

Contribution of the α5 nAChR Subunit and α5SNP to Nicotine-Induced Proliferation and Migration of Human Cancer Cells

 , , , and
, , , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. Drug Treatment
2.3. Proliferation Assay
2.4. Quantitative Real-Time PCR (qPCR)
2.5. Transwell Migration Assay
2.6. Small Interfering RNA Transfection (siRNA)
2.7. Constructs and Nucleofection
2.8. Immune Fluorescence (IF)
3. Results
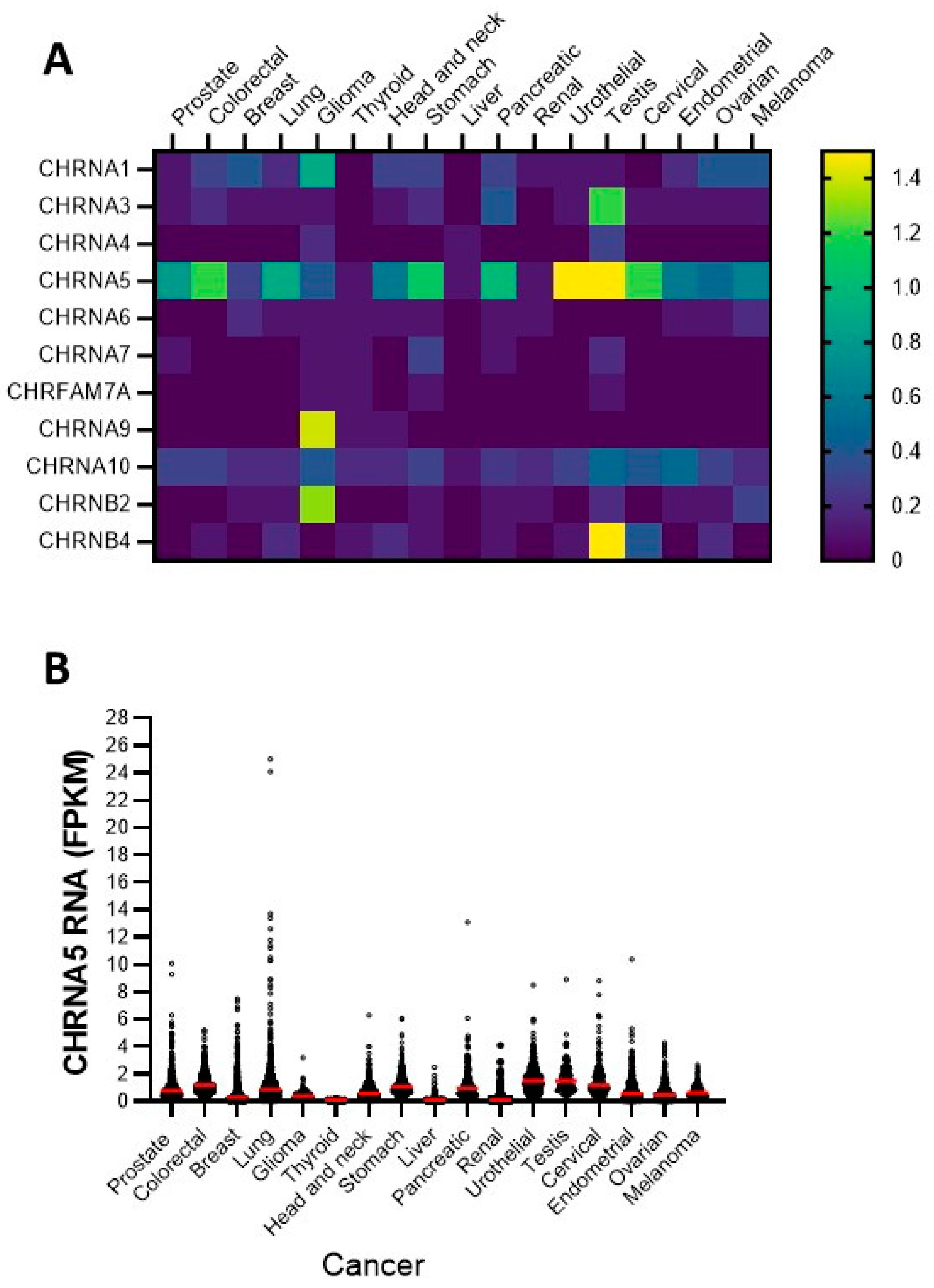
3.1. Expression of nAChR Subunits in Diverse Cancer Cell Lines: Focusing on the α5 Subunit
3.2. Nicotine Increases Proliferation and Migration in Various Human Cancer Cell Lines through nAChRs
3.3. siRNA Based Silencing of the α5 Subunit Reduces Nicotine-Induced Proliferation and Migration in Several Cancer Cell Lines
3.4. Downregulation of the α5 nAChR Subunit Inhibits PDL1, CD47, and EMT Marker Expression Elicited by Nicotine
3.5. The α5SNP Mutation D398N Increases Basal Proliferation and Migration in the DU145 Cell Line
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Millar, N.S.; Gotti, C. Diversity of vertebrate nicotinic acetylcholine receptors. Neuropharmacology 2009, 56, 237–246. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Le Novère, N.; Changeux, J.P. Molecular evolution of the nicotinic acetylcholine receptor: An example of multigene family in excitable cells. J. Mol. Evol. 1995, 40, 155–172. [Google Scholar] [CrossRef]
- Changeux, J.P. Nicotinic receptors and nicotine addiction. Comptes Rendus Biol. 2009, 332, 421–425. [Google Scholar] [CrossRef] [PubMed]
- Corradi, J.; Bouzat, C. Understanding the Bases of Function and Modulation of α7 Nicotinic Receptors: Implications for Drug Discovery. Mol. Pharmacol. 2016, 90, 288–299. [Google Scholar] [CrossRef] [Green Version]
- Wongsamitkul, N.; Maldifassi, M.C.; Simeone, X.; Baur, R.; Ernst, M.; Sigel, E. α subunits in GABAA receptors are dispensable for GABA and diazepam action. Sci. Rep. 2017, 7, 15498. [Google Scholar] [CrossRef] [Green Version]
- Maldifassi, M.C.; Baur, R.; Pierce, D.; Nourmahnad, A.; Forman, S.A.; Sigel, E. Novel positive allosteric modulators of GABAA receptors with anesthetic activity. Sci. Rep. 2016, 6, 25943. [Google Scholar] [CrossRef] [PubMed]
