

The RhoA-ROCK1/ROCK2 Pathway Exacerbates Inflammatory Signaling in Immortalized and Primary Microglia
 , , and
, , and
Abstract
:1. Background
2. Materials and Methods
2.1. Cell Culture Studies
2.1.1. Immortalized Mouse Microglia (IMG)
2.1.2. Primary Microglia (PMg) Studies
2.2. Effects of LPS Challenges and Pharmacological Interventions on ROCK Signaling in Inflammation
2.2.1. Effects of Lipopolysaccharide on IMG and PMg Cells
2.2.2. Pharmacological Interventions Targeting ROCK Signaling in LPS-Induced Inflammation
2.2.3. Effects of Inhibitors of ROCK Signaling on LPS-Activated IMG Cells and PMg
2.2.4. Effects of Nogo-P4 or Narc on LPS-Activated IMG Cells
2.3. Assessment of Cell Viability with Interventions Targeting ROCK Signaling in LPS-Activated IMG and PMg Cells
2.4. Assessment of Media-Secreted Factor Levels following ROCK Signaling Interventions in LPS-Activated IMG and PMg Cells
2.5. Immunochemistry
2.5.1. Immunostaining of Mouse Brain Tissue
2.5.2. IMG Cells
2.6. NF-κB Signal Quantification
2.7. Western Blot Analysis of LPS-Activated IMG Cells
2.8. Assessment of Inflammatory Markers and ROCK Signaling Protein RNA Transcript Levels in LPS-Activated IMG Cells
2.9. siRNA Experiments
2.10. Statistical Analysis of Data
3. Results
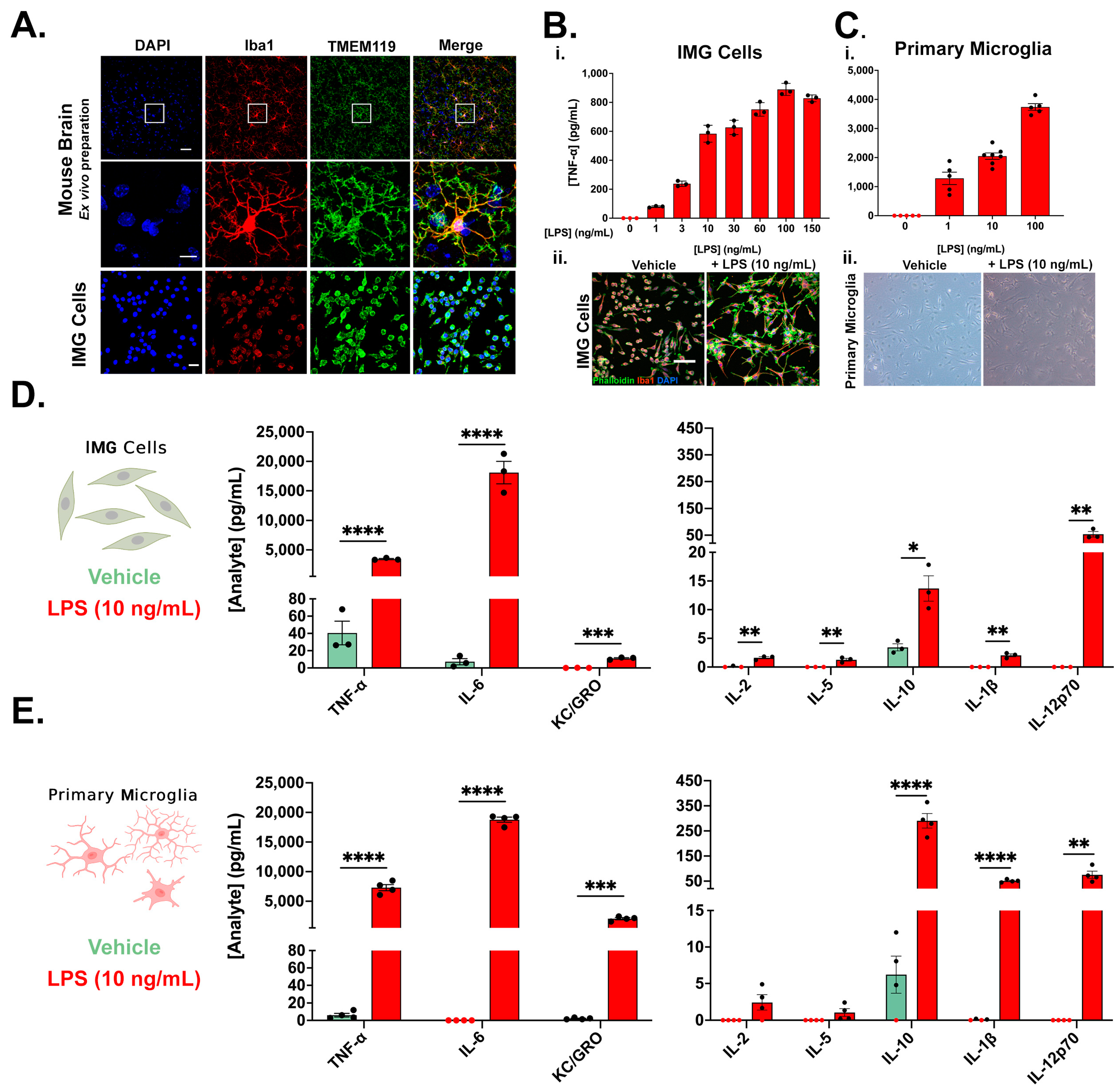
3.1. Immortalized Microglia (IMG Cells) as a Model for Primary Microglia (PMg)
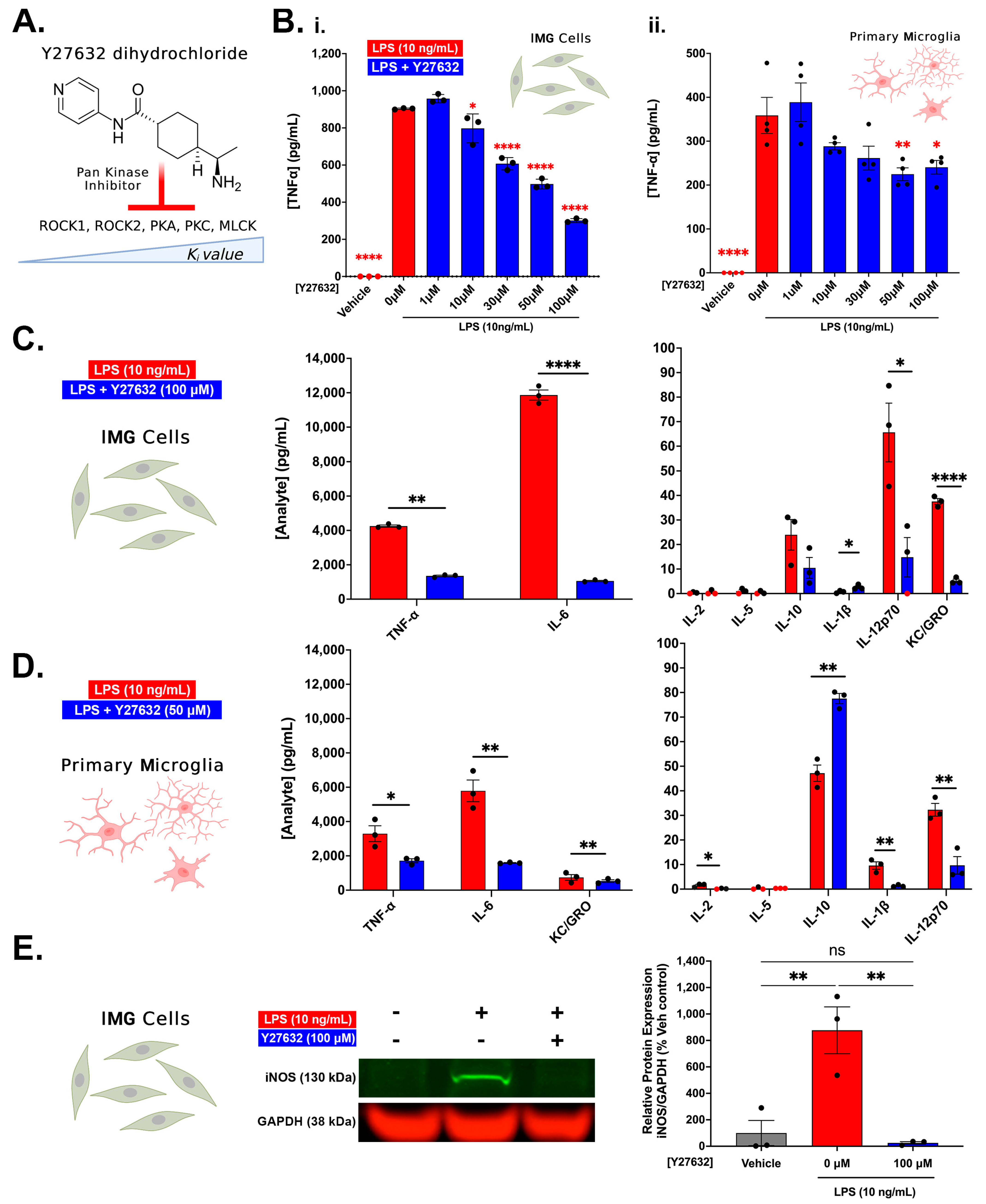
3.2. Pan Kinase Inhibition from Y27632-Dihydrochloride Mitigates Inflammatory Protein Production in IMG and PMg Cells
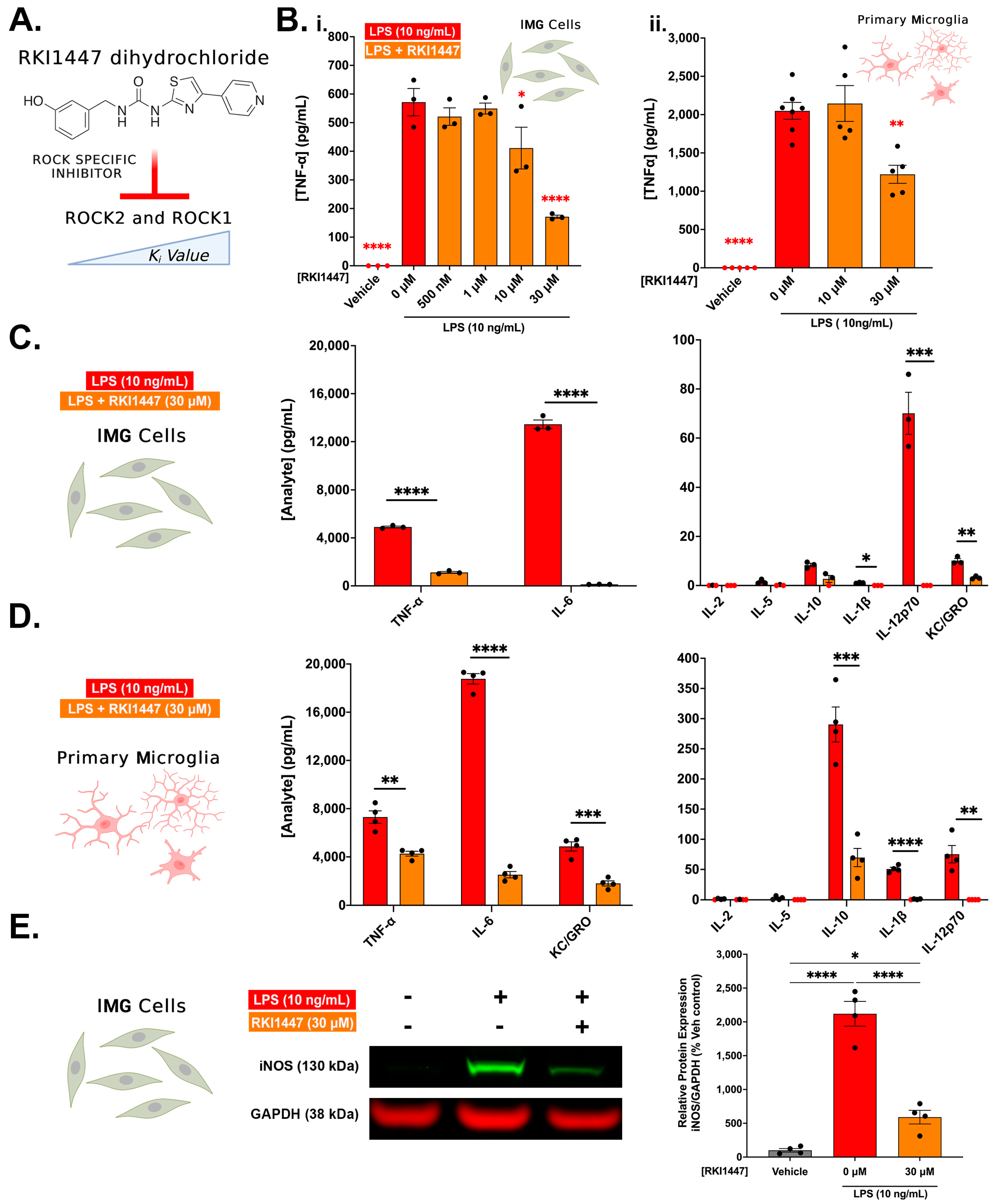
3.3. ROCK1 and ROCK2 Inhibition Is Sufficient to Blunt Inflammatory Protein Production in IMG Cells and PMg
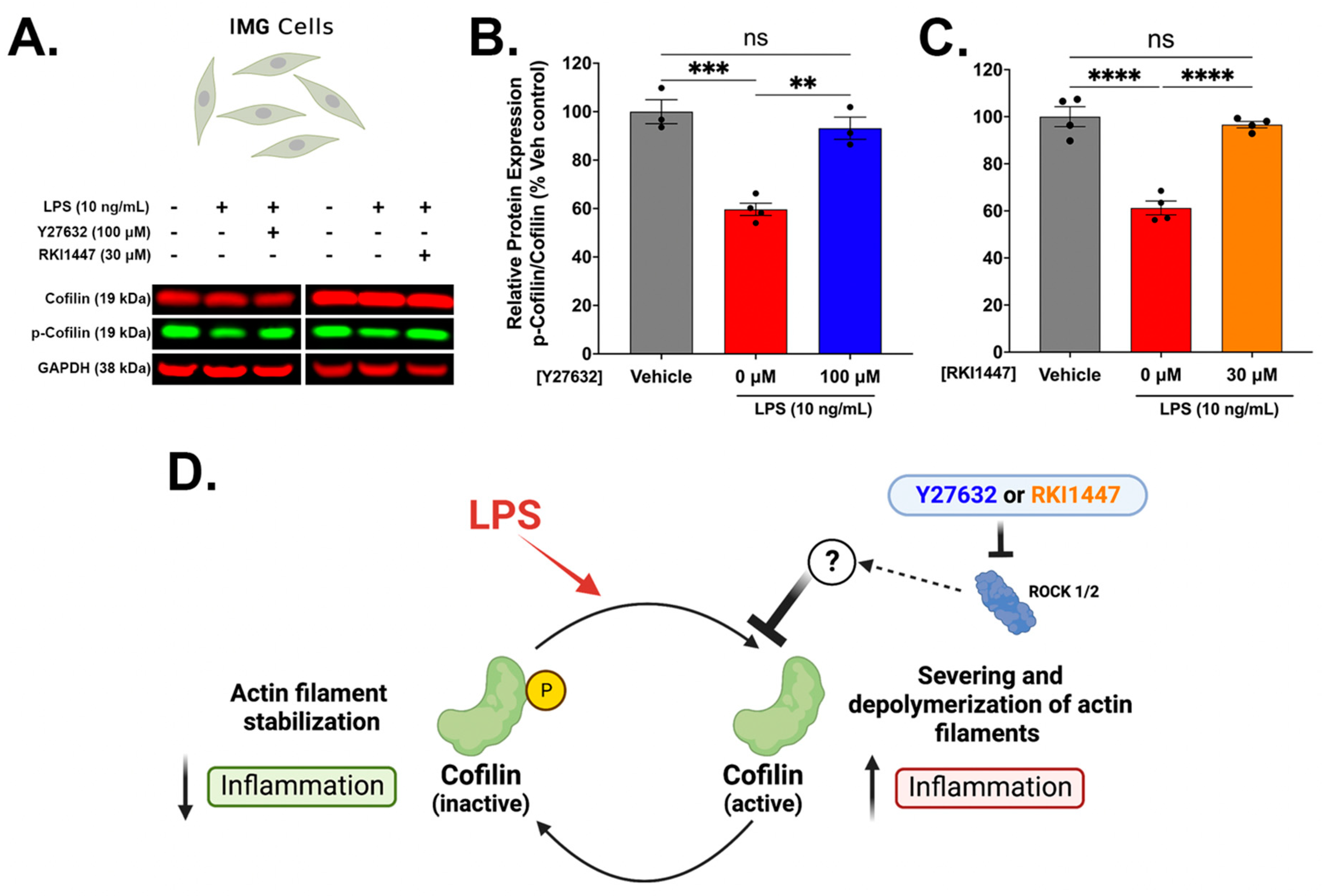
3.4. Y27632 and RKI1447 Restore Ratios of Inactive Cofilin to Total Cofilin in IMG Cells Challenged with LPS
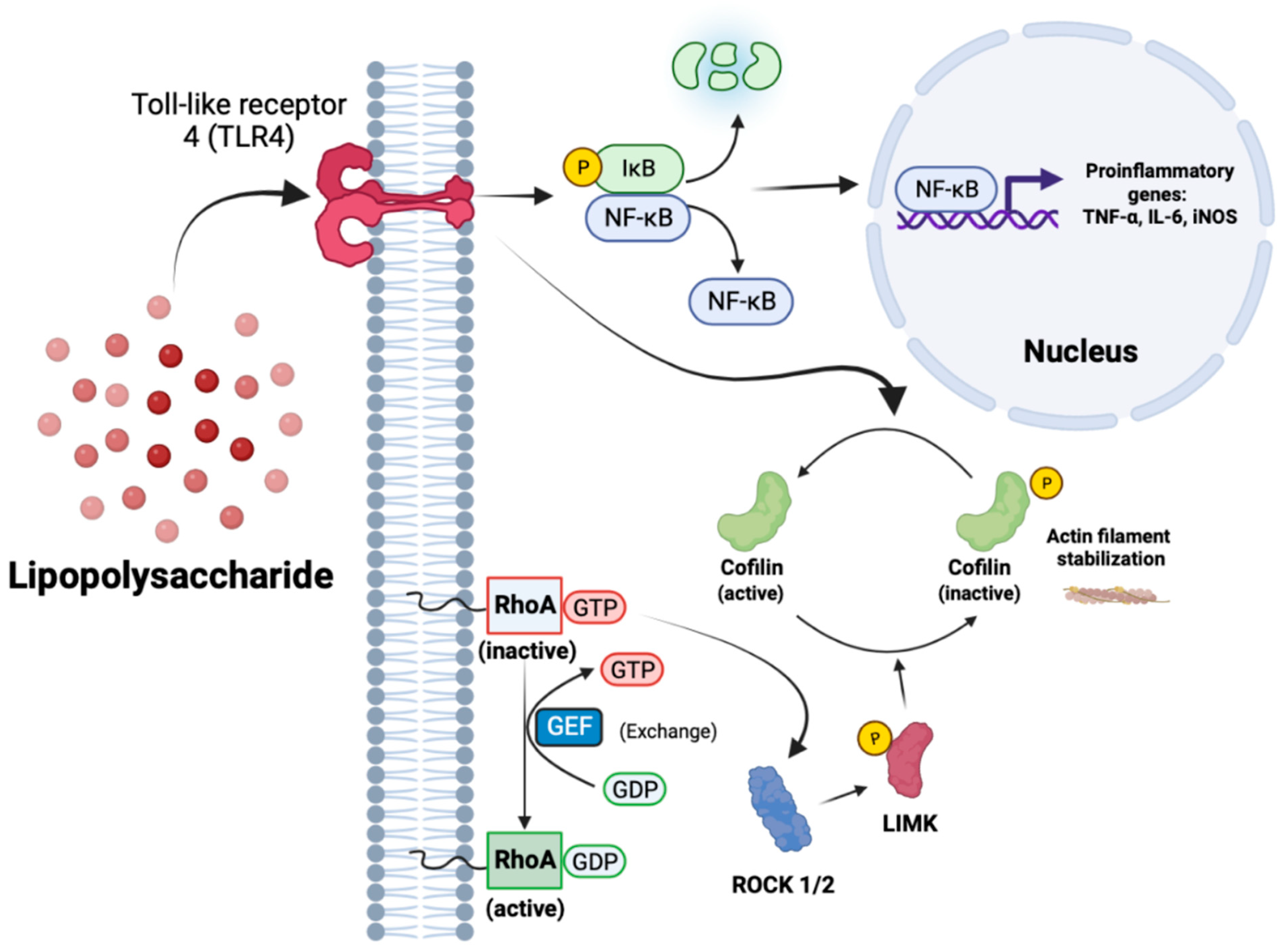
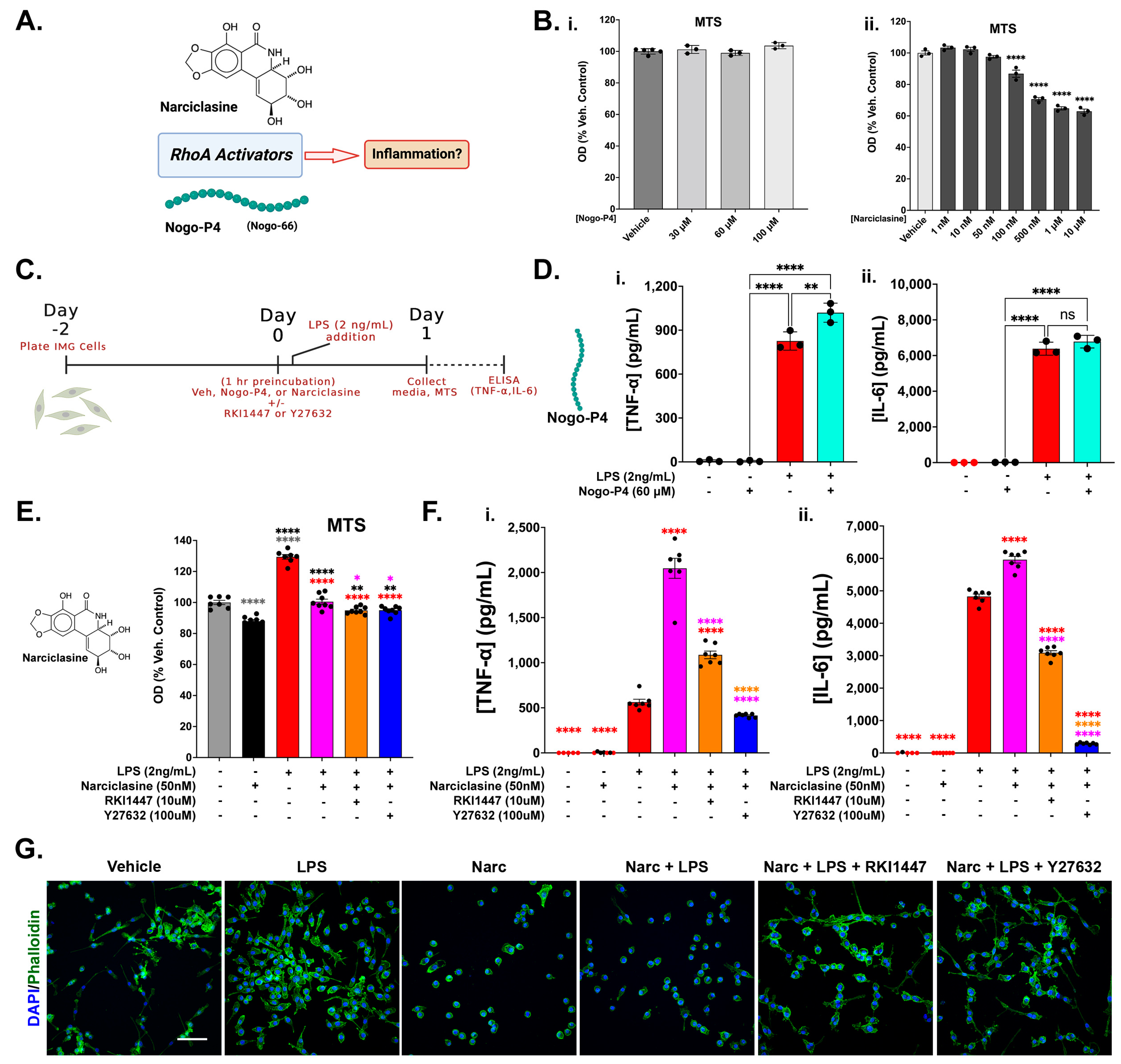
3.5. RhoA Activation Is Not Sufficient to Initiate an Inflammatory Response in IMG Cells but Exacerbates Inflammation in the Presence of a Submaximal LPS Challenge
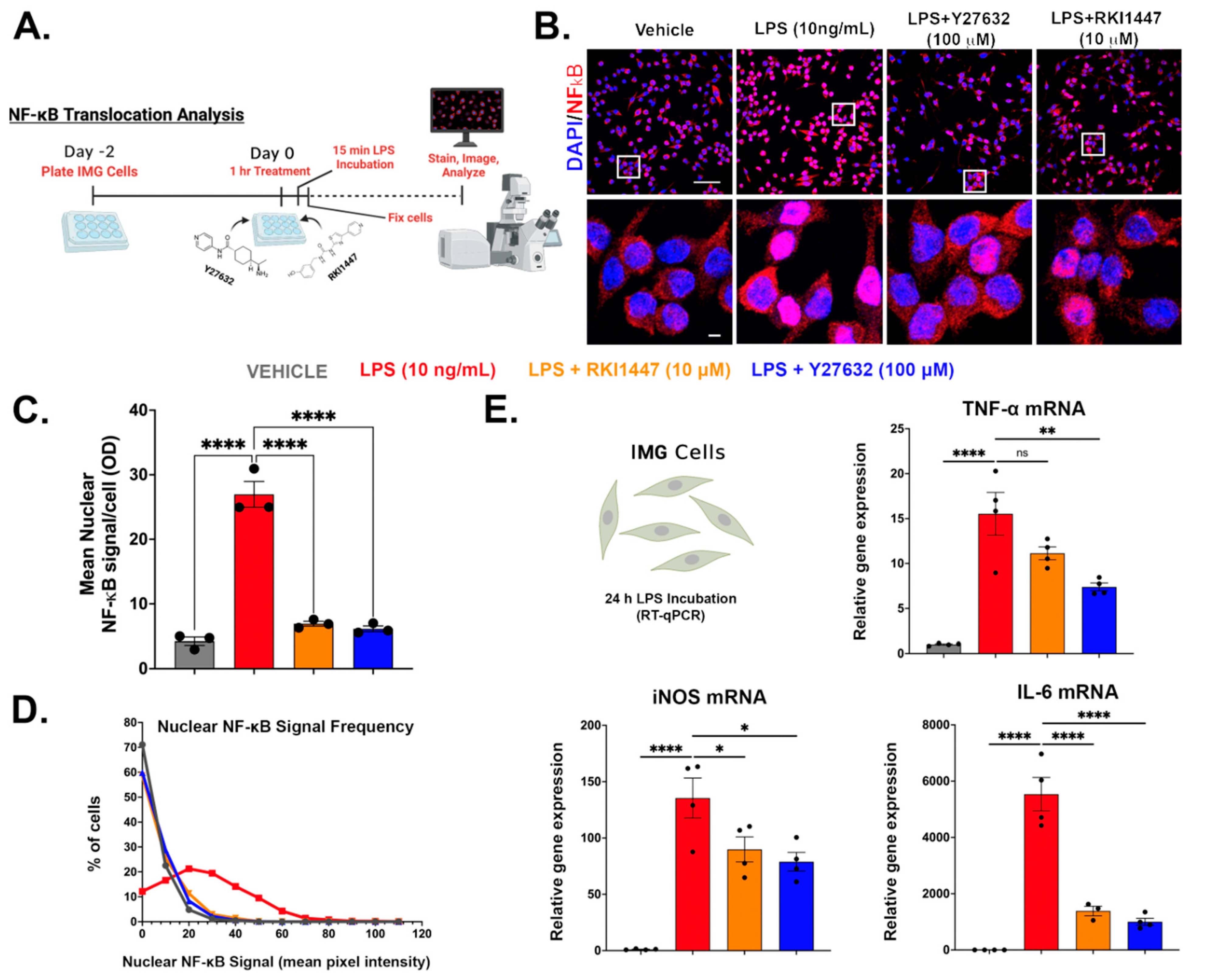
3.6. Y27632 and RKI1447 Block the Nuclear Translocation of NF-κB in IMG Cells
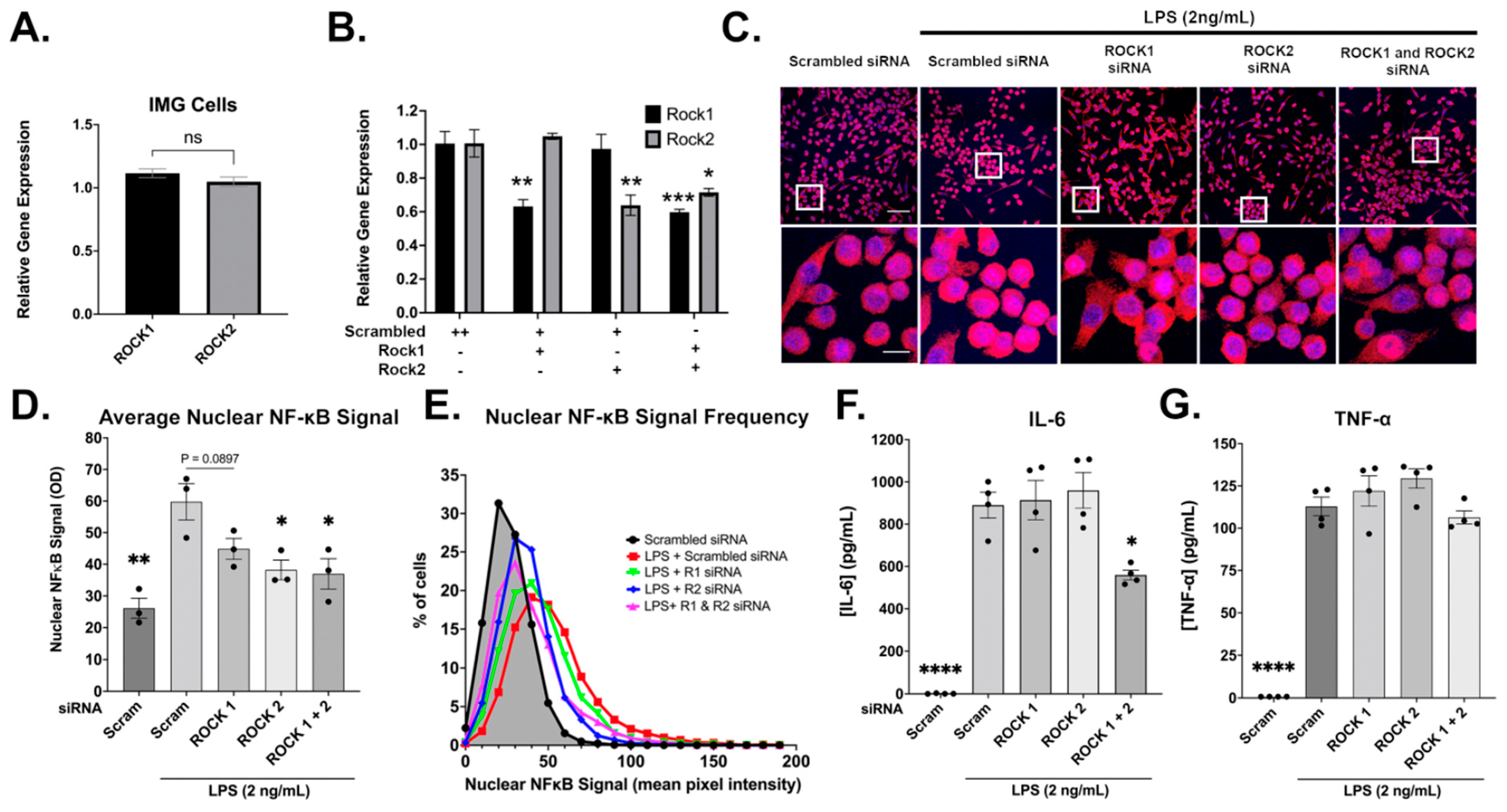
3.7. Evidence for ROCK1 and ROCK2 Roles in LPS-Induced Inflammatory Response
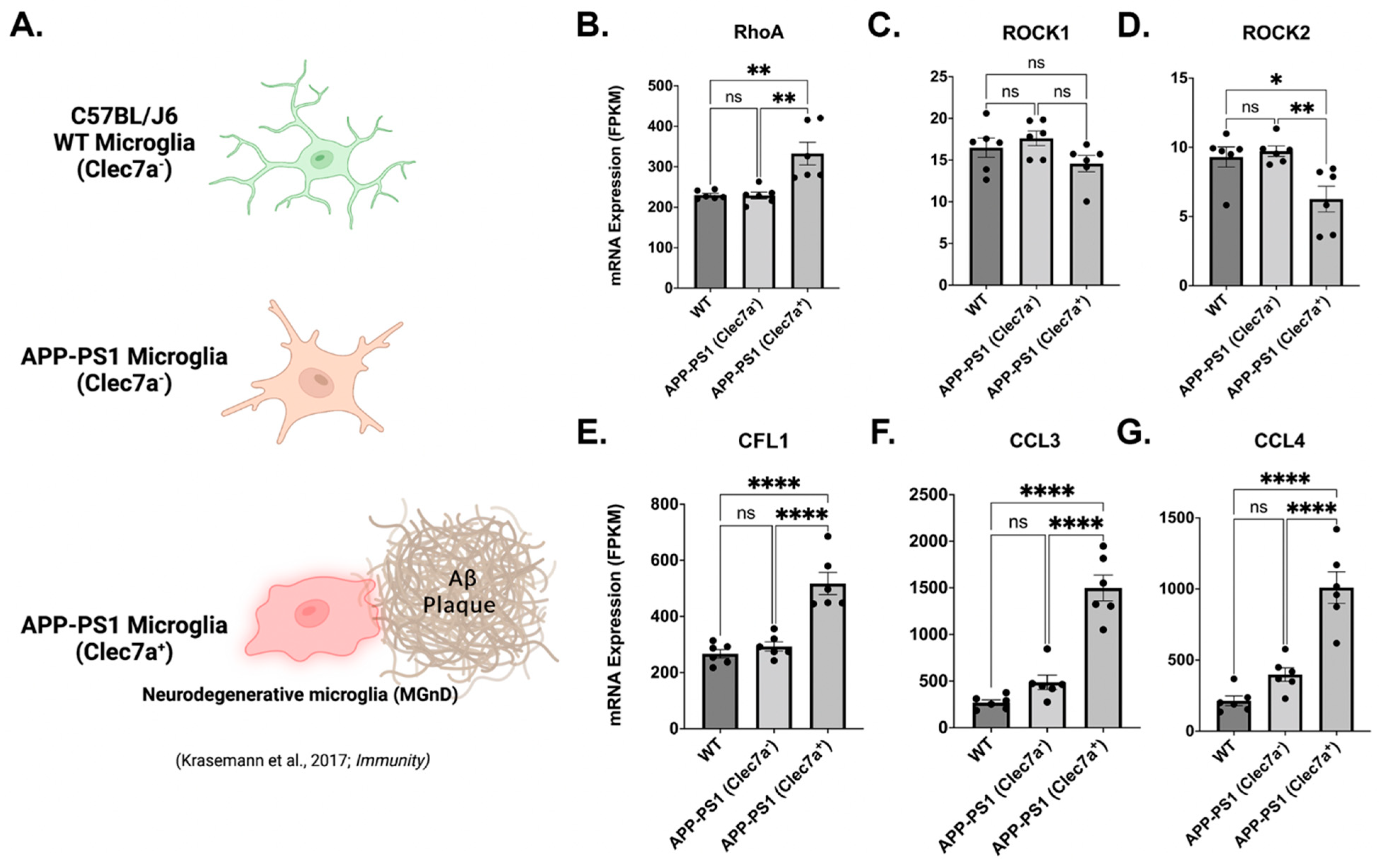
3.8. Neurodegenerative Clec7a+ Microglia Express Increased RhoA, Cofilin, and the Key Modulators of Neuroinflammation
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sierra, A.; Paolicelli, R.C.; Kettenmann, H. Cien Años de Microglía: Milestones in a Century of Microglial Research. Trends Neurosci. 2019, 42, 778–792. [Google Scholar] [CrossRef] [PubMed]
- Tang, Y.; Le, W. Differential Roles of M1 and M2 Microglia in Neurodegenerative Diseases. Mol. Neurobiol. 2016, 53, 1181–1194. [Google Scholar] [CrossRef] [PubMed]
- Paolicelli, R.C.; Sierra, A.; Stevens, B.; Tremblay, M.-E.; Aguzzi, A.; Ajami, B.; Amit, I.; Audinat, E.; Bechmann, I.; Bennett, M.; et al. Microglia states and nomenclature: A field at its crossroads. Neuron 2022, 110, 3458–3483. [Google Scholar] [CrossRef] [PubMed]
- Sastre, A.A.; Montoro, M.L.; Gálvez-Martín, P.; Lacerda, H.M.; Lucia, A.; Llavero, F.; Zugaza, J.L. Small GTPases of the Ras and Rho Families Switch on/off Signaling Pathways in Neurodegenerative Diseases. Int. J. Mol. Sci. 2020, 21, 6312. [Google Scholar] [CrossRef] [PubMed]
- Roser, A.-E.; Tönges, L.; Lingor, P. Modulation of Microglial Activity by Rho-Kinase (ROCK) Inhibition as Therapeutic Strategy in Parkinson’s Disease and Amyotrophic Lateral Sclerosis. Front. Aging Neurosci. 2017, 9, 94. [Google Scholar] [CrossRef]
- Heasman, S.J.; Ridley, A.J. Mammalian Rho GTPases: New insights into their functions from in vivo studies. Nat. Rev. Mol. Cell Biol. 2008, 9, 690–701. [Google Scholar] [CrossRef]
- Wikström, K.; Kavanagh, D.J.; Reid, H.M.; Kinsella, B.T. Differential regulation of RhoA-mediated signaling by the TPα and TPβ isoforms of the human thromboxane A2 receptor: Independent modulation of TPα signaling by prostacyclin and nitric oxide. Cell. Signal. 2008, 20, 1497–1512. [Google Scholar] [CrossRef]
