

Impact of Short-Term (+)-JQ1 Exposure on Mouse Aorta: Unanticipated Inhibition of Smooth Muscle Contractility

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Methods and Materials
2.1. Materials
2.2. Cell Lines and Cell Culture
2.3. Primary Culture of Mouse Aortic SMCs
2.4. Western Blot Analysis
2.5. Myography
2.6. Ca2+ Flux in Isolated Aortic Rings
2.7. Ca2+ Signal Assay
2.8. Generation of Mice for Conditional KO of BRD4 in SMCs
2.9. Non-Invasive Blood Pressure Measurement following (+)-JQ1 Administration
2.10. Neointima Formation in Mouse Carotid Arteries
2.11. Statistical Analysis
3. Results
3.1. (+)-JQ1 Attenuates Mouse Aortic Contractile Responses by KCl and PE
3.2. (+)-JQ1 Inhibition of Aortic Contraction in Endothelium Intact Preparations Involves Activation of eNOS and Guanylyl Cyclase (GC)
3.3. (+)-JQ1 Attenuation of Contractile Responses Is Calcium-Dependent, and through Inhibiting the Contractile Machinery
3.4. (+)-JQ1 Inhibits Ca2+ Influx in Cultured SMCs
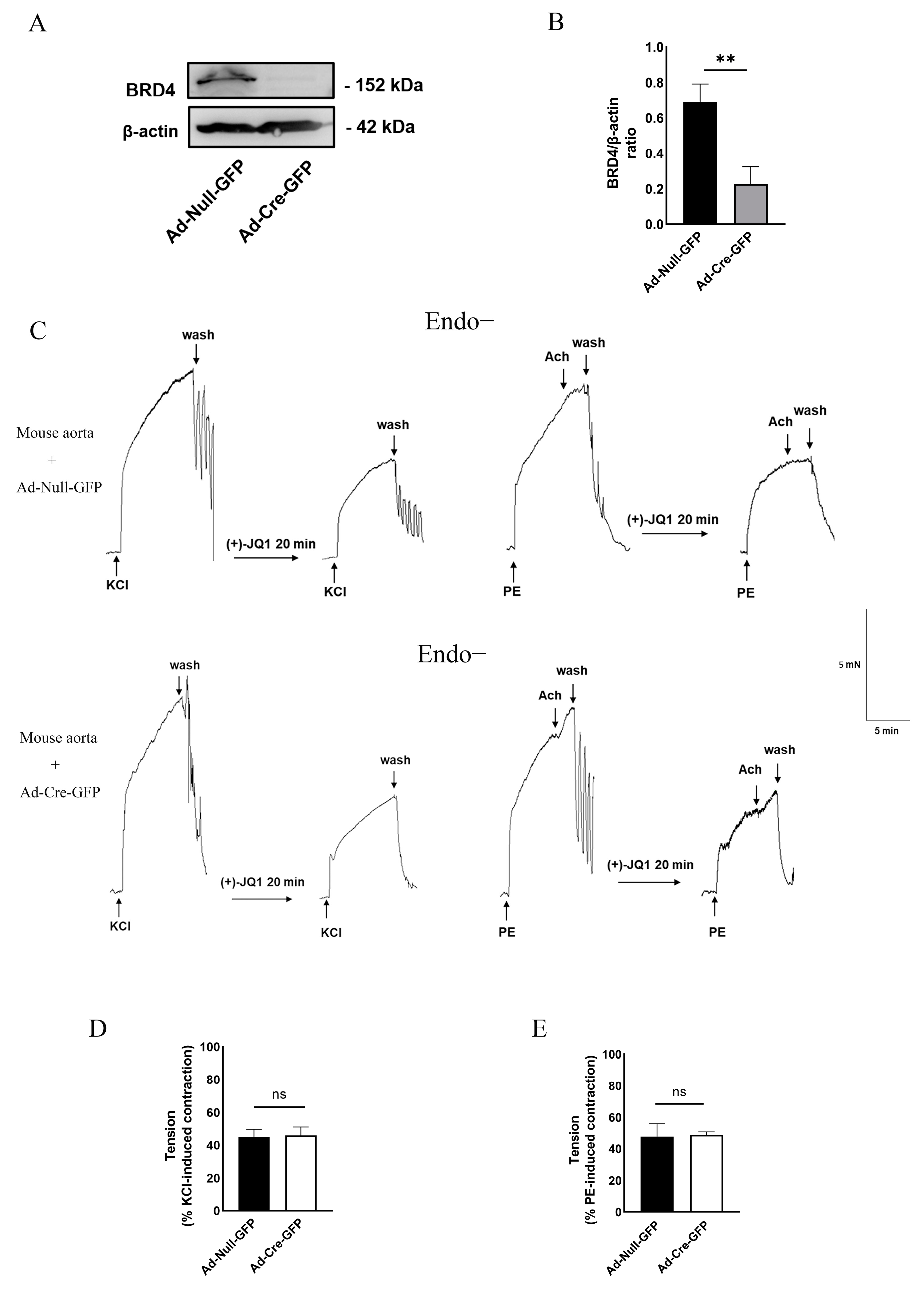
3.5. Direct Inhibition of Smooth Muscle Contraction by (+)-JQ1 Is Independent of BRD4 in SMCs
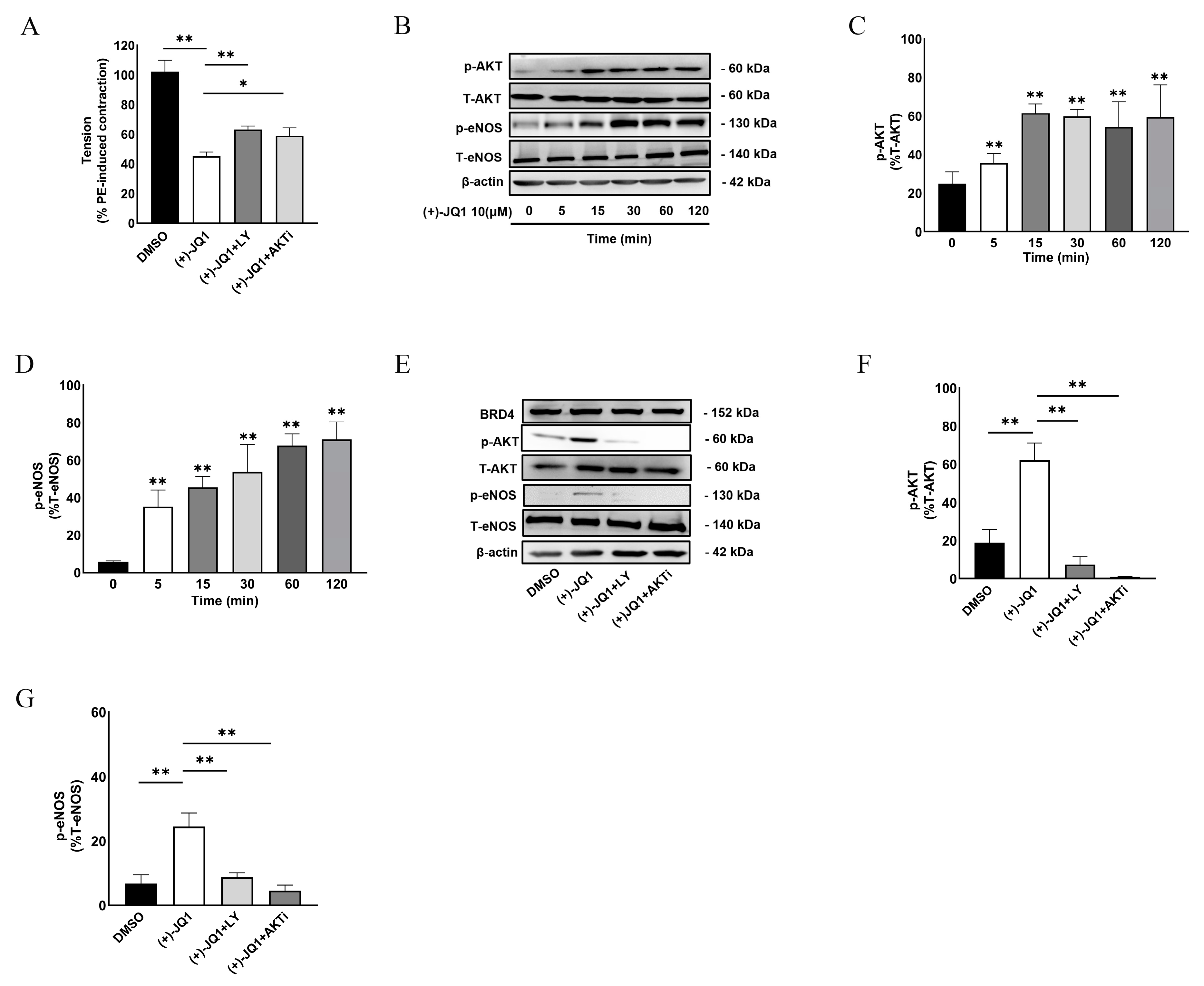
3.6. (+)-JQ1 Activates eNOS through the PI3K/AKT Pathway in Endothelial Cells
3.7. Inhibition of (+)-JQ1 Metabolism by Ketoconazole (KNZ) Does Not Affect (+)-JQ1-Induced Inhibitory Effects on KCl and PE-Induced Contractile Responses
3.8. The Inhibitory Effect of (+)-JQ1 Is Mimicked by (−)-JQ1
3.9. (+)-JQ1 Reduces Systolic Blood Pressure in Mice
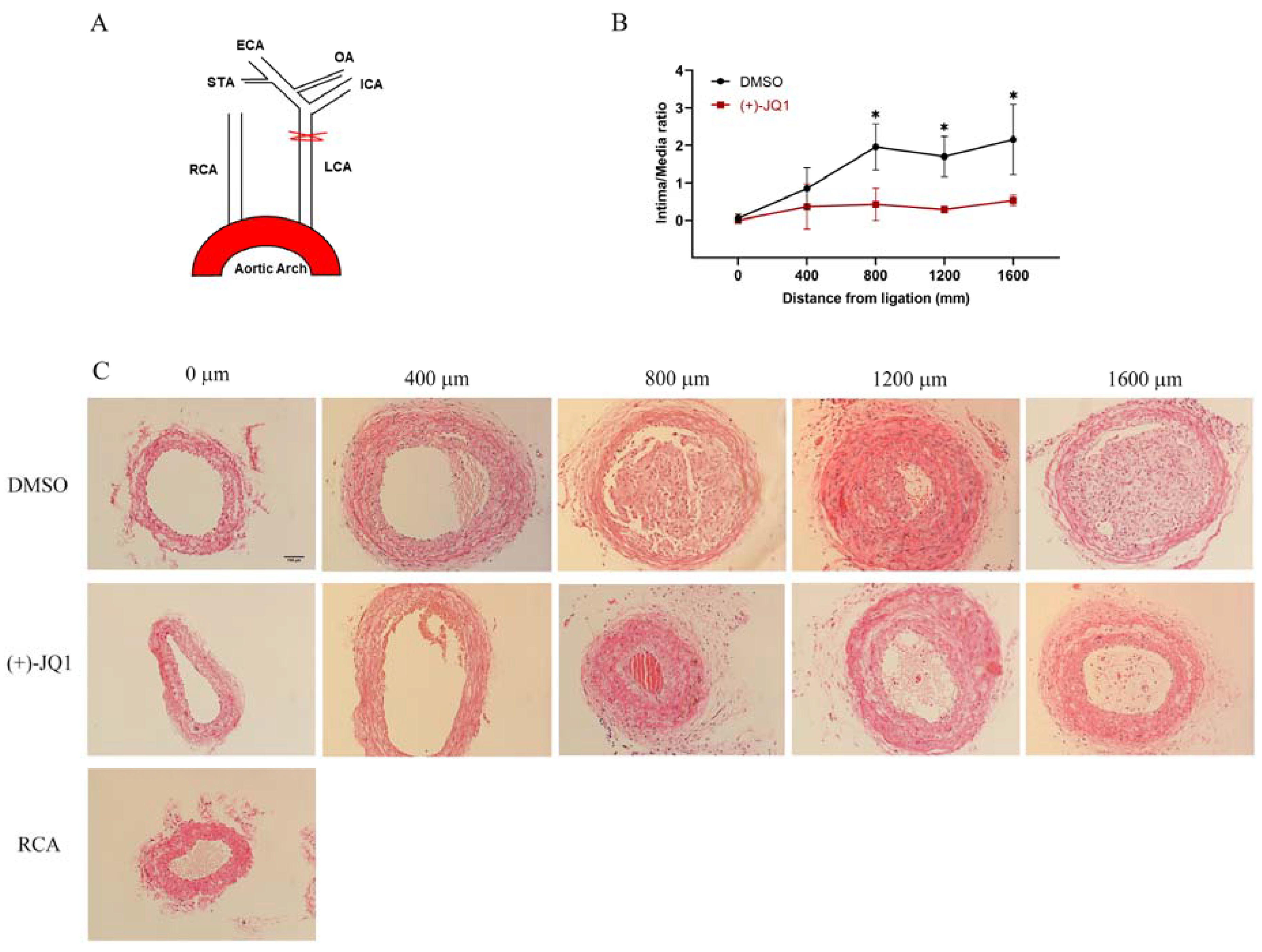
3.10. (+)-JQ1 Administered via Intraperitoneal Injection Ameliorates Neointima Formation in Mouse Carotid Arteries Induced by Complete Carotid Ligation
