

CXCR2 Is Deregulated in ALS Spinal Cord and Its Activation Triggers Apoptosis in Motor Neuron-Like Cells Overexpressing hSOD1-G93A



 , ,
, ,  and
and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Transcriptomic Profiling
2.2. Fluorescent Immunohistochemistry
2.3. Cell Cultures
2.4. Cell Viability
2.5. Western Blot Analysis
2.6. Immunocytofluorescence
2.7. Statistical Analysis
3. Results
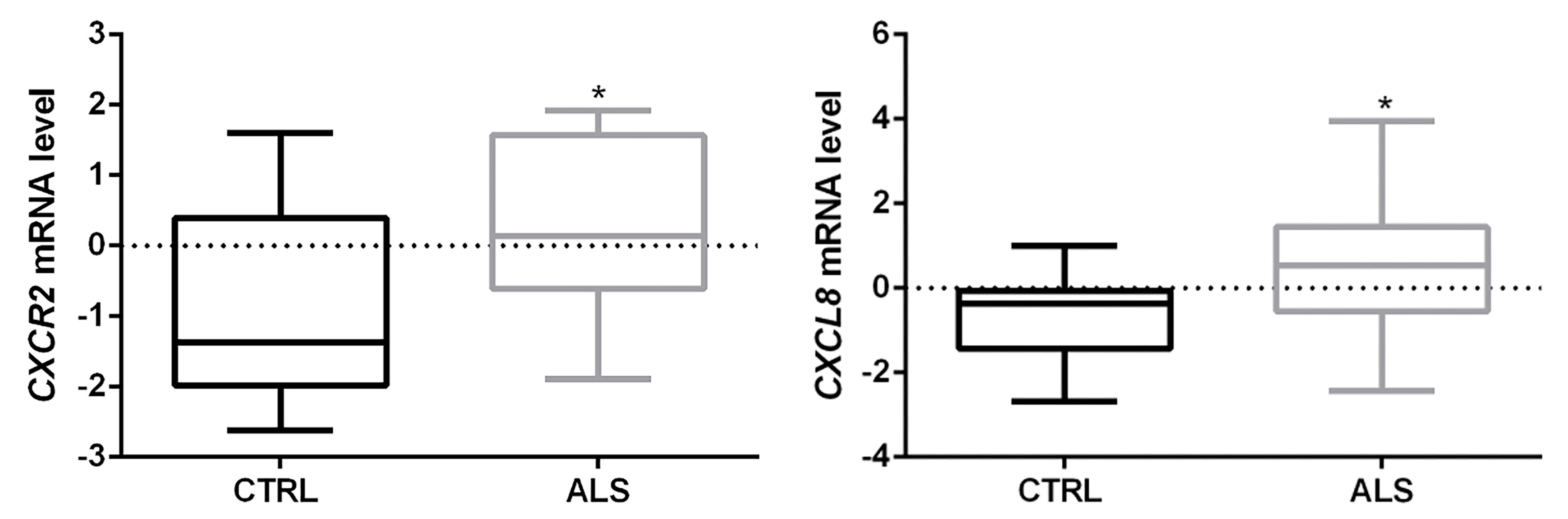
3.1. CXCR2/CXCL8 Expression in Control and Sporadic ALS Spinal Cord Samples
3.2. CXCR2 Activation by GROα and MIP2α Pro-Inflammatory Chemokines Induces Dose-Dependent Cell Death in NSC-34 Cells Overexpressing hSOD1-G93A
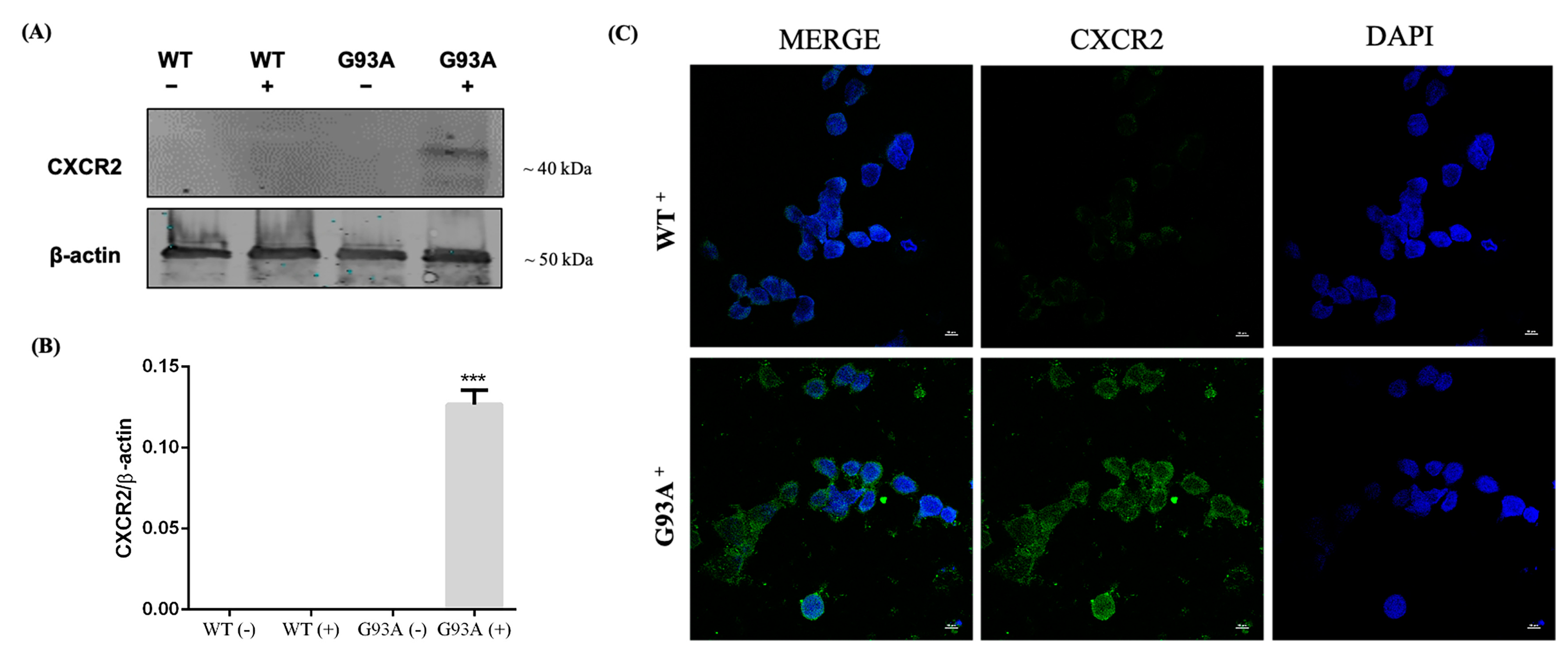
3.3. Activation of CXCR2 Axis by MIP2α Triggers Apoptosis in hSOD1-G93A NSC-34 Cells
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Mathis, S.; Goizet, C.; Soulages, A.; Vallat, J.M.; Masson, G.L. Genetics of amyotrophic lateral sclerosis: A review. J. Neurol. Sci. 2019, 399, 217–226. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, J.D.; Borasio, G.D. Amyotrophic lateral sclerosis. Lancet 2007, 369, 2031–2041. [Google Scholar] [CrossRef] [PubMed]
- Gentile, G.; Morello, G.; La Cognata, V.; Guarnaccia, M.; Conforti, F.L.; Cavallaro, S. Dysregulated miRNAs as Biomarkers and Therapeutical Targets in Neurodegenerative Diseases. J. Pers. Med. 2022, 12, 770. [Google Scholar] [CrossRef] [PubMed]
- Kuuluvainen, L.; Kaivola, K.; Mönkäre, S.; Laaksovirta, H.; Jokela, M.; Udd, B.; Valori, M.; Pasanen, P.; Paetau, A.; Traynor, B.J.; et al. Oligogenic basis of sporadic ALS. Neurol. Genet. 2019, 5, e335. [Google Scholar] [CrossRef] [Green Version]
- Gentile, G.; Perrone, B.; Morello, G.; Simone, I.L.; Ando, S.; Cavallaro, S.; Conforti, F.L. Individual Oligogenic Background in p.D91A-SOD1 Amyotrophic Lateral Sclerosis Patients. Genes 2021, 12, 1843. [Google Scholar] [CrossRef]
- Johnson, S.A.; Fang, T.; De Marchi, F.; Neel, D.; Van Weehaeghe, D.; Berry, J.D.; Paganoni, S. Pharmacotherapy for Amyotrophic Lateral Sclerosis: A Review of Approved and Upcoming Agents. Drugs 2022, 82, 1367–1388. [Google Scholar] [CrossRef]
- Turner, M.R.; Parton, M.J.; Leigh, P.N. Clinical trials in ALS: An overview. Semin. Neurol. 2001, 21, 167–175. [Google Scholar] [CrossRef]
- La Cognata, V.; Morello, G.; Cavallaro, S. Omics Data and Their Integrative Analysis to Support Stratified Medicine in Neurodegenerative Diseases. Int. J. Mol. Sci. 2021, 22, 4820. [Google Scholar] [CrossRef]