- Xiang, S.Y.; Dusaban, S.S.; Brown, J.H. Lysophospholipid receptor activation of RhoA and lipid signaling pathways. Biochim. Biophys. Acta (BBA)—Mol. Cell Biol. Lipids 2013, 1831, 213–222. [Google Scholar] [CrossRef]
- Zhou, X.; Zheng, Y. Cell Type-Specific Signaling Function of Rhoa GTPase: Lessons from Mouse Genetargeting. J. Biol. Chem. 2013, 288, 36179–36188. [Google Scholar] [CrossRef]
- Kempf, A.; Tews, B.; Arzt, M.E.; Weinmann, O.; Obermair, F.J.; Pernet, V.; Zagrebelsky, M.; Delekate, A.; Iobbi, C.; Zemmar, A.; et al. The Sphingolipid Receptor S1PR2 Is a Receptor for Nogo-A Repressing Synaptic Plasticity. PLoS Biol. 2014, 12, e1001763. [Google Scholar] [CrossRef]
- Oertle, T.; van der Haar, M.E.; Bandtlow, C.E.; Robeva, A.; Burfeind, P.; Buss, A.; Huber, A.B.; Simonen, M.; Schnell, L.; Brösamle, C.; et al. Nogo-A Inhibits Neurite Outgrowth and Cell Spreading with Three Discrete Regions. J. Neurosci. 2003, 23, 5393–5406. [Google Scholar] [CrossRef] [PubMed]
- Karlsson, T.E.; Smedfors, G.; Brodin, A.T.S.; Åberg, E.; Mattsson, A.; Högbeck, I.; Wellfelt, K.; Josephson, A.; Brené, S.; Olson, L. NgR1: A Tunable Sensor Regulating Memory Formation, Synaptic, and Dendritic Plasticity. Cereb. Cortex 2016, 26, 1804–1817. [Google Scholar] [CrossRef] [PubMed]
- Cafferty, W.B.J.; Duffy, P.; Huebner, E.; Strittmatter, S.M. MAG and OMgp Synergize with Nogo-A to Restrict Axonal Growth and Neurological Recovery after Spinal Cord Trauma. J. Neurosci. 2010, 30, 6825–6837. [Google Scholar] [CrossRef]
- Kubo, T.; Yamaguchi, A.; Iwata, N.; Yamashita, T. The therapeutic effects of Rho-ROCK inhibitors on CNS disorders. Ther. Clin. Risk Manag. 2008, 4, 605–615. [Google Scholar] [CrossRef] [PubMed]
- Fang, Y.; Yan, J.; Li, C.; Zhou, X.; Yao, L.; Pang, T.; Yan, M.; Zhang, L.; Mao, L.; Liao, H. The Nogo/Nogo Receptor (NgR) Signal Is Involved in Neuroinflammation through the Regulation of Microglial Inflammatory Activation. J. Biol. Chem. 2015, 290, 28901–28914. [Google Scholar] [CrossRef] [PubMed]
- Fang, Y.; Wang, J.; Yao, L.; Li, C.; Wang, J.; Liu, Y.; Tao, X.; Sun, H.; Liao, H. The adhesion and migration of microglia to β-amyloid (Aβ) is decreased with aging and inhibited by Nogo/NgR pathway. J. Neuroinflamm. 2018, 15, 210. [Google Scholar] [CrossRef]
- Leung, T.; Manser, E.; Tan, L.; Lim, L. A Novel Serine/Threonine Kinase Binding the Ras-related RhoA GTPase Which Translocates the Kinase to Peripheral Membranes. J. Biol. Chem. 1995, 270, 29051–29054. [Google Scholar] [CrossRef]
- Sakakibara, T.; Sukigara, S.; Otsuki, T.; Takahashi, A.; Kaneko, Y.; Kaido, T.; Saito, Y.; Sato, N.; Nakagawa, E.; Sugai, K.; et al. Imbalance of interneuron distribution between neocortex and basal ganglia: Consideration of epileptogenesis of focal cortical dysplasia. J. Neurol. Sci. 2012, 323, 128–133. [Google Scholar] [CrossRef]
- Newell-Litwa, K.A.; Badoual, M.; Asmussen, H.; Patel, H.; Whitmore, L.; Horwitz, A.R. ROCK1 and 2 differentially regulate actomyosin organization to drive cell and synaptic polarity. J. Cell Biol. 2015, 210, 225–242. [Google Scholar] [CrossRef]
- Shi, J.; Wu, X.; Surma, M.; Vemula, S.; Zhang, L.; Yang, Y.; Kapur, R.; Wei, L. Distinct roles for ROCK1 and ROCK2 in the regulation of cell detachment. Cell Death Dis. 2013, 4, e483. [Google Scholar] [CrossRef]
- Mertsch, S.; Thanos, S. Opposing Signaling of ROCK1 and ROCK2 Determines the Switching of Substrate Specificity and the Mode of Migration of Glioblastoma Cells. Mol. Neurobiol. 2014, 49, 900–915. [Google Scholar] [CrossRef]
- Greathouse, K.M.; Boros, B.D.; Deslauriers, J.F.; Henderson, B.W.; Curtis, K.A.; Gentry, E.G.; Herskowitz, J.H. Distinct and complementary functions of rho kinase isoforms ROCK1 and ROCK2 in prefrontal cortex structural plasticity. Anat. Embryol. 2018, 223, 4227–4241. [Google Scholar] [CrossRef]
- Feng, Y.; LoGrasso, P.V.; Defert, O.; Li, R. Rho Kinase (ROCK) Inhibitors and Their Therapeutic Potential. J. Med. Chem. 2016, 59, 2269–2300. [Google Scholar] [CrossRef]
- Liu, Y.; Gray, N.S. Rational design of inhibitors that bind to inactive kinase conformations. Nat. Chem. Biol. 2006, 2, 358–364. [Google Scholar] [CrossRef]
