
Effect of Photobiomodulation on Protein Kinase Cδ, Cytochrome C, and Mitochondria in U87 MG Cells





Abstract
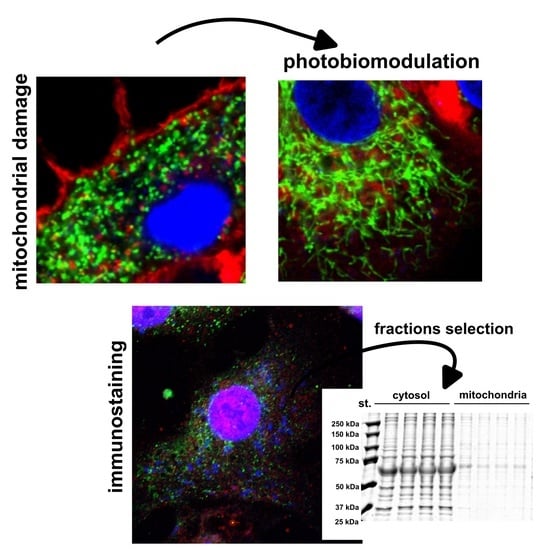
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Cell Cultures and Therapeutical Protocol
- (1)
- Cells were treated with 10 µM rotenone (Sigma-Aldrich, St. Louis, MO, USA) for 24 h. Furthermore, these cells were irradiated with PBM at 808 nm and 1.8 J/cm2 (120 s with an irradiance of 15 mW/cm2). The effect of PBM was evaluated immediately (0 h) after irradiation and 1–5 h after irradiation.
- (2)
- The cells were treated with PBM at 808 nm and 1.8 J/cm2, and subsequently 1 µM PMA (Sigma-Aldrich, St. Louis, MO, USA) was administered to the cells for 5 h.
- (3)
- The cells were treated with PBM at 808 nm and 1.8 J/cm2. The effect of PBM was evaluated immediately (0 h) after irradiation and 1–5 h after irradiation.
2.2. Vital- and Immuno-Fluorescence Confocal Microscopy
2.3. Western Blot Analysis
2.4. Fluorescence Lifetime Imaging of Mitochondrial Oxidative Stress Level
2.5. Statistical Analysis
3. Results and Discussion
3.1. Level of Protein Kinase Cδ and Autophagy in U87MG Cells after PBM
3.2. Morphology of the Mitochondria and Level of Oxidative Stress in the Mitochondria of U87MG Cells after PBM
3.3. Cytochrome c and PKCδ Distribution in U87MG Cells after PBM
3.4. Phosphorylation of PKCδ in U87MG Cells after PBM
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Hamblin, M.R. Mechanisms and Mitochondrial Redox Signaling in Photobiomodulation. Photochem. Photobiol. 2018, 94, 199–212. [Google Scholar] [CrossRef]
- Sroka, R.; Schaffer, M.; Fuchs, C.; Pongratz, T.; Schrader-Reichard, U.; Busch, M.; Schaffer, P.M.; Dühmke, E.; Baumgartner, R. Effects on the mitosis of normal and tumor cells induced by light treatment of different wavelengths. Lasers Surg. Med. 1999, 25, 263–271. [Google Scholar] [CrossRef]
- Murayama, H.; Sadakane, K.; Yamanoha, B.; Kogure, S. Low-power 808-nm laser irradiation inhibits cell proliferation of a human-derived glioblastoma cell line in vitro. Lasers Med. Sci. 2012, 27, 87–93. [Google Scholar] [CrossRef] [PubMed]
- Ravera, S.; Bertola, N.; Pasquale, C.; Bruno, S.; Benedicenti, S.; Ferrando, S.; Zekiy, A.; Arany, P.; Amaroli, A. 808-nm photobiomodulation affects the viability of a head and neck squamous carcinoma cellular model, acting on energy metabolism and oxidative stress production. Biomedicines 2021, 9, 1717. [Google Scholar] [CrossRef]
- Amaroli, A.; Pasquale, C.; Zekiy, A.; Utyuzh, A.; Benedicenti, S.; Signore, A.; Ravera, S. Photobiomodulation and Oxidative Stress: 980 nm Diode Laser Light Regulates Mitochondrial Activity and Reactive Oxygen Species Production. Oxid. Med. Cell. Longev. 2021, 2021, 6626286. [Google Scholar] [CrossRef] [PubMed]
- Kausar, S.; Wang, F.; Cui, H. The Role of Mitochondria in Reactive Oxygen Species Generation and Its Implications for Neurodegenerative Diseases. Cells 2018, 7, 274. [Google Scholar] [CrossRef]
