

A Novel Method for Primary Blood Cell Culturing and Selection in Drosophila melanogaster


 , , and
, , and
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Drosophila Stocks and Maintenance
2.2. Wasp Infestation and Wounding
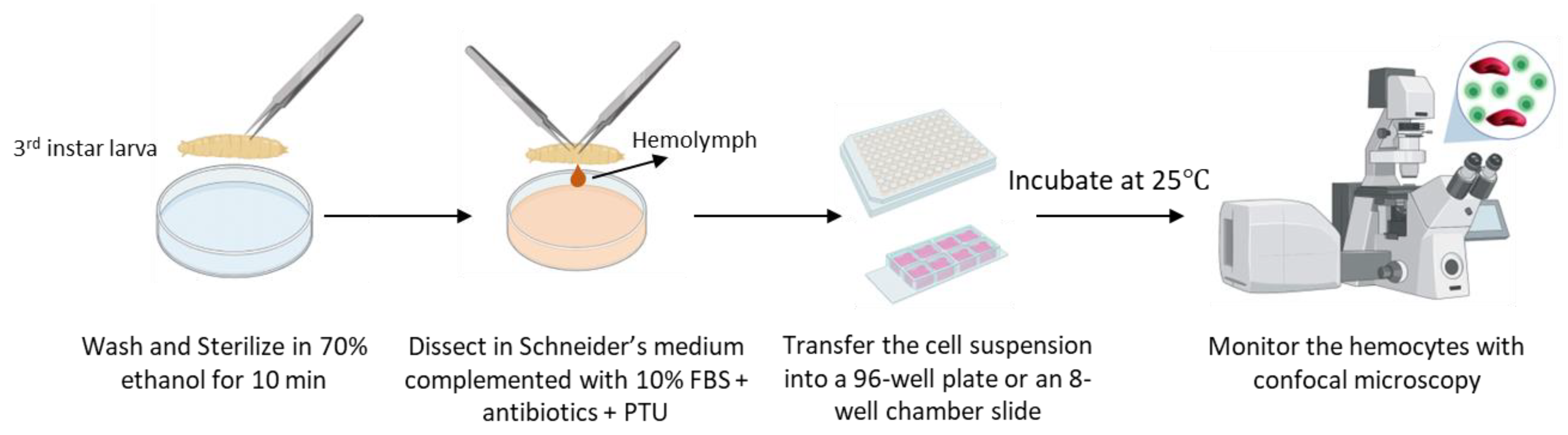
2.3. Ex Vivo Culturing of Isolated Hemocytes
2.4. Video Microscopy
2.5. Generation of the UAS-BsdR Line
2.6. Blasticidin Treatment of Cell Cultures
2.7. Cell Counting, Cell Size Measurement and Statistics
3. Results
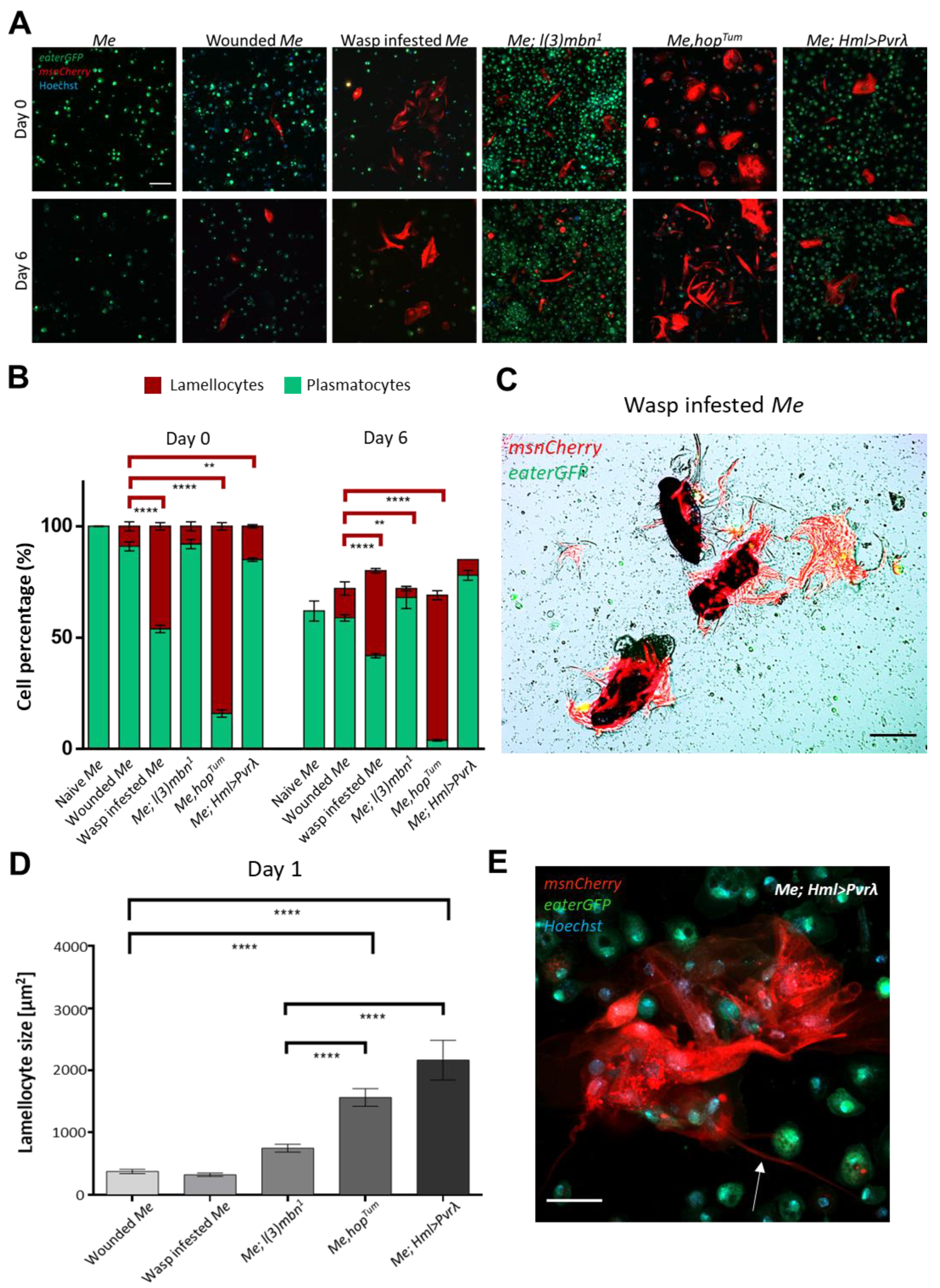
3.1. Hemocyte Composition and Morphology in Primary Cell Cultures Depends on the Genotype and the Immune Condition of the Larva
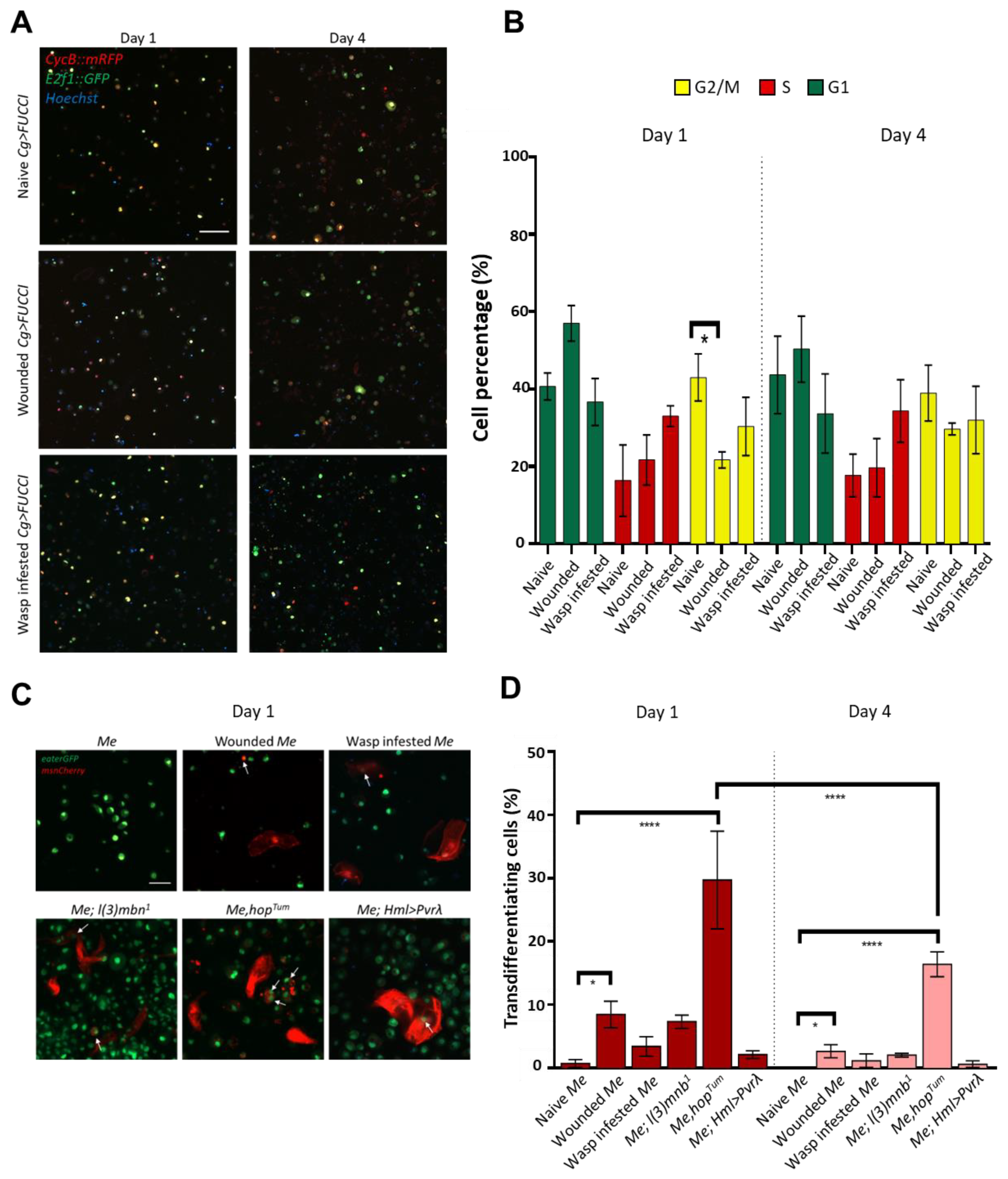
3.2. Cultured Hemocytes Maintain Their Proliferation and Transdifferentiation Capacity
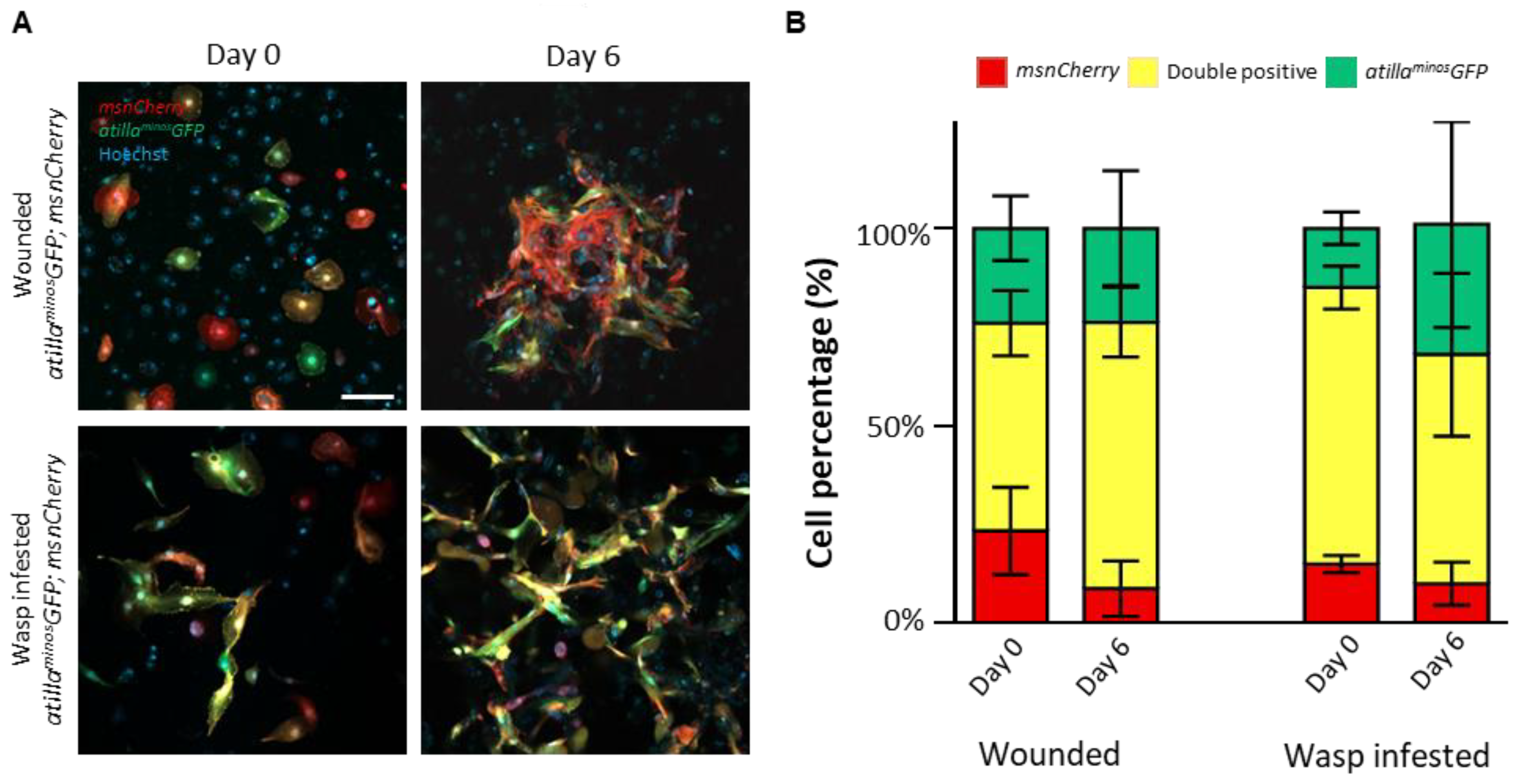
3.3. Primary Hemocyte Cultures Contain Heterogeneous Lamellocyte Subsets
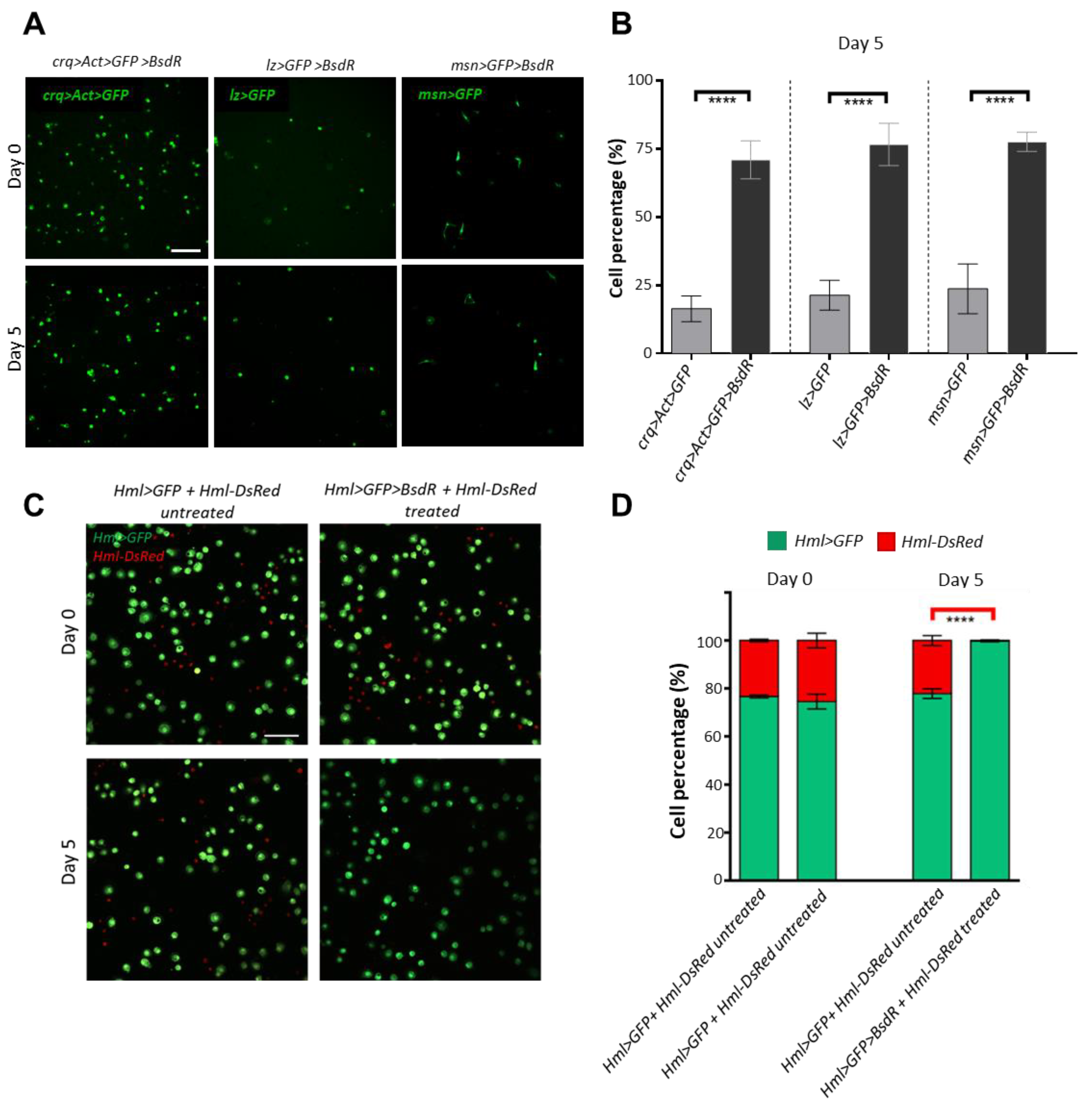
3.4. Construction of a Blasticidin Resistance-Based Blood Cell Selection System
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hultmark, D. Insect Immunology. Ancient Relationships. Nature 1994, 367, 116–117. [Google Scholar] [CrossRef] [PubMed]
- Evans, C.J.; Hartenstein, V.; Banerjee, U. Thicker than Blood: Conserved Mechanisms in Drosophila and Vertebrate Hematopoiesis. Dev. Cell 2003, 5, 673–690. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Akira, S.; Uematsu, S.; Takeuchi, O. Pathogen Recognition and Innate Immunity. Cell 2006, 124, 783–801. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Honti, V.; Csordás, G.; Kurucz, É.; Márkus, R.; Andó, I. The Cell-Mediated Immunity of Drosophila melanogaster: Hemocyte Lineages, Immune Compartments, Microanatomy and Regulation. Dev. Comp. Immunol. 2014, 42, 47–56. [Google Scholar] [CrossRef]
- Kleino, A.; Silverman, N. The Drosophila IMD Pathway in the Activation of the Humoral Immune Response. Dev. Comp. Immunol. 2014, 42, 25–35. [Google Scholar] [CrossRef] [Green Version]
- Banerjee, U.; Girard, J.R.; Goins, L.M.; Spratford, C.M. Drosophila as a Genetic Model for Hematopoiesis. Genetics 2019, 211, 367–417. [Google Scholar] [CrossRef] [Green Version]
- Hultmark, D.; Andó, I. Hematopoietic Plasticity Mapped in Drosophila and Other Insects. eLife 2022, 11, e78906. [Google Scholar] [CrossRef]
- Lanot, R.; Zachary, D.; Holder, F.; Meister, M. Postembryonic Hematopoiesis in Drosophila. Dev. Biol. 2001, 230, 243–257. [Google Scholar] [CrossRef] [Green Version]
- Holz, A.; Bossinger, B.; Strasser, T.; Janning, W.; Klapper, R. The Two Origins of Hemocytes in Drosophila. Development 2003, 130, 4955–4962. [Google Scholar] [CrossRef] [Green Version]
- Roehrborn, G. Drosophila Tumors and the Structure of Larval Lymph Glands. Experientia 1961, 17, 507–509. [Google Scholar] [CrossRef]