- Thany, S.H.; Tricoire-Leignel, H. Emerging Pharmacological Properties of Cholinergic Synaptic Transmission: Comparison between Mammalian and Insect Synaptic and Extrasynaptic Nicotinic Receptors. Curr. Neuropharmacol. 2011, 9, 706–714. [Google Scholar] [CrossRef] [PubMed]
- Nemecz, Á.; Prevost, M.S.; Menny, A.; Corringer, P.J. Emerging Molecular Mechanisms of Signal Transduction in Pentameric Ligand-Gated Ion Channels. Neuron 2016, 90, 452–470. [Google Scholar] [CrossRef]
- Zoli, M.; Pucci, S.; Vilella, A.; Gotti, C. Neuronal and Extraneuronal Nicotinic Acetylcholine Receptors. Curr. Neuropharmacol. 2018, 16, 338–349. [Google Scholar] [CrossRef]
- Dineley, K.T.; Pandya, A.A.; Yakel, J.L. Nicotinic ACh receptors as therapeutic targets in CNS disorders. Trends Pharmacol. Sci. 2015, 36, 96–108. [Google Scholar] [CrossRef] [Green Version]
- Arias, H.R. Positive and negative modulation of nicotinic receptors. Adv. Protein Chem. Struct. Biol. 2010, 80, 153–203. [Google Scholar] [CrossRef]
- Wessler, I.; Kirkpatrick, C.J. Acetylcholine beyond neurons: The non-neuronal cholinergic system in humans. Br. J. Pharmacol. 2008, 154, 1558–1571. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kabbani, N.; Nichols, R.A. Beyond the Channel: Metabotropic Signaling by Nicotinic Receptors. Trends Pharmacol. Sci. 2018, 39, 354–366. [Google Scholar] [CrossRef] [PubMed]
- Rosas-Ballina, M.; Tracey, K.J. The neurology of the immune system: Neural reflexes regulate immunity. Neuron 2009, 64, 28–32. [Google Scholar] [CrossRef] [Green Version]
- Maldifassi, M.C.; Atienza, G.; Arnalich, F.; López-Collazo, E.; Cedillo, J.L.; Martín-Sánchez, C.; Bordas, A.; Renart, J.; Montiel, C. A new IRAK-M-mediated mechanism implicated in the anti-inflammatory effect of nicotine via α7 nicotinic receptors in human macrophages. PLoS ONE 2014, 9, e108397. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Corringer, P.J.; Le Novère, N.; Changeux, J.P. Nicotinic receptors at the amino acid level. Annu. Rev. Pharmacol. Toxicol. 2000, 40, 431–458. [Google Scholar] [CrossRef]
- Grutter, T.; Le Novère, N.; Changeux, J.P. Rational understanding of nicotinic receptors drug binding. Curr. Top Med. Chem. 2004, 4, 645–650. [Google Scholar] [CrossRef]
- Schaal, C.; Chellappan, S.P. Nicotine-mediated cell proliferation and tumor progression in smoking-related cancers. Mol. Cancer Res. 2014, 12, 14–23. [Google Scholar] [CrossRef] [Green Version]