- Tönges, L.; Günther, R.; Suhr, M.; Jansen, J.; Balck, A.; Saal, K.-A.; Barski, E.; Nientied, T.; Götz, A.A.; Koch, J.-C.; et al. Rho kinase inhibition modulates microglia activation and improves survival in a model of amyotrophic lateral sclerosis. Glia 2013, 62, 217–232. [Google Scholar] [CrossRef] [PubMed]
- He, Q.; Li, Y.-H.; Guo, S.-S.; Wang, Y.; Lin, W.; Zhang, Q.; Wang, J.; Ma, C.-G.; Xiao, B.-G. Inhibition of Rho-kinase by Fasudil protects dopamine neurons and attenuates inflammatory response in an intranasal lipopolysaccharide-mediated Parkinson’s model. Eur. J. Neurosci. 2015, 43, 41–52. [Google Scholar] [CrossRef]
- Borrajo, A.; Rodriguez-Perez, A.I.; Villar-Cheda, B.; Guerra, M.J.; Labandeira-Garcia, J.L. Inhibition of the microglial response is essential for the neuroprotective effects of Rho-kinase inhibitors on MPTP-induced dopaminergic cell death. Neuropharmacology 2014, 85, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Villar-Cheda, B.; Dominguez-Meijide, A.; Joglar, B.; Rodriguez-Perez, A.I.; Guerra, M.J.; Labandeira-Garcia, J.L. Involvement of microglial RhoA/Rho-Kinase pathway activation in the dopaminergic neuron death. Role of angiotensin via angiotensin type 1 receptors. Neurobiol. Dis. 2012, 47, 268–279. [Google Scholar] [CrossRef] [PubMed]
- Guo, M.-F.; Zhang, H.-Y.; Li, Y.-H.; Gu, Q.-F.; Wei, W.-Y.; Wang, Y.-Y.; Zhang, X.-J.; Liu, X.-Q.; Song, L.-J.; Chai, Z.; et al. Fasudil inhibits the activation of microglia and astrocytes of transgenic Alzheimer’s disease mice via the downregulation of TLR4/Myd88/NF-κB pathway. J. Neuroimmunol. 2020, 346, 577284. [Google Scholar] [CrossRef]
- Gunther, R.; Saal, K.A.; Suhr, M.; Scheer, D.; Koch, J.C.; Bahr, M.; Lingor, P.; Tonges, L. The rho kinase inhibitor Y-27632 improves motor performance in male SOD1G93A mice. Front. Neurosci. 2014, 8, 304. [Google Scholar] [CrossRef] [PubMed]
- Labandeira-Garcia, J.L.; Rodríguez-Perez, A.I.; Villar-Cheda, B.; Borrajo, A.; Dominguez-Meijide, A.; Guerra, M.J. Rho Kinase and Dopaminergic Degeneration: A Promising Therapeutic Target for Parkinsons Disease. Neuroscientist 2015, 21, 616–629. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Su, Y.; Li, B.; Liu, F.; Ryder, J.W.; Wu, X.; Gonzalez-DeWhitt, P.A.; Gelfanova, V.; Hale, J.E.; May, P.C.; et al. Nonsteroidal Anti-Inflammatory Drugs Can Lower Amyloidogenic Aß42 by Inhibiting Rho. Science 2003, 302, 1215–1217. [Google Scholar] [CrossRef] [PubMed]
- Blair, H.A. Belumosudil: First Approval. Drugs 2021, 81, 1677–1682. [Google Scholar] [CrossRef]
- Berrino, E.; Supuran, C.T. Rho-kinase inhibitors in the management of glaucoma. Expert Opin. Ther. Patents 2019, 29, 817–827. [Google Scholar] [CrossRef]
- Shibuya, M.; Hirai, S.; Seto, M.; Satoh, S.-I.; Ohtomo, E.; Fasudil Ischemic Stroke Study Group. Effects of fasudil in acute ischemic stroke: Results of a prospective placebo-controlled double-blind trial. J. Neurol. Sci. 2005, 238, 31–39. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Zhou, D.; Guo, J.; Ren, Z.; Zhou, L.; Wang, S.; Xu, B.; Wang, R. Effect of Fasudil Hydrochloride, a Protein Kinase Inhibitor, on Cerebral Vasospasm and Delayed Cerebral Ischemic Symptoms After Aneurysmal Subarachnoid Hemorrhage-Results of a Randomized Trial of Fasudil Hydrochloride Versus Nimodipine-. Neurol. Med.-Chir. 2006, 46, 421–428. [Google Scholar] [CrossRef] [PubMed]
- Vicari, R.M.; Chaitman, B.; Keefe, D.; Smith, W.B.; Chrysant, S.G.; Tonkon, M.J.; Bittar, N.; Weiss, R.J.; Morales-Ballejo, H.; Thadani, U. Efficacy and Safety of Fasudil in Patients With Stable Angina: A Double-Blind, Placebo-Controlled, Phase 2 Trial. J. Am. Coll. Cardiol. 2005, 46, 1803–1811. [Google Scholar] [CrossRef]
- Sharif, O.; Bolshakov, V.N.; Raines, S.; Newham, P.; Perkins, N.D. Transcriptional profiling of the LPS induced NF-κB response in macrophages. BMC Immunol. 2007, 8, 1. [Google Scholar] [CrossRef] [PubMed]
- Blackwell, T.S.; Christman, J.W. The Role of Nuclear Factor-ΚB in Cytokine Gene Regulation. Am. J. Respir. Cell Mol. Biol. 1997, 17, 3–9. [Google Scholar] [CrossRef]