- La Cognata, V.; Golini, E.; Iemmolo, R.; Balletta, S.; Morello, G.; De Rosa, C.; Villari, A.; Marinelli, S.; Vacca, V.; Bonaventura, G.; et al. CXCR2 increases in ALS cortical neurons and its inhibition prevents motor neuron degeneration in vitro and improves neuromuscular function in SOD1G93A mice. Neurobiol. Dis. 2021, 160, 105538. [Google Scholar] [CrossRef]
- Morello, G.; Spampinato, A.G.; Conforti, F.L.; D’Agata, V.; Cavallaro, S. Selection and Prioritization of Candidate Drug Targets for Amyotrophic Lateral Sclerosis Through a Meta-Analysis Approach. J. Mol. Neurosci. 2017, 61, 563–580. [Google Scholar] [CrossRef] [Green Version]
- Aronica, E.; Baas, F.; Iyer, A.; ten Asbroek, A.L.; Morello, G.; Cavallaro, S. Molecular classification of amyotrophic lateral sclerosis by unsupervised clustering of gene expression in motor cortex. Neurobiol. Dis. 2015, 74, 359–376. [Google Scholar] [CrossRef] [Green Version]
- Morello, G.; Guarnaccia, M.; Spampinato, A.G.; Salomone, S.; D’Agata, V.; Conforti, F.L.; Aronica, E.; Cavallaro, S. Integrative multi-omic analysis identifies new drivers and pathways in molecularly distinct subtypes of ALS. Sci. Rep. 2019, 9, 9968. [Google Scholar] [CrossRef] [Green Version]
- Morello, G.; Spampinato, A.G.; Cavallaro, S. Molecular Taxonomy of Sporadic Amyotrophic Lateral Sclerosis Using Disease-Associated Genes. Front. Neurol. 2017, 8, 152. [Google Scholar] [CrossRef] [Green Version]
- Semple, B.D.; Kossmann, T.; Morganti-Kossmann, M.C. Role of chemokines in CNS health and pathology: A focus on the CCL2/CCR2 and CXCL8/CXCR2 networks. J. Cereb. Blood Flow Metab. 2010, 30, 459–473. [Google Scholar] [CrossRef] [Green Version]
- Horuk, R.; Martin, A.W.; Wang, Z.; Schweitzer, L.; Gerassimides, A.; Guo, H.; Lu, Z.; Hesselgesser, J.; Perez, H.D.; Kim, J.; et al. Expression of chemokine receptors by subsets of neurons in the central nervous system. J. Immunol. 1997, 158, 2882–2890. [Google Scholar] [CrossRef]
- Nguyen, D.; Stangel, M. Expression of the chemokine receptors CXCR1 and CXCR2 in rat oligodendroglial cells. Brain Res. Dev. Brain Res. 2001, 128, 77–81. [Google Scholar] [CrossRef]
- Popivanova, B.K.; Koike, K.; Tonchev, A.B.; Ishida, Y.; Kondo, T.; Ogawa, S.; Mukaida, N.; Inoue, M.; Yamashima, T. Accumulation of microglial cells expressing ELR motif-positive CXC chemokines and their receptor CXCR2 in monkey hippocampus after ischemia-reperfusion. Brain Res. 2003, 970, 195–204. [Google Scholar] [CrossRef]
- Konrad, F.M.; Reutershan, J. CXCR2 in acute lung injury. Mediat. Inflamm. 2012, 2012, 740987. [Google Scholar] [CrossRef] [Green Version]