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Liu, H.; Wang, L.; Weng, X.; Chen, H.; Du, Y.; Diao, C.; Chen, Z.; Liu, X. Inhibition of Brd4 alleviates renal ischemia/reperfusion injury-induced apoptosis and endoplasmic reticulum stress by blocking FoxO4-mediated oxidative stress. Redox Biol. 2019, 24, 101195. [Google Scholar] [CrossRef] [PubMed]
- Meloche, J.; Potus, F.; Vaillancourt, M.; Bourgeois, A.; Johnson, I.; Deschamps, L.; Chabot, S.; Ruffenach, G.; Henry, S.; Breuils-Bonnet, S.; et al. Bromodomain-Containing Protein 4: The Epigenetic Origin of Pulmonary Arterial Hypertension. Circ. Res. 2015, 117, 525–535. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Zhang, M.; Takayama, T.; Shi, X.; Roenneburg, D.A.; Kent, K.C.; Guo, L.W. BET Bromodomain Blockade Mitigates Intimal Hyperplasia in Rat Carotid Arteries. EBioMedicine 2015, 2, 1650–1661. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.Y.; Zhang, X.; Schiattarella, G.G.; Altamirano, F.; Ramos, T.A.R.; French, K.M.; Jiang, N.; Szweda, P.A.; Evers, B.M.; May, H.I.; et al. Epigenetic Reader BRD4 (Bromodomain-Containing Protein 4) Governs Nucleus-Encoded Mitochondrial Transcriptome to Regulate Cardiac Function. Circulation 2020, 142, 2356–2370. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Zhang, M.; Xu, C.; Chai, D.; Peng, F.; Lin, J. Anti-Diabetic Atherosclerosis by Inhibiting High Glucose-Induced Vascular Smooth Muscle Cell Proliferation via Pin1/BRD4 Pathway. Oxid. Med. Cell. Longev. 2020, 2020, 4196482. [Google Scholar] [CrossRef]
- Filippakopoulos, P.; Qi, J.; Picaud, S.; Shen, Y.; Smith, W.B.; Fedorov, O.; Morse, E.M.; Keates, T.; Hickman, T.T.; Felletar, I.; et al. Selective inhibition of BET bromodomains. Nature 2010, 468, 1067–1073. [Google Scholar] [CrossRef]