- Shrestha, R.; Gateff, E. Ultrastructure and Cytochemistry of the Cell-Types in the Tumorous Hematopoietic Organs and the Hemolymph of the Mutant Lethal (1) Malignant Blood Neoplasm (l(1)Mbn) of Drosophila melanogaster. Dev. Growth Differ. 1982, 24, 83–98. [Google Scholar] [CrossRef]
- Jung, S.-H.; Evans, C.J.; Uemura, C.; Banerjee, U. The Drosophila Lymph Gland as a Developmental Model of Hematopoiesis. Development 2005, 132, 2521–2533. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Márkus, R.; Laurinyecz, B.; Kurucz, E.; Honti, V.; Bajusz, I.; Sipos, B.; Somogyi, K.; Kronhamn, J.; Hultmark, D.; Andó, I. Sessile Hemocytes as a Hematopoietic Compartment in Drosophila Melanogaster. Proc. Natl. Acad. Sci. USA 2009, 106, 4805–4809. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krzemien, J.; Oyallon, J.; Crozatier, M.; Vincent, A. Hematopoietic Progenitors and Hemocyte Lineages in the Drosophila Lymph Gland. Dev. Biol. 2010, 346, 310–319. [Google Scholar] [CrossRef] [Green Version]
- Makhijani, K.; Alexander, B.; Tanaka, T.; Rulifson, E.; Brückner, K. The Peripheral Nervous System Supports Blood Cell Homing and Survival in the Drosophila Larva. Development 2011, 138, 5379–5391. [Google Scholar] [CrossRef] [Green Version]
- Rizki, T.M.; Rizki, R.M. Properties of the Larval Hemocytes of Drosophila melanogaster. Experientia 1980, 36, 1223–1226. [Google Scholar] [CrossRef] [Green Version]
- Letourneau, M.; Lapraz, F.; Sharma, A.; Vanzo, N.; Waltzer, L.; Crozatier, M. Drosophila Hematopoiesis under Normal Conditions and in Response to Immune Stress. FEBS Lett. 2016, 590, 4034–4051. [Google Scholar] [CrossRef]
- Csordás, G.; Gábor, E.; Honti, V. There and Back Again: The Mechanisms of Differentiation and Transdifferentiation in Drosophila Blood Cells. Dev. Biol. 2021, 469, 135–143. [Google Scholar] [CrossRef]
- Brehélin, M. Comparative Study of Structure and Function of Blood Cells from Two Drosophila Species. Cell Tissue Res. 1982, 221, 607–615. [Google Scholar] [CrossRef]
- Dudzic, J.P.; Kondo, S.; Ueda, R.; Bergman, C.M.; Lemaitre, B. Drosophila Innate Immunity: Regional and Functional Specialization of Prophenoloxidases. BMC Biol. 2015, 13, 81. [Google Scholar] [CrossRef]
- Anderl, I.; Vesala, L.; Ihalainen, T.O.; Vanha-Aho, L.-M.; Andó, I.; Rämet, M.; Hultmark, D. Transdifferentiation and Proliferation in Two Distinct Hemocyte Lineages in Drosophila melanogaster Larvae after Wasp Infection. PLoS Pathog. 2016, 12, e1005746. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Avet-Rochex, A.; Boyer, K.; Polesello, C.; Gobert, V.; Osman, D.; Roch, F.; Augé, B.; Zanet, J.; Haenlin, M.; Waltzer, L. An in Vivo RNA Interference Screen Identifies Gene Networks Controlling Drosophila melanogaster Blood Cell Homeostasis. BMC Dev. Biol. 2010, 10, 65. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Honti, V.; Csordás, G.; Márkus, R.; Kurucz, E.; Jankovics, F.; Andó, I. Cell Lineage Tracing Reveals the Plasticity of the Hemocyte Lineages and of the Hematopoietic Compartments in Drosophila melanogaster. Mol. Immunol. 2010, 47, 1997–2004. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stofanko, M.; Kwon, S.Y.; Badenhorst, P. Lineage Tracing of Lamellocytes Demonstrates Drosophila Macrophage Plasticity. PLoS ONE 2010, 5, e14051. [Google Scholar] [CrossRef] [Green Version]
- Liu, Z.-Z.; Sun, G.-Q.; Hu, X.-H.; Kwak-Kim, J.; Liao, A.-H. The Transdifferentiation of Regulatory T and Th17 Cells in Autoimmune/Inflammatory Diseases and Its Potential Implications in Pregnancy Complications. Am. J. Reprod. Immunol. 2017, 78, 2. [Google Scholar] [CrossRef]
- Agalioti, T.; Villablanca, E.J.; Huber, S.; Gagliani, N. TH17 cell Plasticity: The Role of Dendritic Cells and Molecular Mechanisms. J. Autoimmun. 2018, 87, 50–60. [Google Scholar] [CrossRef]
- Cattenoz, P.B.; Sakr, R.; Pavlidaki, A.; Delaporte, C.; Riba, A.; Molina, N.; Hariharan, N.; Mukherjee, T.; Giangrande, A. Temporal Specificity and Heterogeneity of Drosophila Immune Cells. EMBO J. 2020, 39, e104486. [Google Scholar] [CrossRef]
- Tattikota, S.G.; Cho, B.; Liu, Y.; Hu, Y.; Barrera, V.; Steinbaugh, M.J.; Yoon, S.-H.; Comjean, A.; Li, F.; Dervis, F.; et al. A Single-Cell Survey of Drosophila Blood. eLife 2020, 9, e54818. [Google Scholar] [CrossRef]
- Hanratty, W.P.; Ryerse, J.S. A Genetic Melanotic Neoplasm of Drosophila Melanogaster. Dev. Biol. 1981, 83, 238–249. [Google Scholar] [CrossRef]
- Luo, H.; Hanratty, W.P.; Dearolf, C.R. An Amino Acid Substitution in the Drosophila HopTum-l Jak Kinase Causes Leukemia-like Hematopoietic Defects. EMBO J. 1995, 14, 1412–1420. [Google Scholar] [CrossRef]
- Zettervall, C.-J.; Anderl, I.; Williams, M.J.; Palmer, R.; Kurucz, E.; Ando, I.; Hultmark, D. A Directed Screen for Genes Involved in Drosophila Blood Cell Activation. Proc. Natl. Acad. Sci. USA 2004, 101, 14192–14197. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmid, M.R.; Anderl, I.; Vesala, L.; Vanha-aho, L.-M.; Deng, X.-J.; Rämet, M.; Hultmark, D. Control of Drosophila Blood Cell Activation via Toll Signaling in the Fat Body. PLoS ONE 2014, 9, e102568. [Google Scholar] [CrossRef] [Green Version]