- Mennini, T.; Giordano, L.; Mengozzi, M.; Ghezzi, P.; Tonelli, R.; Mantegazza, R.; Silani, V.; Corbo, M.; Lunetta, C.; Beghi, E. Increased Il-8 Levels in the Cerebrospinal Fluid of Patients with Amyotrophic Lateral Sclerosis. Eur. J. Inflamm. 2017, 7, 39–44. [Google Scholar] [CrossRef] [Green Version]
- Cao, M.C.; Cawston, E.E.; Chen, G.; Brooks, C.; Douwes, J.; McLean, D.; Graham, E.S.; Dragunow, M.; Scotter, E.L. Serum biomarkers of neuroinflammation and blood-brain barrier leakage in amyotrophic lateral sclerosis. BMC Neurol. 2022, 22, 216. [Google Scholar] [CrossRef]
- Rusconi, M.; Gerardi, F.; Santus, W.; Lizio, A.; Sansone, V.A.; Lunetta, C.; Zanoni, I.; Granucci, F. Inflammatory role of dendritic cells in Amyotrophic Lateral Sclerosis revealed by an analysis of patients’ peripheral blood. Sci. Rep. 2017, 7, 7853. [Google Scholar] [CrossRef] [PubMed]
- Ehrhart, J.; Smith, A.J.; Kuzmin-Nichols, N.; Zesiewicz, T.A.; Jahan, I.; Shytle, R.D.; Kim, S.H.; Sanberg, C.D.; Vu, T.H.; Gooch, C.L.; et al. Humoral factors in ALS patients during disease progression. J. Neuroinflammation 2015, 12, 127. [Google Scholar] [CrossRef] [Green Version]
- Hu, Y.; Cao, C.; Qin, X.Y.; Yu, Y.; Yuan, J.; Zhao, Y.; Cheng, Y. Increased peripheral blood inflammatory cytokine levels in amyotrophic lateral sclerosis: A meta-analysis study. Sci. Rep. 2017, 7, 9094. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuhle, J.; Lindberg, R.L.; Regeniter, A.; Mehling, M.; Steck, A.J.; Kappos, L.; Czaplinski, A. Increased levels of inflammatory chemokines in amyotrophic lateral sclerosis. Eur. J. Neurol. 2009, 16, 771–774. [Google Scholar] [CrossRef] [PubMed]
- Won, Y.H.; Lee, M.Y.; Choi, Y.C.; Ha, Y.; Kim, H.; Kim, D.Y.; Kim, M.S.; Yu, J.H.; Seo, J.H.; Kim, M.; et al. Elucidation of Relevant Neuroinflammation Mechanisms Using Gene Expression Profiling in Patients with Amyotrophic Lateral Sclerosis. PLoS ONE 2016, 11, e0165290. [Google Scholar] [CrossRef] [Green Version]
- Quek, H.; Cuni-Lopez, C.; Stewart, R.; Colletti, T.; Notaro, A.; Nguyen, T.H.; Sun, Y.; Guo, C.C.; Lupton, M.K.; Roberts, T.L.; et al. ALS monocyte-derived microglia-like cells reveal cytoplasmic TDP-43 accumulation, DNA damage, and cell-specific impairment of phagocytosis associated with disease progression. J. Neuroinflammation 2022, 19, 58. [Google Scholar] [CrossRef]
- Zhang, Z.J.; Jiang, B.C.; Gao, Y.J. Chemokines in neuron-glial cell interaction and pathogenesis of neuropathic pain. Cell. Mol. Life Sci. 2017, 74, 3275–3291. [Google Scholar] [CrossRef]