- Murphy, M.P. How mitochondria produce reactive oxygen species. Biochem. J. 2009, 417, 1–13. [Google Scholar] [CrossRef]
- Chance, B. Reaction of Oxygen with the Respiratory Chain in Cells and Tissues. J. Gen. Physiol. 1965, 49, 163–188. [Google Scholar] [CrossRef]
- Zhao, R.Z.; Jiang, S.; Zhang, L.; Yu, Z. Bin Mitochondrial electron transport chain, ROS generation and uncoupling (Review). Int. J. Mol. Med. 2019, 44, 3–15. [Google Scholar]
- Ighodaro, O.M.; Akinloye, O.A. First line defence antioxidants-superoxide dismutase (SOD), catalase (CAT) and glutathione peroxidase (GPX): Their fundamental role in the entire antioxidant defence grid. Alex. J. Med. 2018, 54, 287–293. [Google Scholar] [CrossRef]
- Zorov, D.B.; Juhaszova, M.; Sollott, S.J. Mitochondrial Reactive Oxygen Species (ROS) and ROS-Induced ROS Release. Physiol. Rev. 2014, 94, 909–950. [Google Scholar] [CrossRef] [PubMed]
- Zorov, D.B.; Filburn, C.R.; Klotz, L.O.; Zweier, J.L.; Sollott, S.J. Reactive oxygen species (ROS)-induced ROS release: A new phenomenon accompanying induction of the mitochondrial permeability transition in cardiac myocytes. J. Exp. Med. 2000, 192, 1001–1014. [Google Scholar] [CrossRef] [PubMed]
- Wan, J.; Kalpage, H.A.; Vaishnav, A.; Liu, J.; Lee, I.; Mahapatra, G.; Turner, A.A.; Zurek, M.P.; Ji, Q.; Moraes, C.T.; et al. Regulation of Respiration and Apoptosis by Cytochrome c Threonine 58 Phosphorylation. Sci. Rep. 2019, 9, 15815. [Google Scholar] [CrossRef] [PubMed]
- Garrido, C.; Galluzzi, L.; Brunet, M.; Puig, P.E.; Didelot, C.; Kroemer, G. Mechanisms of cytochrome c release from mitochondria. Cell Death Differ. 2006, 13, 1423–1433. [Google Scholar] [CrossRef]
- Cereghetti, G.M.; Scorrano, L. The many shapes of mitochondrial death. Oncogene 2006, 25, 4717–4724. [Google Scholar] [CrossRef] [PubMed]
- Martinou, J.C.; Youle, R.J. Mitochondria in Apoptosis: Bcl-2 Family Members and Mitochondrial Dynamics. Dev. Cell 2011, 21, 92–101. [Google Scholar] [CrossRef]
- Ott, M.; Robertson, J.D.; Gogvadze, V.; Zhivotovsky, B.; Orrenius, S. Cytochrome c release from mitochondria proceeds by a two-step process. Proc. Natl. Acad. Sci. USA 2002, 99, 1259–1263. [Google Scholar] [CrossRef]
- Zamzami, N.; El Hamel, C.; Maisse, C.; Brenner, C.; Mũoz-Pinedo, C.; Belzacq, A.S.; Costantini, P.; Vieira, H.; Loeffler, M.; Molle, G.; et al. Bid acts on the permeability transition pore complex to induce apoptosis. Oncogene 2000, 19, 6342–6350. [Google Scholar] [CrossRef]
- Hayes, J.D.; Dinkova-Kostova, A.T.; Tew, K.D. Oxidative Stress in Cancer. Cancer Cell 2020, 38, 167–197. [Google Scholar] [CrossRef]
- Lenkavska, L.; Tomkova, S.; Horvath, D.; Huntosova, V. Searching for combination therapy by clustering methods: Stimulation of PKC in Golgi apparatus combined with hypericin induced PDT. Photodiagnosis Photodyn. Ther. 2020, 31, 101813. [Google Scholar] [CrossRef]
