] [CrossRef] [Green Version]
- Slade, L.; Chalker, J.; Kuksal, N.; Young, A.; Gardiner, D.; Mailloux, R.J. Examination of the superoxide/hydrogen peroxide forming and quenching potential of mouse liver mitochondria. Biochim. Biophys. Acta (BBA)—Gen. Subj. 2017, 1861, 1960–1969. [Google Scholar] [CrossRef]
- Humphries, K.M.; Szweda, L.I. Selective Inactivation of α-Ketoglutarate Dehydrogenase and Pyruvate Dehydrogenase: Re-action of Lipoic Acid with 4-Hydroxy-2-nonenal. Biochemistry 1998, 37, 15835–15841. [Google Scholar] [CrossRef]
- McLain, A.L.; Szweda, P.A.; Szweda, L.I. α-Ketoglutarate dehydrogenase: A mitochondrial redox sensor. Free Radic. Res. 2010, 45, 29–36. [Google Scholar] [CrossRef] [Green Version]
- Tretter, L.; Adam-Vizi, V. Alpha-ketoglutarate dehydrogenase: A target and generator of oxidative stress. Philos. Trans. R. Soc. B Biol. Sci. 2005, 360, 2335–2345. [Google Scholar] [CrossRef] [Green Version]
- McLain, A.L.; Cormier, P.J.; Kinter, M.; Szweda, L.I. Glutathionylation of α-ketoglutarate dehydrogenase: The chemical nature and relative susceptibility of the cofactor lipoic acid to modification. Free Radic. Biol. Med. 2013, 61, 161–169. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nulton-Persson, A.C.; Starke, D.W.; Mieyal, J.J.; Szweda, L.I. Reversible Inactivation of α-Ketoglutarate Dehydrogenase in Response to Alterations in the Mitochondrial Glutathione Status. Biochemistry 2003, 42, 4235–4242. [Google Scholar] [CrossRef]
- Applegate, M.A.B.; Humphries, K.M.; Szweda, L.I. Reversible Inhibition of α-Ketoglutarate Dehydrogenase by Hydrogen Peroxide: Glutathionylation and Protection of Lipoic Acid. Biochemistry 2008, 47, 473–478. [Google Scholar] [CrossRef] [PubMed]
- Wagner, M.; Bertero, E.; Nickel, A.; Kohlhaas, M.; Gibson, G.E.; Heggermont, W.; Heymans, S.; Maack, C. Selective NADH communication from α-ketoglutarate dehydrogenase to mitochondrial transhydrogenase prevents reactive oxygen species formation under reducing conditions in the heart. Basic Res. Cardiol. 2020, 115, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Fisher-Wellman, K.H.; Lin, C.-T.; Ryan, T.E.; Reese, L.R.; Gilliam, L.A.A.; Cathey, B.L.; Lark, D.; Smith, C.D.; Muoio, D.; Neufer, P.D. Pyruvate dehydrogenase complex and nicotinamide nucleotide transhydrogenase constitute an energy-consuming redox circuit. Biochem. J. 2015, 467, 271–280. [Google Scholar] [CrossRef]
- Smith, C.D.; Schmidt, C.A.; Lin, C.-T.; Fisher-Wellman, K.H.; Neufer, P.D. Flux through mitochondrial redox circuits linked to nicotinamide nucleotide transhydrogenase generates counterbalance changes in energy expenditure. J. Biol. Chem. 2020, 295, 16207–16216. [Google Scholar] [CrossRef] [PubMed]
- Figueira, T.R.; Francisco, A.; Treberg, J.R.; Castilho, R.F. Can NAD(P)+ transhydrogenase (NNT) mediate a physiologically meaningful increase in energy expenditure by mitochondria during H2O2 removal? J. Biol. Chem. 2021, 296, 100377. [Google Scholar] [CrossRef] [PubMed]
- Kampjut, D.; Sazanov, L.A. Structure and mechanism of mitochondrial proton-translocating transhydrogenase. Nature 2019, 573, 291–295. [Google Scholar] [CrossRef]
- Zachar, Z.; Marecek, J.; Maturo, C.; Gupta, S.; Stuart, S.D.; Howell, K.; Schauble, A.; Lem, J.; Piramzadian, A.; Karnik, S.; et al. Non-redox-active lipoate derivates disrupt cancer cell mitochondrial metabolism and are potent anticancer agents in vivo. J. Mol. Med. 2011, 89, 1137–1148. [Google Scholar] [CrossRef]
- Koufos, O.; Mailloux, R.J. Protein S-glutathionylation and sex dimorphic effects on hydrogen peroxide production by dihy-droorotate dehydrogenase in liver mitochondria. Free Radic. Biol. Med. 2022, 194, 123–130. [Google Scholar] [CrossRef]
- Zhou, Y.; Tao, L.; Zhou, X.; Zuo, Z.; Gong, J.; Liu, X.; Zhou, Y.; Liu, C.; Na Sang, N.; Liu, H.; et al. DHODH and cancer: Promising prospects to be explored. Cancer Metab. 2021, 9, 1–25. [Google Scholar] [CrossRef] [PubMed]
- Hey-Mogensen, M.; Goncalves, R.L.; Orr, A.L.; Brand, M.D. Production of superoxide/H2O2 by dihydroorotate dehydrogenase in rat skeletal muscle mitochondria. Free Radic. Biol. Med. 2014, 72, 149–155. [Google Scholar] [CrossRef] [PubMed]
- Mallay, S.; Gill, R.; Young, A.; Mailloux, R.J. Sex-dependent Differences in the Bioenergetics of Liver and Muscle Mitochondria from Mice Containing a Deletion for glutaredoxin-2. Antioxidants 2019, 8, 245. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pfefferle, A.; Mailloux, R.J.; Adjeitey, C.N.-K.; Harper, M.-E. Glutathionylation of UCP2 sensitizes drug resistant leukemia cells to chemotherapeutics. Biochim. Biophys. Acta (BBA)—Mol. Cell Res. 2013, 1833, 80–89. [Google Scholar] [CrossRef]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mailloux, R.J.; Grayson, C.; Koufos, O. Regulation of Mitochondrial Hydrogen Peroxide Availability by Protein S-glutathionylation. Cells 2023, 12, 107. https://doi.org/10.3390/cells12010107
Mailloux RJ, Grayson C, Koufos O. Regulation of Mitochondrial Hydrogen Peroxide Availability by Protein S-glutathionylation. Cells. 2023; 12(1):107. https://doi.org/10.3390/cells12010107
Chicago/Turabian StyleMailloux, Ryan J., Cathryn Grayson, and Olivia Koufos. 2023. "Regulation of Mitochondrial Hydrogen Peroxide Availability by Protein S-glutathionylation" Cells 12, no. 1: 107. https://doi.org/10.3390/cells12010107
APA StyleMailloux, R. J., Grayson, C., & Koufos, O. (2023). Regulation of Mitochondrial Hydrogen Peroxide Availability by Protein S-glutathionylation. Cells, 12(1), 107. https://doi.org/10.3390/cells12010107