- La Cognata, V.; Gentile, G.; Aronica, E.; Cavallaro, S. Splicing Players Are Differently Expressed in Sporadic Amyotrophic Lateral Sclerosis Molecular Clusters and Brain Regions. Cells 2020, 9, 159. [Google Scholar] [CrossRef] [Green Version]
- Cashman, N.R.; Durham, H.D.; Blusztajn, J.K.; Oda, K.; Tabira, T.; Shaw, I.T.; Dahrouge, S.; Antel, J.P. Neuroblastoma × spinal cord (NSC) hybrid cell lines resemble developing motor neurons. Dev. Dyn. 1992, 194, 209–221. [Google Scholar] [CrossRef]
- Maugeri, G.; D’Amico, A.G.; Rasa, D.M.; Federico, C.; Saccone, S.; Morello, G.; La Cognata, V.; Cavallaro, S.; D’Agata, V. Molecular mechanisms involved in the protective effect of pituitary adenylate cyclase-activating polypeptide in an in vitro model of amyotrophic lateral sclerosis. J. Cell. Physiol. 2019, 234, 5203–5214. [Google Scholar] [CrossRef]
- D’Amico, A.G.; Maugeri, G.; Saccone, S.; Federico, C.; Cavallaro, S.; Reglodi, D.; D’Agata, V. PACAP Modulates the Autophagy Process in an In Vitro Model of Amyotrophic Lateral Sclerosis. Int. J. Mol. Sci. 2020, 21, 2943. [Google Scholar] [CrossRef] [Green Version]
- D’Amico, A.G.; Scuderi, S.; Maugeri, G.; Cavallaro, S.; Drago, F.; D’Agata, V. NAP reduces murine microvascular endothelial cells proliferation induced by hyperglycemia. J. Mol. Neurosci. 2014, 54, 405–413. [Google Scholar] [CrossRef]
- La Cognata, V.; Maugeri, G.; D’Amico, A.G.; Saccone, S.; Federico, C.; Cavallaro, S.; D’Agata, V. Differential expression of PARK2 splice isoforms in an in vitro model of dopaminergic-like neurons exposed to toxic insults mimicking Parkinson’s disease. J. Cell. Biochem. 2018, 119, 1062–1073. [Google Scholar] [CrossRef]
- Bonaventura, G.; La Cognata, V.; Iemmolo, R.; Zimbone, M.; Contino, A.; Maccarrone, G.; Failla, B.; Barcellona, M.L.; Conforti, F.L.; D’Agata, V.; et al. Ag-NPs induce apoptosis, mitochondrial damages and MT3/OSGIN2 expression changes in an in vitro model of human dental-pulp-stem-cells-derived neurons. Neurotoxicology 2018, 67, 84–93. [Google Scholar] [CrossRef]
- Laudani, S.; La Cognata, V.; Iemmolo, R.; Bonaventura, G.; Villaggio, G.; Saccone, S.; Barcellona, M.L.; Cavallaro, S.; Sinatra, F. Effect of a Bone Marrow-Derived Extracellular Matrix on Cell Adhesion and Neural Induction of Dental Pulp Stem Cells. Front. Cell Dev. Biol. 2020, 8, 100. [Google Scholar] [CrossRef] [Green Version]