- Tomkova, S.; Misuth, M.; Lenkavska, L.; Miskovsky, P.; Huntosova, V. In vitro identification of mitochondrial oxidative stress production by time-resolved fluorescence imaging of glioma cells. Biochim. Biophys. Acta—Mol. Cell Res. 2018, 1865, 616–628. [Google Scholar] [CrossRef] [PubMed]
- Misuth, M.; Joniova, J.; Horvath, D.; Dzurova, L.; Nichtova, Z.; Novotova, M.; Miskovsky, P.; Stroffekova, K.; Huntosova, V. The flashlights on a distinct role of protein kinase C δ: Phosphorylation of regulatory and catalytic domain upon oxidative stress in glioma cells. Cell. Signal. 2017, 34, 11–22. [Google Scholar] [CrossRef] [PubMed]
- Blumberg, P.M. Protein Kinase C as the Receptor for the Phorbol Ester Tumor Promoters: Sixth Rhoads Memorial Award Lecture. Cancer Res. 1986, 48, 1–8. [Google Scholar]
- Gonzalez-Guerrico, A.M.; Kazanietz, M.G. Phorbol ester-induced apoptosis in prostate cancer cells via autocrine activation of the extrinsic apoptotic cascade: A key role for protein kinase Cδ. J. Biol. Chem. 2005, 280, 38982–38991. [Google Scholar] [CrossRef] [PubMed]
- De Vente, J.E.; Kukoly, C.A.; Bryant, W.O.; Posekany, K.J.; Chen, J.; Fletcher, D.J.; Parker, P.J.; Pettit, G.J.; Lozano, G.; Cook, P.P.; et al. Phorbol esters induce death in MCF-7 breast cancer cells with altered expression of protein kinase C isoforms: Role for p53-independent induction of gadd45 in initiating death. J. Clin. Investig. 1995, 96, 1874–1886. [Google Scholar] [CrossRef]
- Bond, J.A.; Gescher, A.J.; Verschoyle, R.D.; Lemoine, N.R.; Errington, R.; Wiltshire, M.; Smith, P.J.; Wynford-Thomas, D. Cytotoxic action of phorbol esters on human pancreatic cancer cells. Int. J. Cancer 2007, 121, 1445–1454. [Google Scholar] [CrossRef]
- Wang, Y.; Biswas, G.; Prabu, S.K.; Avadhani, N.G. Modulation of mitochondrial metabolic function by phorbol 12-myristate 13-acetate through increased mitochondrial translocation of protein kinase Cα in C2C12 myocytes. Biochem. Pharmacol. 2006, 72, 881–892. [Google Scholar] [CrossRef]
- Huang, Y.Y.; Nagata, K.; Tedford, C.E.; Hamblin, M.R. Low-level laser therapy (810 nm) protects primary cortical neurons against excitotoxicity in vitro. J. Biophotonics 2014, 7, 656–664. [Google Scholar] [CrossRef]
- Santulli, G.; Marks, A. Essential Roles of Intracellular Calcium Release Channels in Muscle, Brain, Metabolism, and Aging. Curr. Mol. Pharmacol. 2015, 8, 206–222. [Google Scholar] [CrossRef]
- Dikalov, S.I.; Li, W.; Doughan, A.K.; Blanco, R.R.; Zafari, A.M. Mitochondrial reactive oxygen species and calcium uptake regulate activation of phagocytic NADPH oxidase. Am. J. Physiol. Integr. Comp. Physiol. 2012, 302, R1134–R1142. [Google Scholar] [CrossRef]
- Sato, T.; O’Rourke, B.; Marbán, E. Modulation of mitochondrial ATP-dependent K+ channels by protein kinase C. Circ. Res. 1998, 83, 110–114. [Google Scholar] [CrossRef] [PubMed]