- Bordas, A.; Cedillo, J.L.; Arnalich, F.; Esteban-Rodriguez, I.; Guerra-Pastrián, L.; de Castro, J.; Martín-Sánchez, C.; Atienza, G.; Fernández-Capitan, C.; Rios, J.J.; et al. Expression patterns for nicotinic acetylcholine receptor subunit genes in smoking-related lung cancers. Oncotarget 2017, 8, 67878–67890. [Google Scholar] [CrossRef] [Green Version]
- Dasgupta, P.; Rizwani, W.; Pillai, S.; Kinkade, R.; Kovacs, M.; Rastogi, S.; Banerjee, S.; Carless, M.; Kim, E.; Coppola, D.; et al. Nicotine induces cell proliferation, invasion and epithelial-mesenchymal transition in a variety of human cancer cell lines. Int. J. Cancer 2009, 124, 36–45. [Google Scholar] [CrossRef] [Green Version]
- Mucchietto, V.; Fasoli, F.; Pucci, S.; Moretti, M.; Benfante, R.; Maroli, A.; Di Lascio, S.; Bolchi, C.; Pallavicini, M.; Dowell, C.; et al. α9- and α7-containing receptors mediate the pro-proliferative effects of nicotine in the A549 adenocarcinoma cell line. Br. J. Pharmacol. 2018, 175, 1957–1972. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- West, K.A.; Brognard, J.; Clark, A.S.; Linnoila, I.R.; Yang, X.; Swain, S.M.; Harris, C.; Belinsky, S.; Dennis, P.A. Rapid Akt activation by nicotine and a tobacco carcinogen modulates the phenotype of normal human airway epithelial cells. J. Clin. Investig. 2003, 111, 81–90. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, C.; Ding, X.P.; Zhao, Q.N.; Yang, X.J.; An, S.M.; Wang, H.; Xu, L.; Zhu, L.; Chen, H.Z. Role of α7-nicotinic acetylcholine receptor in nicotine-induced invasion and epithelial-to-mesenchymal transition in human non-small cell lung cancer cells. Oncotarget 2016, 7, 59199–59208. [Google Scholar] [CrossRef] [Green Version]
- Lian, S.; Xie, R.; Ye, Y.; Lu, Y.; Cheng, Y.; Xie, X.; Li, S.; Jia, L. Dual blockage of both PD-L1 and CD47 enhances immunotherapy against circulating tumor cells. Sci. Rep. 2019, 9, 4532. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Jia, Y.; Li, P.; Li, H.; Xiao, D.; Wang, Y.; Ma, X. Reciprocal activation of α5-nAChR and STAT3 in nicotine-induced human lung cancer cell proliferation. J. Genet. Genom. 2017, 44, 355–362. [Google Scholar] [CrossRef]