- van Es, M.A.; Hardiman, O.; Chio, A.; Al-Chalabi, A.; Pasterkamp, R.J.; Veldink, J.H.; van den Berg, L.H. Amyotrophic lateral sclerosis. Lancet 2017, 390, 2084–2098. [Google Scholar] [CrossRef]
- Perrone, B.; La Cognata, V.; Sprovieri, T.; Ungaro, C.; Conforti, F.L.; Ando, S.; Cavallaro, S. Alternative Splicing of ALS Genes: Misregulation and Potential Therapies. Cell. Mol. Neurobiol. 2020, 40, 1–14. [Google Scholar] [CrossRef]
- Morello, G.; Salomone, S.; D’Agata, V.; Conforti, F.L.; Cavallaro, S. From Multi-Omics Approaches to Precision Medicine in Amyotrophic Lateral Sclerosis. Front. Neurosci. 2020, 14, 577755. [Google Scholar] [CrossRef]
- De Marchi, F.; Munitic, I.; Amedei, A.; Berry, J.D.; Feldman, E.L.; Aronica, E.; Nardo, G.; Van Weehaeghe, D.; Niccolai, E.; Prtenjaca, N.; et al. Interplay between immunity and amyotrophic lateral sclerosis: Clinical impact. Neurosci. Biobehav. Rev. 2021, 127, 958–978. [Google Scholar] [CrossRef]
- Lall, D.; Baloh, R.H. Microglia and C9orf72 in neuroinflammation and ALS and frontotemporal dementia. J. Clin. Investig. 2017, 127, 3250–3258. [Google Scholar] [CrossRef]
- McCauley, M.E.; Baloh, R.H. Inflammation in ALS/FTD pathogenesis. Acta Neuropathol. 2019, 137, 715–730. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walz, A.; Peveri, P.; Aschauer, H.; Baggiolini, M. Purification and amino acid sequencing of NAF, a novel neutrophil-activating factor produced by monocytes. Biochem. Biophys. Res. Commun. 1987, 149, 755–761. [Google Scholar] [CrossRef] [PubMed]
- Bonecchi, R.; Facchetti, F.; Dusi, S.; Luini, W.; Lissandrini, D.; Simmelink, M.; Locati, M.; Bernasconi, S.; Allavena, P.; Brandt, E.; et al. Induction of functional IL-8 receptors by IL-4 and IL-13 in human monocytes. J. Immunol. 2000, 164, 3862–3869. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nilsson, G.; Mikovits, J.A.; Metcalfe, D.D.; Taub, D.D. Mast Cell Migratory Response to Interleukin-8 Is Mediated through Interaction with Chemokine Receptor CXCR2/Interleukin-8RB. Blood 1999, 93, 2791–2797. [Google Scholar] [CrossRef]
- Lippert, U.; Zachmann, K.; Henz, B.M.; Neumann, C. Human T lymphocytes and mast cells differentially express and regulate extra- and intracellular CXCR1 and CXCR2. Exp. Dermatol. 2004, 13, 520–525. [Google Scholar] [CrossRef]
- Nirodi, C.S.; Devalaraja, R.; Nanney, L.B.; Arrindell, S.; Russell, S.; Trupin, J.; Richmond, A. Chemokine and chemokine receptor expression in keloid and normal fibroblasts. Wound Repair Regen. 2000, 8, 371–382. [Google Scholar] [CrossRef]