- Majumder, P.K.; Pandey, P.; Sun, X.; Cheng, K.; Datta, R.; Saxena, S.; Kharbanda, S.; Kufe, D. Mitochondrial translocation of protein kinase C δ in phorbol ester-induced cytochrome c release and apoptosis. J. Biol. Chem. 2000, 275, 21793–21796. [Google Scholar] [CrossRef] [PubMed]
- Pevna, V.; Wagnières, G.; Huntosova, V. Autophagy and apoptosis induced in u87 mg glioblastoma cells by hypericin-mediated photodynamic therapy can be photobiomodulated with 808 nm light. Biomedicines 2021, 9, 1703. [Google Scholar] [CrossRef]
- Schneider, C.A.; Rasband, W.S.; Eliceiri, K.W. NIH Image to ImageJ: 25 years of image analysis. Nat. Methods 2012, 9, 671–675. [Google Scholar] [CrossRef] [PubMed]
- Tatmatsu-Rocha, J.C.; Tim, C.R.; Avo, L.; Bernardes-Filho, R.; Brassolatti, P.; Kido, H.W.; Hamblin, M.R.; Parizotto, N.A. Mitochondrial dynamics (fission and fusion) and collagen production in a rat model of diabetic wound healing treated by photobiomodulation: Comparison of 904 nm laser and 850 nm light-emitting diode (LED). J. Photochem. Photobiol. B Biol. 2018, 187, 41–47. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; Dong, Y.; Lu, Y.; Zhang, W.; Brann, D.W.; Zhang, Q. Photobiomodulation for Global Cerebral Ischemia: Targeting Mitochondrial Dynamics and Functions. Mol. Neurobiol. 2019, 56, 1852–1869. [Google Scholar] [CrossRef]
- Chernivec, E.; Cooper, J.; Naylor, K. Exploring the effect of rotenone—A known inducer of Parkinson’s disease—On mitochondrial dynamics in dictyostelium discoideum. Cells 2018, 7, 201. [Google Scholar] [CrossRef]
- Jin, J.; Davis, J.; Zhu, D.; Kashima, D.T.; Leroueil, M.; Pan, C.; Montine, K.S.; Zhang, J. Identification of novel proteins affected by rotenone in mitochondria of dopaminergic cells. BMC Neurosci. 2007, 8, 67. [Google Scholar] [CrossRef]
- Datta, R.; Heaster, T.M.; Sharick, J.T.; Gillette, A.A.; Skala, M.C. Fluorescence lifetime imaging microscopy: Fundamentals and advances in instrumentation, analysis, and applications. J. Biomed. Opt. 2020, 25, 1–43. [Google Scholar] [CrossRef]
- Silveira, P.C.L.; Ferreira, G.K.; Zaccaron, R.P.; Glaser, V.; Remor, A.P.; Mendes, C.; Pinho, R.A.; Latini, A. Effects of photobiomodulation on mitochondria of brain, muscle, and C6 astroglioma cells. Med. Eng. Phys. 2019, 71, 108–113. [Google Scholar] [CrossRef]
- Dzurová, L.; Petrovajova, D.; Nadova, Z.; Huntosova, V.; Miskovsky, P.; Stroffekova, K. The role of anti-apoptotic protein kinase Cα in response to hypericin photodynamic therapy in U-87 MG cells. Photodiagnosis Photodyn. Ther. 2014, 11, 213–226. [Google Scholar] [CrossRef] [PubMed]
- Misuth, M.; Joniova, J.; Belej, D.; Hrivnak, S.; Horvath, D.; Huntosova, V. Estimation of PKCδ autophosphorylation in U87 MG glioma cells: Combination of experimental, conceptual and numerical approaches. J. Biophotonics 2017, 10, 423–432. [Google Scholar] [CrossRef] [PubMed]