- McCarthy, R.C.; Lu, D.-Y.; Alkhateeb, A.; Gardeck, A.M.; Lee, C.-H.; Wessling-Resnick, M. Characterization of a novel adult murine immortalized microglial cell line and its activation by amyloid-beta. J. Neuroinflammation 2016, 13, 21. [Google Scholar] [CrossRef]
- Krasemann, S.; Madore, C.; Cialic, R.; Baufeld, C.; Calcagno, N.; El Fatimy, R.; Beckers, L.; O’Loughlin, E.; Xu, Y.; Fanek, Z.; et al. The TREM2-APOE Pathway Drives the Transcriptional Phenotype of Dysfunctional Microglia in Neurodegenerative Diseases. Immunity 2017, 47, 566–581.E9. [Google Scholar] [CrossRef] [PubMed]
- Hammond, T.R.; Dufort, C.; Dissing-Olesen, L.; Giera, S.; Young, A.; Wysoker, A.; Walker, A.J.; Gergits, F.; Segel, M.; Nemesh, J.; et al. Single-Cell RNA Sequencing of Microglia throughout the Mouse Lifespan and in the Injured Brain Reveals Complex Cell-State Changes. Immunity 2019, 50, 253–271.E6. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.K.; Tansey, M.G. Microglia Isolation from Adult Mouse Brain. In Microglia; Joseph, B., Venero, J., Eds.; Methods in Molecular Biology; Humana Press: Totowa, NJ, USA, 2013; Volume 1041, pp. 17–23. [Google Scholar]
- Davies, S.P.; Reddy, H.; Caivano, M.; Cohen, P. Specificity and mechanism of action of some commonly used protein kinase inhibitors. Biochem. J. 2000, 351, 95–105. [Google Scholar] [CrossRef]
- Patel, R.A.; Forinash, K.D.; Pireddu, R.; Sun, Y.; Sun, N.; Martin, M.P.; Schönbrunn, E.; Lawrence, N.J.; Sebti, S.M. RKI-1447 Is a Potent Inhibitor of the Rho-Associated ROCK Kinases with Anti-Invasive and Antitumor Activities in Breast Cancer. Cancer Res. 2012, 72, 5025–5034. [Google Scholar] [CrossRef]
- Lefranc, F.; Sauvage, S.; Van Goietsenoven, G.; Mégalizzi, V.; Lamoral-Theys, D.; Debeir, O.; Spiegl-Kreinecker, S.; Berger, W.; Mathieu, V.; Decaestecker, C.; et al. Narciclasine, a plant growth modulator, activates Rho and stress fibers in glioblastoma cells. Mol. Cancer Ther. 2009, 8, 1739–1750. [Google Scholar] [CrossRef] [PubMed]
- Yan, J.; Zhou, X.; Guo, J.-J.; Mao, L.; Wang, Y.-J.; Sun, J.; Sun, L.-X.; Zhang, L.-Y.; Zhou, X.-F.; Liao, H. Nogo-66 inhibits adhesion and migration of microglia via GTPase Rho pathway in vitro. J. Neurochem. 2011, 120, 721–731. [Google Scholar] [CrossRef]
- Schindelin, J.; Arganda-Carreras, I.; Frise, E.; Kaynig, V.; Longair, M.; Pietzsch, T.; Preibisch, S.; Rueden, C.; Saalfeld, S.; Schmid, B.; et al. Fiji: An open-source platform for biological-image analysis. Nat. Methods 2012, 9, 676–682. [Google Scholar] [CrossRef]
- Babagana, M.; Oh, K.-S.; Chakraborty, S.; Pacholewska, A.; Aqdas, M.; Sung, M.-H. Hedgehog dysregulation contributes to tissue-specific inflammaging of resident macrophages. Aging 2021, 13, 19207–19229. [Google Scholar] [CrossRef]
- Wang, J.-Y.; Huang, Y.-N.; Chiu, C.-C.; Tweedie, D.; Luo, W.; Pick, C.G.; Chou, S.-Y.; Luo, Y.; Hoffer, B.J.; Greig, N.H.; et al. Pomalidomide mitigates neuronal loss, neuroinflammation, and behavioral impairments induced by traumatic brain injury in rat. J. Neuroinflamm. 2016, 13, 168. [Google Scholar] [CrossRef]
- Kanazawa, H.; Ohsawa, K.; Sasaki, Y.; Kohsaka, S.; Imai, Y. Macrophage/Microglia-specific Protein Iba1 Enhances Membrane Ruffling and Rac Activation via Phospholipase C-γ-dependent Pathway. J. Biol. Chem. 2002, 277, 20026–20032. [Google Scholar] [CrossRef]
- Bennett, M.L.; Bennett, F.C.; Liddelow, S.A.; Ajami, B.; Zamanian, J.L.; Fernhoff, N.B.; Mulinyawe, S.B.; Bohlen, C.J.; Adil, A.; Tucker, A.; et al. New tools for studying microglia in the mouse and human CNS. Proc. Natl. Acad. Sci. USA 2016, 113, E1738–E1746. [Google Scholar] [CrossRef]
- Li, Y.; Glotfelty, E.J.; Karlsson, T.; Fortuno, L.V.; Harvey, B.K.; Greig, N.H. The metabolite GLP-1 (9-36) is neuroprotective and anti-Inflammatory in cellular models of neurodegeneration. J. Neurochem. 2021, 159, 867–886. [Google Scholar] [CrossRef] [PubMed]
- Abd-El-Basset, E.; Fedoroff, S. Effect of bacterial wall lipopolysaccharide (LPS) on morphology, motility, and cytoskeletal organization of microglia in cultures. J. Neurosci. Res. 1995, 41, 222–237. [Google Scholar] [CrossRef] [PubMed]