- Kurucz, E.; Zettervall, C.-J.; Sinka, R.; Vilmos, P.; Pivarcsi, A.; Ekengren, S.; Hegedüs, Z.; Ando, I.; Hultmark, D. Hemese, a Hemocyte-Specific Transmembrane Protein, Affects the Cellular Immune Response in Drosophila. Proc. Natl. Acad. Sci. USA 2003, 100, 2622–2627. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sinenko, S.A.; Mathey-Prevot, B. Increased expression of Drosophila tetraspanin, Tsp68C, suppresses the abnormal proliferation of ytr-deficient and Ras/Raf-activated hemocytes. Oncogene 2004, 23, 9120–9128. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kurucz, E.; Márkus, R.; Zsámboki, J.; Folkl-Medzihradszky, K.; Darula, Z.; Vilmos, P.; Udvardy, A.; Krausz, I.; Lukacsovich, T.; Gateff, E.; et al. Nimrod, a Putative Phagocytosis Receptor with EGF Repeats in Drosophila Plasmatocytes. Curr. Biol. 2007, 17, 649–654. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kurucz, E.; Váczi, B.; Márkus, R.; Laurinyecz, B.; Vilmos, P.; Zsámboki, J.; Csorba, K.; Gateff, E.; Hultmark, D.; Andó, I. Definition of Drosophila Hemocyte Subsets by Cell-Type Specific Antigens. Acta Biol. Hung. 2007, 58, 95–111. [Google Scholar] [CrossRef] [Green Version]
- Honti, V.; Kurucz, E.; Csordás, G.; Laurinyecz, B.; Márkus, R.; Andó, I. In Vivo Detection of Lamellocytes in Drosophila Melanogaster. Immunol. Lett. 2009, 126, 83–84. [Google Scholar] [CrossRef] [Green Version]
- Tokusumi, T.; Shoue, D.A.; Tokusumi, Y.; Stoller, J.R.; Schulz, R.A. New Hemocyte-Specific Enhancer-Reporter Transgenes for the Analysis of Hematopoiesis in Drosophila. Genesis 2009, 47, 771–774. [Google Scholar] [CrossRef]
- Tokusumi, T.; Tokusumi, Y.; Brahier, M.S.; Lam, V.; Stoller-Conrad, J.R.; Kroeger, P.T.; Schulz, R.A. Screening and Analysis of Janelia FlyLight Project Enhancer-Gal4 Strains Identifies Multiple Gene Enhancers Active During Hematopoiesis in Normal and Wasp-Challenged Drosophila Larvae. G3 Genes Genomes Genet. 2017, 7, 437–448. [Google Scholar] [CrossRef] [Green Version]
- Szkalisity, A.; Piccinini, F.; Beleon, A.; Balassa, T.; Varga, I.G.; Migh, E.; Molnar, C.; Paavolainen, L.; Timonen, S.; Banerjee, I.; et al. Regression Plane Concept for Analysing Continuous Cellular Processes with Machine Learning. Nat. Commun. 2021, 12, 2532. [Google Scholar] [CrossRef]
- Li, M.Z.; Elledge, S.J. SLIC: A Method for Sequence- and Ligation-Independent Cloning. Methods Mol. Biol. 2012, 852, 51–59. [Google Scholar] [CrossRef] [PubMed]
- Konrad, L.; Becker, G.; Schmidt, A.; Klöckner, T.; Kaufer-Stillger, G.; Dreschers, S.; Edström, J.E.; Gateff, E. Cloning, Structure, Cellular Localization, and Possible Function of the Tumor Suppressor Gene Lethal(3)Malignant Blood Neoplasm-1 of Drosophila Melanogaster. Dev. Biol. 1994, 163, 98–111. [Google Scholar] [CrossRef] [PubMed]
- Márkus, R.; Kurucz, E.; Rus, F.; Andó, I. Sterile Wounding Is a Minimal and Sufficient Trigger for a Cellular Immune Response in Drosophila Melanogaster. Immunol. Lett. 2005, 101, 108–111. [Google Scholar] [CrossRef]