- Dutzmann, J.; Haertlé, M.; Daniel, J.M.; Kloss, F.; Musmann, R.J.; Kalies, K.; Knöpp, K.; Pilowski, C.; Sirisko, M.; Sieweke, J.T.; et al. BET bromodomain-containing epigenetic reader proteins regulate vascular smooth muscle cell proliferation and neointima formation. Cardiovasc. Res. 2021, 117, 850–862. [Google Scholar] [CrossRef]
- Huang, M.; Qiu, Q.; Xiao, Y.; Zeng, S.; Zhan, M.; Shi, M.; Zou, Y.; Ye, Y.; Liang, L.; Yang, X.; et al. BET Bromodomain Suppression Inhibits VEGF-induced Angiogenesis and Vascular Permeability by Blocking VEGFR2-mediated Activation of PAK1 and eNOS. Sci. Rep. 2016, 6, 23770. [Google Scholar] [CrossRef]
- Yang, Y.M.; Shi, R.H.; Xu, C.X.; Li, L. BRD4 expression in patients with essential hypertension and its effect on blood pressure in spontaneously hypertensive rats. J. Am. Soc. Hypertens. 2018, 12, e107–e117. [Google Scholar] [CrossRef]
- Guo, Y.; Tang, Z.; Yan, B.; Yin, H.; Tai, S.; Peng, J.; Cui, Y.; Gui, Y.; Belke, D.; Zhou, S.; et al. PCSK9 (Proprotein Convertase Subtilisin/Kexin Type 9) Triggers Vascular Smooth Muscle Cell Senescence and Apoptosis: Implication of Its Direct Role in Degenerative Vascular Disease. Arterioscler. Thromb. Vasc. Biol. 2022, 42, 67–86. [Google Scholar] [CrossRef]
- Takenaga, M.; Kawasaki, H.; Wada, A.; Eto, T. Calcitonin gene-related peptide mediates acetylcholine-induced endothelium-independent vasodilation in mesenteric resistance blood vessels of the rat. Circ. Res. 1995, 76, 935–941. [Google Scholar] [CrossRef]
- Saifeddine, M.; El-Daly, M.; Mihara, K.; Bunnett, N.W.; McIntyre, P.; Altier, C.; Hollenberg, M.D.; Ramachandran, R. GPCR-mediated EGF receptor transactivation regulates TRPV4 action in the vasculature. Br. J. Pharmacol. 2015, 172, 2493–2506. [Google Scholar] [CrossRef]
- Bol, V.; Desjardins, F.; Reusens, B.; Balligand, J.L.; Remacle, C. Does early mismatched nutrition predispose to hypertension and atherosclerosis, in male mice? PLoS ONE 2010, 5, e12656. [Google Scholar] [CrossRef]