- Hsueh, S.C.; Luo, W.; Tweedie, D.; Kim, D.S.; Kim, Y.K.; Hwang, I.; Gil, J.-E.; Han, B.-S.; Chiang, Y.-H.; Selman, W.; et al. N-Adamantyl Phthalimidine: A New Thalidomide-like Drug That Lacks Cereblon Binding and Mitigates Neuronal and Synaptic Loss, Neuroinflammation, and Behavioral Deficits in Traumatic Brain Injury and LPS Challenge. ACS Pharmacol. Transl. Sci. 2021, 4, 980–1000. [Google Scholar] [CrossRef] [PubMed]
- Horvath, R.J.; Nutile-McMenemy, N.; Alkaitis, M.S.; DeLeo, J.A. Differential migration, LPS-induced cytokine, chemokine, and NO expression in immortalized BV-2 and HAPI cell lines and primary microglial cultures. J. Neurochem. 2008, 107, 557–569. [Google Scholar] [CrossRef] [PubMed]
- Stansley, B.; Post, J.; Hensley, K. A comparative review of cell culture systems for the study of microglial biology in Alzheimer’s disease. J. Neuroinflamm. 2012, 9, 115. [Google Scholar] [CrossRef] [PubMed]
- Narumiya, S.; Ishizaki, T.; Ufhata, M. Use and properties of ROCK-specific inhibitor Y-27632. In Methods in Enzymology; Academic Press: Cambridge, MA, USA, 2000; Volume 325, pp. 273–284. [Google Scholar] [CrossRef]
- Aktan, F. INOS-Mediated Nitric Oxide Production and Its Regulation. Life Sci. 2004, 75, 639–653. [Google Scholar] [CrossRef]
- Mizuno, K. Signaling mechanisms and functional roles of cofilin phosphorylation and dephosphorylation. Cell. Signal. 2013, 25, 457–469. [Google Scholar] [CrossRef]
- Alhadidi, Q.; Shah, Z.A. Cofilin Mediates LPS-Induced Microglial Cell Activation and Associated Neurotoxicity Through Activation of NF-κB and JAK–STAT Pathway. Mol. Neurobiol. 2018, 55, 1676–1691. [Google Scholar] [CrossRef]
- Alhadidi, Q.; Nash, K.M.; Alaqel, S.; Bin Sayeed, M.S.; Shah, Z.A. Cofilin Knockdown Attenuates Hemorrhagic Brain Injury-induced Oxidative Stress and Microglial Activation in Mice. Neuroscience 2018, 383, 33–45. [Google Scholar] [CrossRef]
- Niederöst, B.; Oertle, T.; Fritsche, J.; McKinney, R.A.; Bandtlow, C.E. Nogo-A and Myelin-Associated Glycoprotein Mediate Neurite Growth Inhibition by Antagonistic Regulation of RhoA and Rac1. J. Neurosci. 2002, 22, 10368–10376. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Song, S.; Zhang, Y.; Ge, Y.; Fang, X.; Huang, T.; Du, J.; Gao, J. Inhibition of the Rho/Rho kinase pathway prevents lipopolysaccharide-induced hyperalgesia and the release of TNF-α and IL-1β in the mouse spinal cord. Sci. Rep. 2015, 5, 14553. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, S.; Tahara, M.; Ogata, S.; Hashimoto, K.; Morishige, K.; Tasaka, K.; Murata, Y. Involvement of nuclear factor-kB activation through RhoA/Rho-kinase pathway in LPS-induced IL-8 production in human cervical stromal cells. Mol. Hum. Reprod. 2007, 13, 181–187. [Google Scholar] [CrossRef] [PubMed]
- Hsueh, S.C.; Scerba, M.T.; Tweedie, D.; Lecca, D.; Kim, D.S.; Baig, A.M.; Kim, Y.K.; Hwang, I.; Kim, S.; Selman, W.R.; et al. Activity of a Novel Anti-Inflammatory Agent F-3,6′-dithiopomalidomide as a Treatment for Traumatic Brain Injury. Biomedicines 2022, 10, 2449. [Google Scholar] [CrossRef] [PubMed]
- Lier, J.; Streit, W.J.; Bechmann, I. Beyond Activation: Characterizing Microglial Functional Phenotypes. Cells 2021, 10, 2236. [Google Scholar] [CrossRef] [PubMed]
- Tak, P.P.; Firestein, G.S. NF-ΚB: A Key Role in Inflammatory Diseases. J. Clin. Investig. 2001, 107, 7–11. [Google Scholar] [CrossRef]
- Bagaev, A.V.; Garaeva, A.Y.; Lebedeva, E.S.; Pichugin, A.V.; Ataullakhanov, R.I.; Ataullakhanov, F.I. Elevated pre-activation basal level of nuclear NF-κB in native macrophages accelerates LPS-induced translocation of cytosolic NF-κB into the cell nucleus. Sci. Rep. 2019, 9, 4563. [Google Scholar] [CrossRef]
- Zambrano, S.; De Toma, I.; Piffer, A.; Bianchi, M.E.; Agresti, A. NF-κB oscillations translate into functionally related patterns of gene expression. Elife 2016, 5, e09100. [Google Scholar] [CrossRef] [PubMed]
- Sung, M.-H.; Salvatore, L.; De Lorenzi, R.; Indrawan, A.; Pasparakis, M.; Hager, G.L.; Bianchi, M.E.; Agresti, A. Sustained Oscillations of NF-κB Produce Distinct Genome Scanning and Gene Expression Profiles. PLoS ONE 2009, 4, e7163. [Google Scholar] [CrossRef]