- Asha, H.; Nagy, I.; Kovacs, G.; Stetson, D.; Ando, I.; Dearolf, C.R. Analysis of Ras-Induced Overproliferation in Drosophila Hemocytes. Genetics 2003, 163, 203–215. [Google Scholar] [CrossRef] [PubMed]
- Zielke, N.; Korzelius, J.; van Straaten, M.; Bender, K.; Schuhknecht, G.F.P.; Dutta, D.; Xiang, J.; Edgar, B.A. Fly-FUCCI: A Versatile Tool for Studying Cell Proliferation in Complex Tissues. Cell Rep. 2014, 7, 588–598. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Evans, C.J.; Liu, T.; Banerjee, U. Drosophila Hematopoiesis: Markers and Methods for Molecular Genetic Analysis. Methods 2014, 68, 242–251. [Google Scholar] [CrossRef] [Green Version]
- Verfaillie, C.M. Can Human Hematopoietic Stem Cells Be Cultured Ex Vivo? Stem Cells 1994, 12, 466–476. [Google Scholar] [CrossRef]
- Amsellem, S.; Fichelson, S. Ex vivo expansion of human hematopoietic stem cells by passive transduction of the HOXB4 homeoprotein. J. Soc. Biol. 2006, 200, 235–241. [Google Scholar] [CrossRef]
- Centenera, M.M.; Raj, G.V.; Knudsen, K.E.; Tilley, W.D.; Butler, L.M. Ex Vivo Culture of Human Prostate Tissue and Drug Development. Nat. Rev. Urol. 2013, 10, 483–487. [Google Scholar] [CrossRef]
- Dumont, J.; Euwart, D.; Mei, B.; Estes, S.; Kshirsagar, R. Human Cell Lines for Biopharmaceutical Manufacturing: History, Status, and Future Perspectives. Crit. Rev. Biotechnol. 2016, 36, 1110–1122. [Google Scholar] [CrossRef]
- Sugimoto, N.; Eto, K. Platelet Production from Induced Pluripotent Stem Cells. J. Thromb. Haemost. 2017, 15, 1717–1727. [Google Scholar] [CrossRef] [PubMed]
- Zonari, E.; Desantis, G.; Petrillo, C.; Boccalatte, F.E.; Lidonnici, M.R.; Kajaste-Rudnitski, A.; Aiuti, A.; Ferrari, G.; Naldini, L.; Gentner, B. Efficient Ex Vivo Engineering and Expansion of Highly Purified Human Hematopoietic Stem and Progenitor Cell Populations for Gene Therapy. Stem Cell Rep. 2017, 8, 977–990. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Demers, I.; Donkers, J.; Kremer, B.; Speel, E.J. Ex Vivo Culture Models to Indicate Therapy Response in Head and Neck Squamous Cell Carcinoma. Cells 2020, 9, 2527. [Google Scholar] [CrossRef]
- Walsh, A.J.; Poole, K.M.; Duvall, C.L.; Skala, M.C. Ex Vivo Optical Metabolic Measurements from Cultured Tissue Reflect in Vivo Tissue Status. J. Biomed. Opt. 2012, 17, 116015. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Housden, B.E.; Valvezan, A.J.; Kelley, C.; Sopko, R.; Hu, Y.; Roesel, C.; Lin, S.; Buckner, M.; Tao, R.; Yilmazel, B.; et al. Identification of Potential Drug Targets for Tuberous Sclerosis Complex by Synthetic Screens Combining CRISPR-Based Knockouts with RNAi. Sci. Signal 2015, 8, rs9. [Google Scholar] [CrossRef] [Green Version]
- Kunzelmann, S.; Böttcher, R.; Schmidts, I.; Förstemann, K. A Comprehensive Toolbox for Genome Editing in Cultured Drosophila Melanogaster Cells. G3 Genes Genomes Genet. 2016, 6, 1777–1785. [Google Scholar] [CrossRef] [Green Version]