- Wang, Y.; Thatcher, S.E.; Cassis, L.A. Measuring Blood Pressure Using a Noninvasive Tail Cuff Method in Mice. Methods Mol. Biol. 2017, 1614, 69–73. [Google Scholar] [CrossRef]
- Pfeiffer, S.; Leopold, E.; Schmidt, K.; Brunner, F.; Mayer, B. Inhibition of nitric oxide synthesis by NG-nitro-L-arginine methyl ester (L-NAME): Requirement for bioactivation to the free acid, NG-nitro-L-arginine. Br. J. Pharmacol. 1996, 118, 1433–1440. [Google Scholar] [CrossRef] [PubMed]
- Garthwaite, J.; Southam, E.; Boulton, C.L.; Nielsen, E.B.; Schmidt, K.; Mayer, B. Potent and selective inhibition of nitric oxide-sensitive guanylyl cyclase by 1H-[1,2,4]oxadiazolo[4,3-a]quinoxalin-1-one. Mol. Pharmacol. 1995, 48, 184–188. [Google Scholar]
- Ubl, J.J.; Reiser, G. Activity of protein kinase C is necessary for sustained thrombin-induced [Ca2+]i oscillations in rat glioma cells. Pflugers Arch. 1997, 433, 312–320. [Google Scholar] [CrossRef]
- Hollenberg, M.D.; Saifeddine, M.; al-Ani, B.; Kawabata, A. Proteinase-activated receptors: Structural requirements for activity, receptor cross-reactivity, and receptor selectivity of receptor-activating peptides. Can. J. Physiol. Pharmacol. 1997, 75, 832–841. [Google Scholar] [CrossRef]
- McNulty, S.; Morgan, P.J.; Thompson, M.; Davidson, G.; Lawson, W.; Hastings, M.H. Phospholipases and melatonin signal transduction in the ovine pars tuberalis. Mol. Cell. Endocrinol. 1994, 99, 73–79. [Google Scholar] [CrossRef]
- Hao, K.; Jiang, W.; Zhou, M.; Li, H.; Chen, Y.; Jiang, F.; Hu, Q. Targeting BRD4 prevents acute gouty arthritis by regulating pyroptosis. Int. J. Biol. Sci. 2020, 16, 3163–3173. [Google Scholar] [CrossRef]
- Li, F.; MacKenzie, K.R.; Jain, P.; Santini, C.; Young, D.W.; Matzuk, M.M. Metabolism of JQ1, an inhibitor of bromodomain and extra terminal bromodomain proteins, in human and mouse liver microsomes. Biol. Reprod. 2020, 103, 427–436. [Google Scholar] [CrossRef] [PubMed]