- Keren-Shaul, H.; Spinrad, A.; Weiner, A.; Matcovitch-Natan, O.; Dvir-Szternfeld, R.; Ulland, T.K.; David, E.; Baruch, K.; Lara-Astaiso, D.; Toth, B.; et al. A Unique Microglia Type Associated with Restricting Development of Alzheimer’s Disease. Cell 2017, 169, 1276–1290.e17. [Google Scholar] [CrossRef]
- Marschallinger, J.; Iram, T.; Zardeneta, M.; Lee, S.E.; Lehallier, B.; Haney, M.S.; Pluvinage, J.V.; Mathur, V.; Hahn, O.; Morgens, D.W.; et al. Lipid-droplet-accumulating microglia represent a dysfunctional and proinflammatory state in the aging brain. Nat. Neurosci. 2020, 23, 194–208. [Google Scholar] [CrossRef] [PubMed]
- Clayton, K.; Delpech, J.C.; Herron, S.; Iwahara, N.; Ericsson, M.; Saito, T.; Saido, T.C.; Ikezu, S.; Ikezu, T. Plaque associated microglia hyper-secrete extracellular vesicles and accelerate tau propagation in a humanized APP mouse model. Mol. Neurodegener. 2021, 16, 18. [Google Scholar] [CrossRef] [PubMed]
- Jana, M.; Palencia, C.A.; Pahan, K. Fibrillar Amyloid-β Peptides Activate Microglia via TLR2: Implications for Alzheimer’s Disease. J. Immunol. 2008, 181, 7254–7262. [Google Scholar] [CrossRef] [PubMed]
- Timmerman, R.; Burm, S.M.; Bajramovic, J.J. An Overview of in vitro Methods to Study Microglia. Front. Cell. Neurosci. 2018, 12, 242. [Google Scholar] [CrossRef] [PubMed]
- Sarkar, S.; Malovic, E.; Sarda, D.; Lawana, V.; Rokad, D.; Jin, H.; Anantharam, V.; Kanthasamy, A.; Kanthasamy, A.G. Characterization and comparative analysis of a new mouse microglial cell model for studying neuroinflammatory mechanisms during neurotoxic insults. Neurotoxicology 2018, 67, 129–140. [Google Scholar] [CrossRef]
- Bohlen, C.J.; Bennett, F.C.; Tucker, A.F.; Collins, H.Y.; Mulinyawe, S.B.; Barres, B.A. Diverse Requirements for Microglial Survival, Specification, and Function Revealed by Defined-Medium Cultures. Neuron 2017, 94, 759–773.e8. [Google Scholar] [CrossRef]
- Bohlen, C.J.; Bennett, F.C.; Bennett, M.L. Isolation and Culture of Microglia. Curr. Protoc. Immunol. 2018, 125, e70. [Google Scholar] [CrossRef]
- Cui, G.; Zuo, T.; Zhao, Q.; Hu, J.; Jin, P.; Zhao, H.; Jing, J.; Zhu, J.; Chen, H.; Liu, B.; et al. ROCK mediates the inflammatory response in thrombin induced microglia. Neurosci. Lett. 2013, 554, 82–87. [Google Scholar] [CrossRef]
- Shinozaki, Y.; Danjo, Y.; Koizumi, S. Microglial ROCK is essential for chronic methylmercury-Induced neurodegeneration. J. Neurochem. 2019, 151, 64–78. [Google Scholar] [CrossRef]
- Scheiblich, H.; Bicker, G. Regulation of Microglial Phagocytosis by RhoA/ROCK-Inhibiting Drugs. Cell. Mol. Neurobiol. 2017, 37, 461–473. [Google Scholar] [CrossRef]
- Zhang, Y.; Wang, X.; Jiang, C.; Chen, Z.; Ni, S.; Fan, H.; Wang, Z.; Tian, F.; An, J.; Yang, H.; et al. Rho Kinase Inhibitor Y27632 Improves Recovery After Spinal Cord Injury by Shifting Astrocyte Phenotype and Morphology via the ROCK/NF-κB/C3 Pathway. Neurochem. Res. 2022, 47, 3733–3744. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Pan, Z.; Ma, X.; Cui, J.; Gao, J.; Miao, Q.; Zhu, Z.; Chen, X.; Su, S. ROCK inhibitor fasudil reduces the expression of inflammatory factors in LPS-induced rat pulmonary microvascular endothelial cells via ROS/NF-κB pathway. BMC Pharmacol. Toxicol. 2022, 23, 24. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Sun, Z.; Jin, M.; Tu, Y.; Wang, S.; Yang, X.; Chen, Q.; Zhang, X.; Han, Y.; Pi, R. Inhibition of AGEs/RAGE/Rho/ROCK pathway suppresses non-specific neuroinflammation by regulating BV2 microglial M1/M2 polarization through the NF-κB pathway. J. Neuroimmunol. 2017, 305, 108–114. [Google Scholar] [CrossRef] [PubMed]
- Namme, J.N.; Bepari, A.K.; Takebayashi, H. Cofilin Signaling in the CNS Physiology and Neurodegeneration. Int. J. Mol. Sci. 2021, 22, 10727. [Google Scholar] [CrossRef] [PubMed]
- Alsegiani, A.S.; Shah, Z.A. The role of cofilin in age-related neuroinflammation. Neural Regen. Res. 2020, 15, 1451–1459. [Google Scholar] [CrossRef] [PubMed]
- Endo, M.; Ohashi, K.; Sasaki, Y.; Goshima, Y.; Niwa, R.; Uemura, T.; Mizuno, K. Control of Growth Cone Motility and Morphology by LIM Kinase and Slingshot via Phosphorylation and Dephosphorylation of Cofilin. J. Neurosci. 2003, 23, 2527–2537. [Google Scholar] [CrossRef] [PubMed]