- Singh, R.K.; Kumar, S.; Gautam, P.K.; Tomar, M.S.; Verma, P.K.; Singh, S.P.; Kumar, S.; Acharya, A. Protein kinase C-α and the regulation of diverse cell responses. Biomol. Concepts 2017, 8, 143–153. [Google Scholar] [CrossRef] [PubMed]
- Kaul, S.; Kanthasamy, A.; Kitazawa, M.; Anantharam, V.; Kanthasamy, A.G. Caspase-3 dependent proteolytic activation of protein kinase Cδ mediates and regulates 1-methyl-4-phenylpyridinium (MPP+)-induced apoptotic cell death in dopaminergic cells: Relevance to oxidative stress in dopaminergic degeneration. Eur. J. Neurosci. 2003, 18, 1387–1401. [Google Scholar] [CrossRef] [PubMed]
- Ghayur, T.; Hugunin, M.; Talanian, R.V.; Ratnofsky, S.; Quinlan, C.; Emoto, Y.; Pandey, P.; Datta, R.; Huang, Y.; Kharbanda, S.; et al. Proteolytic activation of protein kinase C δ by an ICE/CED 3-like protease induces characteristics of apoptosis. J. Exp. Med. 1996, 184, 2399–2404. [Google Scholar] [CrossRef]
- Cross, T.; Griffiths, G.; Deacon, E.; Sallis, R.; Gough, M.; Watters, D.; Lord, J.M. PKC-δ is an apoptotic lamin kinase. Oncogene 2000, 19, 2331–2337. [Google Scholar] [CrossRef]
- Larroque-Cardoso, P.; Swiader, A.; Ingueneau, C.; Nègre-Salvayre, A.; Elbaz, M.; Reyland, M.E.; Salvayre, R.; Vindis, C. Role of protein kinase C δ in ER stress and apoptosis induced by oxidized LDL in human vascular smooth muscle cells. Cell Death Dis. 2013, 4, e520. [Google Scholar] [CrossRef] [PubMed]
- Rybin, V.O.; Guo, J.; Sabri, A.; Elouardighi, H.; Schaefer, E.; Steinberg, S.F. Stimulus-specific Differences in Protein Kinase Cδ Localization and Activation Mechanisms in Cardiomyocytes. J. Biol. Chem. 2004, 279, 19350–19361. [Google Scholar] [CrossRef]
- Konishi, H.; Yamauchi, E.; Taniguchi, H.; Yamamoto, T.; Matsuzaki, H.; Takemura, Y.; Ohmae, K.; Kikkawa, U.; Nishizuka, Y. Phosphorylation sites of protein kinase C δ in H2O2-treated cells and its activation by tyrosine kinase in vitro. Proc. Natl. Acad. Sci. USA 2001, 98, 6587–6592. [Google Scholar] [CrossRef]
- Kostyak, J.C.; Mauri, B.; Patel, A.; Dangelmaier, C.; Reddy, H.; Kunapuli, S.P. Phosphorylation of protein kinase Cδ Tyr311 positively regulates thromboxane generation in platelets. J. Biol. Chem. 2021, 296, 100720. [Google Scholar] [CrossRef]
- Nakashima, H.; Frank, G.D.; Shirai, H.; Hinoki, A.; Higuchi, S.; Ohtsu, H.; Eguchi, K.; Sanjay, A.; Reyland, M.E.; Dempsey, P.J.; et al. Novel role of protein kinase C-δ Tyr311 phosphorylation in vascular smooth muscle cell hypertrophy by angiotensin II. Hypertension 2008, 51, 232–238. [Google Scholar] [CrossRef] [PubMed]
- Gavish, L.; Gilon, D.; Beeri, R.; Zuckerman, A.; Nachman, D.; Gertz, S.D. Photobiomodulation and estrogen stabilize mitochondrial membrane potential in angiotensin–II challenged porcine aortic smooth muscle cells. J. Biophotonics 2021, 14, e202000329. [Google Scholar] [CrossRef] [PubMed]