- Franz, A.; Brunner, E.; Basler, K. Generation of Genome-Modified Drosophila Cell Lines Using SwAP. Fly 2017, 11, 303–311. [Google Scholar] [CrossRef] [Green Version]
- Meijer, T.G.; Naipal, K.A.; Jager, A.; van Gent, D.C. Ex Vivo Tumor Culture Systems for Functional Drug Testing and Therapy Response Prediction. Future Sci. OA 2017, 3, FSO190. [Google Scholar] [CrossRef] [Green Version]
- Zhao, E.; Jiang, X.; Cui, H. Bombyx mori Dihydroorotate Dehydrogenase: Knockdown Inhibits Cell Growth and Proliferation via Inducing Cell Cycle Arrest. Int. J. Mol. Sci. 2018, 19, 2581. [Google Scholar] [CrossRef] [Green Version]
- Shi, D.; Mi, G.; Wang, M.; Webster, T.J. In Vitro and Ex Vivo Systems at the Forefront of Infection Modeling and Drug Discovery. Biomaterials 2019, 198, 228–249. [Google Scholar] [CrossRef]
- Kayukawa, T.; Furuta, K.; Nagamine, K.; Shinoda, T.; Yonesu, K.; Okabe, T. Identification of a Juvenile-Hormone Signaling Inhibitor via High-Throughput Screening of a Chemical Library. Sci. Rep. 2020, 10, 18413. [Google Scholar] [CrossRef] [PubMed]
- Obaldía, N.; Nuñez, M. On the Survival of 48 h Plasmodium Vivax Aotus Monkey-Derived Ex Vivo Cultures: The Role of Leucocytes Filtration and Chemically Defined Lipid Concentrate Media Supplementation. Malar. J. 2020, 19, 278. [Google Scholar] [CrossRef] [PubMed]
- Mbye, H.; Bojang, F.; Jawara, A.S.; Njie, B.; Mohammed, N.I.; Okebe, J.; D’Alessandro, U.; Amambua-Ngwa, A. Tolerance of Gambian Plasmodium Falciparum to Dihydroartemisinin and Lumefantrine Detected by Ex Vivo Parasite Survival Rate Assay. Antimicrob. Agents Chemother. 2020, 65, e00720-20. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, K.; Yoshiyama, M.; Akiduki, G.; Yokoi, K.; Hoshida, H.; Kayukawa, T.; Kimura, K.; Hatakeyama, M. A Simple Method for Ex Vivo Honey Bee Cell Culture Capable of in Vitro Gene Expression Analysis. PLoS ONE 2021, 16, e0257770. [Google Scholar] [CrossRef] [PubMed]
- Wilkinson, A.C.; Ishida, R.; Nakauchi, H.; Yamazaki, S. Long-Term Ex Vivo Expansion of Mouse Hematopoietic Stem Cells. Nat. Protoc. 2020, 15, 628–648. [Google Scholar] [CrossRef]
- Echalier, G.; Ohanessian, A. Isolation, in tissue culture, of Drosophila melangaster cell lines. C. R. Acad. Hebd. Seances Acad. Sci. D 1969, 268, 1771–1773. [Google Scholar]
- Samakovlis, C.; Asling, B.; Boman, H.G.; Gateff, E.; Hultmark, D. In Vitro Induction of Cecropin Genes--an Immune Response in a Drosophila Blood Cell Line. Biochem. Biophys. Res. Commun. 1992, 188, 1169–1175. [Google Scholar] [CrossRef]
- Ui, K.; Nishihara, S.; Sakuma, M.; Togashi, S.; Ueda, R.; Miyata, Y.; Miyake, T. Newly Established Cell Lines from Drosophila Larval CNS Express Neural Specific Characteristics. In Vitro Cell. Dev. Biol. Anim. 1994, 30A, 209–216. [Google Scholar] [CrossRef]