- Alaqel, S.I.; Dlamini, S.; Almarghalani, D.A.; Shettigar, A.; Alhadidi, Q.; Kodithuwakku, S.H.; Stary, C.; Tillekeratne, L.M.V.; Shah, Z.A. Synthesis and Development of a Novel First-in-Class Cofilin Inhibitor for Neuroinflammation in Hemorrhagic Brain Injury. ACS Chem. Neurosci. 2022, 13, 1014–1029. [Google Scholar] [CrossRef]
- Zhang, Z.; Ottens, A.; Larner, S.F.; Kobeissy, F.; Williams, M.L.; Hayes, R.L.; Wang, K.K.W. Direct Rho-associated kinase inhibiton induces cofilin dephosphorylation and neurite outgrowth in PC-12 cells. Cell. Mol. Biol. Lett. 2006, 11, 12–29. [Google Scholar] [CrossRef]
- Bi, D.; Wang, Y.; Gao, Y.; Li, X.; Chu, Q.; Cui, J.; Xu, T. Recognition of Lipopolysaccharide and Activation of NF-κB by Cytosolic Sensor NOD1 in Teleost Fish. Front. Immunol. 2018, 9, 1413. [Google Scholar] [CrossRef]
- Bielig, H.; Lautz, K.; Braun, P.R.; Menning, M.; Machuy, N.; Brügmann, C.; Barisic, S.; Eisler, S.A.; Andrée, M.; Zurek, B.; et al. The Cofilin Phosphatase Slingshot Homolog 1 (SSH1) Links NOD1 Signaling to Actin Remodeling. PLoS Pathog. 2014, 10, e1004351. [Google Scholar] [CrossRef]
- Duan, Q.; Jia, Y.; Qin, Y.; Jin, Y.; Hu, H.; Chen, J. Narciclasine attenuates LPS-induced acute lung injury in neonatal rats through suppressing inflammation and oxidative stress. Bioengineered 2020, 11, 801–810. [Google Scholar] [CrossRef] [PubMed]
- Zhao, D.; Zhang, L.J.; Huang, T.Q.; Kim, J.; Gu, M.-Y.; Yang, H.O. Narciclasine inhibits LPS-induced neuroinflammation by modulating the Akt/IKK/NF-κB and JNK signaling pathways. Phytomedicine 2021, 85, 153540. [Google Scholar] [CrossRef]
- Kingsley, M.K.; Bhat, B.V.; Badhe, B.A.; Dhas, B.B.; Parija, S.C. Narciclasine improves outcome in sepsis among neonatal rats via inhibition of calprotectin and alleviating inflammatory responses. Sci. Rep. 2020, 10, 2947. [Google Scholar] [CrossRef] [PubMed]
- Shen, C.-Y.; Xu, X.-L.; Yang, L.-J.; Jiang, J.-G. Identification of narciclasine from Lycoris radiata (L’Her.) Herb. and its inhibitory effect on LPS-induced inflammatory responses in macrophages. Food Chem. Toxicol. 2019, 125, 605–613. [Google Scholar] [CrossRef] [PubMed]
- Fessler, M.B.; Arndt, P.G.; Just, I.; Nick, J.A.; Malcolm, K.C.; Worthen, G.S. Dual role for RhoA in suppression and induction of cytokines in the human neutrophil. Blood 2007, 109, 1248–1256. [Google Scholar] [CrossRef]
- Schwartz, M. Rho signalling at a glance. J. Cell Sci. 2004, 117, 5457–5458. [Google Scholar] [CrossRef]
- Socodato, R.; Portugal, C.C.; Canedo, T.; Rodrigues, A.; Almeida, T.O.; Henriques, J.F.; Vaz, S.H.; Magalhães, J.; Silva, C.M.; Baptista, F.I.; et al. Microglia Dysfunction Caused by the Loss of Rhoa Disrupts Neuronal Physiology and Leads to Neurodegeneration. Cell Rep. 2020, 31, 107796. [Google Scholar] [CrossRef]
- Gil, V.; Nicolas, O.; Mingorance, A.; Ureña, J.M.; Tang, B.L.; Hirata, T.; Sáez-Valero, J.; Ferrer, I.; Soriano, E.; Del Río, J.A. Nogo-A Expression in the Human Hippocampus in Normal Aging and in Alzheimer Disease. J. Neuropathol. Exp. Neurol. 2006, 65, 433–444. [Google Scholar] [CrossRef]
- Satoh, J.-I.; Onoue, H.; Arima, K.; Yamamura, T. Nogo-A and Nogo Receptor Expression in Demyelinating Lesions of Multiple Sclerosis. J. Neuropathol. Exp. Neurol. 2005, 64, 129–138. [Google Scholar] [CrossRef]
- Bandtlow, C.E.; Dlaska, M.; Pirker, S.; Czech, T.; Baumgartner, C.; Sperk, G. Increased expression of Nogo-A in hippocampal neurons of patients with temporal lobe epilepsy. Eur. J. Neurosci. 2004, 20, 195–206. [Google Scholar] [CrossRef]
- Trifunovski, A.; Josephson, A.; Bickford, P.; Olson, L.; Brené, S. Selective decline of Nogo mRNA in the aging brain. Neuroreport 2006, 17, 913–916. [Google Scholar] [CrossRef] [PubMed]
- Smedfors, G.; Olson, L.; Karlsson, T.E. A Nogo-Like Signaling Perspective from Birth to Adulthood and in Old Age: Brain Expression Patterns of Ligands, Receptors and Modulators. Front. Mol. Neurosci. 2018, 11, 42. [Google Scholar] [CrossRef] [PubMed]
- Tsai, S.-Y.; Papadopoulos, C.M.; Schwab, M.E.; Kartje, G.L. Delayed Anti-Nogo-A Therapy Improves Function After Chronic Stroke in Adult Rats. Stroke 2011, 42, 186–190. [Google Scholar] [CrossRef] [PubMed]