- Ayajiki, K.; Fujioka, H.; Toda, N.; Okada, S.; Minamiyama, Y.; Imaoka, S.; Funae, Y.; Watanabe, S.; Nakamura, A.; Okamura, T. Mediation of arachidonic acid metabolite(s) produced by endothelial cytochrome P-450 3A4 in monkey arterial relaxation. Hypertens. Res. 2003, 26, 237–243. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Xu, M.; Kao, C.Y.; Tsai, S.Y.; Tsai, M.J. Small molecule JQ1 promotes prostate cancer invasion via BET-independent inactivation of FOXA1. J. Clin. Investig. 2020, 130, 1782–1792. [Google Scholar] [CrossRef] [PubMed]
- Davignon, J.; Ganz, P. Role of endothelial dysfunction in atherosclerosis. Circulation 2004, 109, III27-32. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Youn, J.Y.; Cai, H. Mechanisms and consequences of endothelial nitric oxide synthase dysfunction in hypertension. J. Hypertens. 2015, 33, 1128–1136. [Google Scholar] [CrossRef]
- Wang, H.; Fu, H.; Zhu, R.; Wu, X.; Ji, X.; Li, X.; Jiang, H.; Lin, Z.; Tang, X.; Sun, S.; et al. BRD4 contributes to LPS-induced macrophage senescence and promotes progression of atherosclerosis-associated lipid uptake. Aging (Albany N. Y.) 2020, 12, 9240–9259. [Google Scholar] [CrossRef]
- Ding, N.; Hah, N.; Yu, R.T.; Sherman, M.H.; Benner, C.; Leblanc, M.; He, M.; Liddle, C.; Downes, M.; Evans, R.M. BRD4 is a novel therapeutic target for liver fibrosis. Proc. Natl. Acad. Sci. USA 2015, 112, 15713–15718. [Google Scholar] [CrossRef]
- Niu, Q.; Liu, Z.; Alamer, E.; Fan, X.; Chen, H.; Endsley, J.; Gelman, B.B.; Tian, B.; Kim, J.H.; Michael, N.L.; et al. Structure-guided drug design identifies a BRD4-selective small molecule that suppresses HIV. J. Clin. Investig. 2019, 129, 3361–3373. [Google Scholar] [CrossRef]