- Echalier, G. Drosophila Cells in Culture; Academic Press: Cambridge, MA, USA, 1997. [Google Scholar]
- Niki, Y.; Yamaguchi, T.; Mahowald, A.P. Establishment of Stable Cell Lines of Drosophila Germ-Line Stem Cells. Proc. Natl. Acad. Sci. USA 2006, 103, 16325–16330. [Google Scholar] [CrossRef] [Green Version]
- Cherbas, L.; Gong, L. Cell Lines. Methods 2014, 68, 74–81. [Google Scholar] [CrossRef]
- Ishizu, H.; Sumiyoshi, T.; Siomi, M.C. Use of the CRISPR-Cas9 System for Genome Editing in Cultured Drosophila Ovarian Somatic Cells. Methods 2017, 126, 186–192. [Google Scholar] [CrossRef] [PubMed]
- Luhur, A.; Klueg, K.M.; Zelhof, A.C. Generating and Working with Drosophila Cell Cultures: Current Challenges and Opportunities. Wiley Interdiscip. Rev. Dev. Biol. 2019, 8, e339. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Zheng, Y.; Yin, F.; Yu, J.; Silverman, N.; Pan, D. Toll Receptor-Mediated Hippo Signaling Controls Innate Immunity in Drosophila. Cell 2016, 164, 406–419. [Google Scholar] [CrossRef] [Green Version]
- Nonaka, S.; Ando, Y.; Kanetani, T.; Hoshi, C.; Nakai, Y.; Nainu, F.; Nagaosa, K.; Shiratsuchi, A.; Nakanishi, Y. Signaling Pathway for Phagocyte Priming upon Encounter with Apoptotic Cells. J. Biol. Chem. 2017, 292, 8059–8072. [Google Scholar] [CrossRef] [Green Version]
- Evans, C.J.; Liu, T.; Girard, J.R.; Banerjee, U. Injury-Induced Inflammatory Signaling and Hematopoiesis in Drosophila. Proc. Natl. Acad. Sci. USA 2022, 119, e2119109119. [Google Scholar] [CrossRef] [PubMed]
- Dubuffet, A.; Colinet, D.; Anselme, C.; Dupas, S.; Carton, Y.; Poirié, M. Variation of Leptopilina Boulardi Success in Drosophila Hosts: What Is inside the Black Box? Adv. Parasitol. 2009, 70, 147–188. [Google Scholar] [CrossRef]
- Heavner, M.E.; Hudgins, A.D.; Rajwani, R.; Morales, J.; Govind, S. Harnessing the Natural Drosophila-Parasitoid Model for Integrating Insect Immunity with Functional Venomics. Curr. Opin. Insect Sci. 2014, 6, 61–67. [Google Scholar] [CrossRef] [Green Version]
- Boulet, M.; Renaud, Y.; Lapraz, F.; Benmimoun, B.; Vandel, L.; Waltzer, L. Characterization of the Drosophila Adult Hematopoietic System Reveals a Rare Cell Population With Differentiation and Proliferation Potential. Front. Cell Dev. Biol. 2021, 9, 739357. [Google Scholar] [CrossRef]
- Izumi, M.; Miyazawa, H.; Kamakura, T.; Yamaguchi, I.; Endo, T.; Hanaoka, F. Blasticidin S-Resistance Gene (Bsr): A Novel Selectable Marker for Mammalian Cells. Exp. Cell Res. 1991, 197, 229–233. [Google Scholar] [CrossRef] [Green Version]