- Starkey, M.L.; Schwab, M.E. Anti-Nogo-A and Training: Can One plus One Equal Three? Exp. Neurol. 2012, 235, 53–61. [Google Scholar] [CrossRef] [PubMed]
- Kulczyńska-Przybik, A.; Dulewicz, M.; Słowik, A.; Borawska, R.; Kułakowska, A.; Kochanowicz, J.; Mroczko, B. The Clinical Significance of Cerebrospinal Fluid Reticulon 4 (RTN4) Levels in the Differential Diagnosis of Neurodegenerative Diseases. J. Clin. Med. 2021, 10, 5281. [Google Scholar] [CrossRef] [PubMed]
- Nakagawa, O.; Fujisawa, K.; Ishizaki, T.; Saito, Y.; Nakao, K.; Narumiya, S. ROCK-I and ROCK-II, two isoforms of Rho-associated coiled-coil forming protein serine/threonine kinase in mice. FEBS Lett. 1996, 392, 189–193. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Sloan, S.A.; Clarke, L.E.; Caneda, C.; Plaza, C.A.; Blumenthal, P.D.; Vogel, H.; Steinberg, G.K.; Edwards, M.S.; Li, G.; et al. Purification and Characterization of Progenitor and Mature Human Astrocytes Reveals Transcriptional and Functional Differences with Mouse. Neuron 2016, 89, 37–53. [Google Scholar] [CrossRef] [PubMed]
- Glotfelty, E.; Olson, L.; Karlsson, T.E.; Li, Y.; Greig, N.H. Glucagon-like peptide-1 (GLP-1)-based receptor agonists as a treatment for Parkinson’s disease. Expert Opin. Investig. Drugs 2020, 29, 595–602. [Google Scholar] [CrossRef] [PubMed]
- Gunosewoyo, H.; Yu, L.; Munoz, L.; Kassiou, M. Kinase targets in CNS drug discovery. Future Med. Chem. 2017, 9, 303–314. [Google Scholar] [CrossRef]
- Sellers, K.J.; Elliott, C.; Jackson, J.; Ghosh, A.; Ribe, E.; Rojo, A.I.; Jarosz-Griffiths, H.H.; Watson, I.A.; Xia, W.; Semenov, M.; et al. Amyloid β synaptotoxicity is Wnt-PCP dependent and blocked by fasudil. Alzheimer’s Dement. 2017, 14, 306–317. [Google Scholar] [CrossRef]
- Takanashi, Y.; Ishida, T.; Kirchmeier, M.J.; Shuaib, A.; Allen, T.M. Neuroprotection by Intrathecal Application of Liposome-entrapped Fasudil in a Rat Model of Ischemia. Neurol. Med.-Chir. 2001, 41, 107–114. [Google Scholar] [CrossRef] [PubMed]
- Wadman, M. FDA no longer has to require animal testing for new drugs. Science 2023, 379, 127–128. [Google Scholar] [CrossRef] [PubMed]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Probe Name | 5′-3′ Sequence | Source | |
|---|---|---|---|
| ROCK1 | Fw | GCTCATCTCTGTGTGACTCT | NM_009071.2 |
| Rv | TACGGAAAGCAAGTCAGACC | ||
| ROCK2 | Fw | GGTCAATCAGCTCCAGAAAC | NM_009072.2 |
| Rv | GTTTGGAACTTTCTGCCTGG | ||
| TNF-α | Fw | GGCAGGTCTACTTTGGAGTCATTG | [49] |
| Rv | ACATTCGAGGCTCCAGTGAATTCGG | ||
| iNOS | Fw | TACTCCATCAGCTCCTCCCA | NM_010927.4 |
| Rv | GTTCCTGATCCAAGTGCTGC | ||
| IL-6 | Fw | TTCTTGGGACTGATGTTGTTGAC | [50] |
| Rv | AATTAAGCCTCCGACTTGTGAAG | ||
| β-actin | Fw | TGAGAGGGAAATCGTGCGTGAC | NM_007393.4 |
| Rv | CCGCTCGTTGCCAATAGTGATG |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Glotfelty, E.J.; Tovar-y-Romo, L.B.; Hsueh, S.-C.; Tweedie, D.; Li, Y.; Harvey, B.K.; Hoffer, B.J.; Karlsson, T.E.; Olson, L.; Greig, N.H. The RhoA-ROCK1/ROCK2 Pathway Exacerbates Inflammatory Signaling in Immortalized and Primary Microglia. Cells 2023, 12, 1367. https://doi.org/10.3390/cells12101367
Glotfelty EJ, Tovar-y-Romo LB, Hsueh S-C, Tweedie D, Li Y, Harvey BK, Hoffer BJ, Karlsson TE, Olson L, Greig NH. The RhoA-ROCK1/ROCK2 Pathway Exacerbates Inflammatory Signaling in Immortalized and Primary Microglia. Cells. 2023; 12(10):1367. https://doi.org/10.3390/cells12101367
Chicago/Turabian StyleGlotfelty, Elliot J., Luis B. Tovar-y-Romo, Shih-Chang Hsueh, David Tweedie, Yazhou Li, Brandon K. Harvey, Barry J. Hoffer, Tobias E. Karlsson, Lars Olson, and Nigel H. Greig. 2023. "The RhoA-ROCK1/ROCK2 Pathway Exacerbates Inflammatory Signaling in Immortalized and Primary Microglia" Cells 12, no. 10: 1367. https://doi.org/10.3390/cells12101367
APA StyleGlotfelty, E. J., Tovar-y-Romo, L. B., Hsueh, S. -C., Tweedie, D., Li, Y., Harvey, B. K., Hoffer, B. J., Karlsson, T. E., Olson, L., & Greig, N. H. (2023). The RhoA-ROCK1/ROCK2 Pathway Exacerbates Inflammatory Signaling in Immortalized and Primary Microglia. Cells, 12(10), 1367. https://doi.org/10.3390/cells12101367