- Mu, J.; Zhang, D.; Tian, Y.; Xie, Z.; Zou, M.H. BRD4 inhibition by JQ1 prevents high-fat diet-induced diabetic cardiomyopathy by activating PINK1/Parkin-mediated mitophagy in vivo. J. Mol. Cell. Cardiol. 2020, 149, 1–14. [Google Scholar] [CrossRef]
- Zhou, Z.; Li, X.; Liu, Z.; Huang, L.; Yao, Y.; Li, L.; Chen, J.; Zhang, R.; Zhou, J.; Wang, L.; et al. A Bromodomain-Containing Protein 4 (BRD4) Inhibitor Suppresses Angiogenesis by Regulating AP-1 Expression. Front. Pharmacol. 2020, 11, 1043. [Google Scholar] [CrossRef]










Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yan, B.; Gui, Y.; Guo, Y.; Sun, J.; Saifeddine, M.; Deng, J.; Hill, J.A.; Hollenberg, M.D.; Jiang, Z.-S.; Zheng, X.-L. Impact of Short-Term (+)-JQ1 Exposure on Mouse Aorta: Unanticipated Inhibition of Smooth Muscle Contractility. Cells 2023, 12, 1461. https://doi.org/10.3390/cells12111461
Yan B, Gui Y, Guo Y, Sun J, Saifeddine M, Deng J, Hill JA, Hollenberg MD, Jiang Z-S, Zheng X-L. Impact of Short-Term (+)-JQ1 Exposure on Mouse Aorta: Unanticipated Inhibition of Smooth Muscle Contractility. Cells. 2023; 12(11):1461. https://doi.org/10.3390/cells12111461
Chicago/Turabian StyleYan, Binjie, Yu Gui, Yanan Guo, Jiaxing Sun, Mahmoud Saifeddine, Jingti Deng, Joseph A. Hill, Morley D. Hollenberg, Zhi-Sheng Jiang, and Xi-Long Zheng. 2023. "Impact of Short-Term (+)-JQ1 Exposure on Mouse Aorta: Unanticipated Inhibition of Smooth Muscle Contractility" Cells 12, no. 11: 1461. https://doi.org/10.3390/cells12111461
APA StyleYan, B., Gui, Y., Guo, Y., Sun, J., Saifeddine, M., Deng, J., Hill, J. A., Hollenberg, M. D., Jiang, Z. -S., & Zheng, X. -L. (2023). Impact of Short-Term (+)-JQ1 Exposure on Mouse Aorta: Unanticipated Inhibition of Smooth Muscle Contractility. Cells, 12(11), 1461. https://doi.org/10.3390/cells12111461