- Keszler, A.; Lindemer, B.; Broeckel, G.; Weihrauch, D.; Gao, Y.; Lohr, N.L. In Vivo Characterization of a Red Light-Activated Vasodilation: A Photobiomodulation Study. Front. Physiol. 2022, 13, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Yang, Q.; Langston, J.C.; Tang, Y.; Kiani, M.F.; Kilpatrick, L.E. The role of tyrosine phosphorylation of protein kinase C delta in infection and inflammation. Int. J. Mol. Sci. 2019, 20, 1498. [Google Scholar] [CrossRef] [PubMed]
- Kikkawa, U.; Matsuzaki, H.; Yamamoto, T. Protein kinase Cδ (PKCδ): Activation mechanisms and functions. J. Biochem. 2002, 132, 831–839. [Google Scholar] [CrossRef]
- Rahaman, S.O.; Harbor, P.C.; Chernova, O.; Barnett, G.H.; Vogelbaum, M.A.; Haque, S.J. Inhibition of constitutively active Stat3 suppresses proliferation and induces apoptosis in glioblastoma multiforme cells. Oncogene 2002, 21, 8404–8413. [Google Scholar] [CrossRef]
- Shen, S.; Niso-Santano, M.; Adjemian, S.; Takehara, T.; Malik, S.A.; Minoux, H.; Souquere, S.; Mariño, G.; Lachkar, S.; Senovilla, L.; et al. Cytoplasmic STAT3 Represses Autophagy by Inhibiting PKR Activity. Mol. Cell 2012, 48, 667–680. [Google Scholar] [CrossRef]
- Pevna, V.; Horvath, D.; Wagnieres, G.; Huntosova, V. Photobiomodulation and photodynamic therapy-induced switching of autophagy and apoptosis in human dermal fibroblasts. J. Photochem. Photobiol. B Biol. 2022, 234, 112539. [Google Scholar] [CrossRef]
- Pacifico, A.; Damiani, G.; Iacovelli, P.; Conic, R.R.Z.; Scarabello, A.; Filoni, A.; Malagoli, P.; Bragazzi, N.L.; Pigatto, P.D.M.; Morrone, A. Photoadaptation to ultraviolet B TL01 in psoriatic patients. J. Eur. Acad. Dermatol. Venereol. 2020, 34, 1750–1754. [Google Scholar] [CrossRef]
- Pacifico, A.; Conic, R.R.Z.; Cristaudo, A.; Garbarino, S.; Ardigò, M.; Morrone, A.; Iacovelli, P.; Di Gregorio, S.; Pigatto, P.D.M.; Grada, A.; et al. Diet-related phototoxic reactions in psoriatic patients undergoing phototherapy: Results from a multicenter prospective study. Nutrients 2021, 13, 2934. [Google Scholar] [CrossRef]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pevná, V.; Wagnières, G.; Jancura, D.; Huntošová, V. Effect of Photobiomodulation on Protein Kinase Cδ, Cytochrome C, and Mitochondria in U87 MG Cells. Cells 2023, 12, 1441. https://doi.org/10.3390/cells12101441
Pevná V, Wagnières G, Jancura D, Huntošová V. Effect of Photobiomodulation on Protein Kinase Cδ, Cytochrome C, and Mitochondria in U87 MG Cells. Cells. 2023; 12(10):1441. https://doi.org/10.3390/cells12101441
Chicago/Turabian StylePevná, Viktória, Georges Wagnières, Daniel Jancura, and Veronika Huntošová. 2023. "Effect of Photobiomodulation on Protein Kinase Cδ, Cytochrome C, and Mitochondria in U87 MG Cells" Cells 12, no. 10: 1441. https://doi.org/10.3390/cells12101441
APA StylePevná, V., Wagnières, G., Jancura, D., & Huntošová, V. (2023). Effect of Photobiomodulation on Protein Kinase Cδ, Cytochrome C, and Mitochondria in U87 MG Cells. Cells, 12(10), 1441. https://doi.org/10.3390/cells12101441