- Karreman, C. New Positive/Negative Selectable Markers for Mammalian Cells on the Basis of Blasticidin Deaminase-Thymidine Kinase Fusions. Nucleic Acids Res. 1998, 26, 2508–2510. [Google Scholar] [CrossRef] [Green Version]
- Ruan, J.; Ping, C.-Y.; Sun, S.; Cheng, X.; Han, P.-Y.; Zhang, Y.-G.; Sun, D.-X. Construction of a Replication-Competent Hepatitis B Virus Vector Carrying Secreted Luciferase Transgene and Establishment of New Hepatitis B Virus Replication and Expression Cell Lines. World J. Gastroenterol. 2019, 25, 5961–5972. [Google Scholar] [CrossRef]
- Li, Y.-M.; Wang, M.; Wang, T.-Y.; Wei, Y.-G.; Guo, X.; Mi, C.-L.; Zhao, C.-P.; Cao, X.-X.; Dou, Y.-Y. Effects of Different 2A Peptides on Transgene Expression Mediated by Tricistronic Vectors in Transfected CHO Cells. Mol. Biol. Rep. 2020, 47, 469–475. [Google Scholar] [CrossRef] [PubMed]
- Tsai, S.J.; Ai, Y.; Guo, C.; Gould, S.J. Degron Tagging of BleoR and Other Antibiotic-Resistance Genes Selects for Higher Expression of Linked Transgenes and Improved Exosome Engineering. J. Biol. Chem. 2022, 298, 101846. [Google Scholar] [CrossRef]
- Fu, Y.; Huang, X.; Zhang, P.; van de Leemput, J.; Han, Z. Single-Cell RNA Sequencing Identifies Novel Cell Types in Drosophila Blood. J. Genet. Genom. 2020, 47, 175–186. [Google Scholar] [CrossRef]
- Kharrat, B.; Csordás, G.; Honti, V. Peeling Back the Layers of Lymph Gland Structure and Regulation. Int. J. Mol. Sci. 2022, 23, 7767. [Google Scholar] [CrossRef] [PubMed]
- Duchek, P.; Somogyi, K.; Jékely, G.; Beccari, S.; Rørth, P. Guidance of Cell Migration by the Drosophila PDGF/VEGF Receptor. Cell 2001, 107, 17–26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lam, V.K.; Tokusumi, T.; Cerabona, D.; Schulz, R.A. Specific Cell Ablation in Drosophila Using the Toxic Viral Protein M2(H37A). Fly 2010, 4, 338–343. [Google Scholar] [CrossRef]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kúthy-Sutus, E.; Kharrat, B.; Gábor, E.; Csordás, G.; Sinka, R.; Honti, V. A Novel Method for Primary Blood Cell Culturing and Selection in Drosophila melanogaster. Cells 2023, 12, 24. https://doi.org/10.3390/cells12010024
Kúthy-Sutus E, Kharrat B, Gábor E, Csordás G, Sinka R, Honti V. A Novel Method for Primary Blood Cell Culturing and Selection in Drosophila melanogaster. Cells. 2023; 12(1):24. https://doi.org/10.3390/cells12010024
Chicago/Turabian StyleKúthy-Sutus, Enikő, Bayan Kharrat, Erika Gábor, Gábor Csordás, Rita Sinka, and Viktor Honti. 2023. "A Novel Method for Primary Blood Cell Culturing and Selection in Drosophila melanogaster" Cells 12, no. 1: 24. https://doi.org/10.3390/cells12010024
APA StyleKúthy-Sutus, E., Kharrat, B., Gábor, E., Csordás, G., Sinka, R., & Honti, V. (2023). A Novel Method for Primary Blood Cell Culturing and Selection in Drosophila melanogaster. Cells, 12(1), 24. https://doi.org/10.3390/cells12010024