- Strieter, R.M.; Polverini, P.J.; Arenberg, D.A.; Kunkel, S.L. The role of CXC chemokines as regulators of angiogenesis. Shock 1995, 4, 155–160. [Google Scholar] [CrossRef]
- Morello, G.; Conforti, F.L.; Parenti, R.; D’Agata, V.; Cavallaro, S. Selection of Potential Pharmacological Targets in ALS Based on Whole-Genome Expression Profiling. Curr. Med. Chem. 2015, 22, 2004–2021. [Google Scholar] [CrossRef]
- Morello, G.; Spampinato, A.G.; Cavallaro, S. Neuroinflammation and ALS: Transcriptomic Insights into Molecular Disease Mechanisms and Therapeutic Targets. Mediat. Inflamm. 2017, 2017, 7070469. [Google Scholar] [CrossRef] [Green Version]
- Nardo, G.; Trolese, M.C.; Tortarolo, M.; Vallarola, A.; Freschi, M.; Pasetto, L.; Bonetto, V.; Bendotti, C. New Insights on the Mechanisms of Disease Course Variability in ALS from Mutant SOD1 Mouse Models. Brain Pathol. 2016, 26, 237–247. [Google Scholar] [CrossRef]
- De Paola, M.; Buanne, P.; Biordi, L.; Bertini, R.; Ghezzi, P.; Mennini, T. Chemokine MIP-2/CXCL2, acting on CXCR2, induces motor neuron death in primary cultures. Neuroimmunomodulation 2007, 14, 310–316. [Google Scholar] [CrossRef]








Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
La Cognata, V.; D’Amico, A.G.; Maugeri, G.; Morello, G.; Guarnaccia, M.; Magrì, B.; Aronica, E.; D’Agata, V.; Cavallaro, S. CXCR2 Is Deregulated in ALS Spinal Cord and Its Activation Triggers Apoptosis in Motor Neuron-Like Cells Overexpressing hSOD1-G93A. Cells 2023, 12, 1813. https://doi.org/10.3390/cells12141813
La Cognata V, D’Amico AG, Maugeri G, Morello G, Guarnaccia M, Magrì B, Aronica E, D’Agata V, Cavallaro S. CXCR2 Is Deregulated in ALS Spinal Cord and Its Activation Triggers Apoptosis in Motor Neuron-Like Cells Overexpressing hSOD1-G93A. Cells. 2023; 12(14):1813. https://doi.org/10.3390/cells12141813
Chicago/Turabian StyleLa Cognata, Valentina, Agata Grazia D’Amico, Grazia Maugeri, Giovanna Morello, Maria Guarnaccia, Benedetta Magrì, Eleonora Aronica, Velia D’Agata, and Sebastiano Cavallaro. 2023. "CXCR2 Is Deregulated in ALS Spinal Cord and Its Activation Triggers Apoptosis in Motor Neuron-Like Cells Overexpressing hSOD1-G93A" Cells 12, no. 14: 1813. https://doi.org/10.3390/cells12141813
APA StyleLa Cognata, V., D’Amico, A. G., Maugeri, G., Morello, G., Guarnaccia, M., Magrì, B., Aronica, E., D’Agata, V., & Cavallaro, S. (2023). CXCR2 Is Deregulated in ALS Spinal Cord and Its Activation Triggers Apoptosis in Motor Neuron-Like Cells Overexpressing hSOD1-G93A. Cells, 12(14), 1813. https://doi.org/10.3390/cells12141813