- Wu, S.Y.; Xing, F.; Sharma, S.; Wu, K.; Tyagi, A.; Liu, Y.; Zhao, D.; Deshpande, R.P.; Shiozawa, Y.; Ahmed, T.; et al. Nicotine promotes brain metastasis by polarizing microglia and suppressing innate immune function. J. Exp. Med. 2020, 217, e20191131. [Google Scholar] [CrossRef]
- Zhu, P.; Jin, Z.; Kang, G.; Jia, Y.; Liu, D.; Zhang, Q.; Guo, F.; Jia, Y.; Jiao, Y.; Li, J.; et al. Alpha5 nicotinic acetylcholine receptor mediated immune escape of lung adenocarcinoma via STAT3/Jab1-PD-L1 signalling. Cell Commun. Signal. 2022, 20, 121. [Google Scholar] [CrossRef]
- Nguyen, H.D.; Liao, Y.C.; Ho, Y.S.; Chen, L.C.; Chang, H.W.; Cheng, T.C.; Liu, D.; Lee, W.R.; Shen, S.C.; Wu, C.H.; et al. The α9 Nicotinic Acetylcholine Receptor Mediates Nicotine-Induced PD-L1 Expression and Regulates Melanoma Cell Proliferation and Migration. Cancers 2019, 11, 1991. [Google Scholar] [CrossRef] [Green Version]
- Kwok, H.H.; Gao, B.; Chan, K.H.; Ip, M.S.; Minna, J.D.; Lam, D.C. Nicotinic Acetylcholine Receptor Subunit α7 Mediates Cigarette Smoke-Induced PD-L1 Expression in Human Bronchial Epithelial Cells. Cancers 2021, 13, 5345. [Google Scholar] [CrossRef] [PubMed]
- Amos, C.I.; Wu, X.; Broderick, P.; Gorlov, I.P.; Gu, J.; Eisen, T.; Dong, Q.; Zhang, Q.; Gu, X.; Vijayakrishnan, J.; et al. Genome-wide association scan of tag SNPs identifies a susceptibility locus for lung cancer at 15q25.1. Nat. Genet. 2008, 40, 616–622. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hung, R.J.; McKay, J.D.; Gaborieau, V.; Boffetta, P.; Hashibe, M.; Zaridze, D.; Mukeria, A.; Szeszenia-Dabrowska, N.; Lissowska, J.; Rudnai, P.; et al. A susceptibility locus for lung cancer maps to nicotinic acetylcholine receptor subunit genes on 15q25. Nature 2008, 452, 633–637. [Google Scholar] [CrossRef] [Green Version]
- Routhier, J.; Pons, S.; Freidja, M.L.; Dalstein, V.; Cutrona, J.; Jonquet, A.; Lalun, N.; Mérol, J.C.; Lathrop, M.; Stitzel, J.A.; et al. An innate contribution of human nicotinic receptor polymorphisms to COPD-like lesions. Nat. Commun. 2021, 12, 6384. [Google Scholar] [CrossRef] [PubMed]
- Ma, X.; Jia, Y.; Zu, S.; Li, R.; Jia, Y.; Zhao, Y.; Xiao, D.; Dang, N.; Wang, Y. α5 Nicotinic acetylcholine receptor mediates nicotine-induced HIF-1α and VEGF expression in non-small cell lung cancer. Toxicol. Appl. Pharmacol. 2014, 278, 172–179. [Google Scholar] [CrossRef] [PubMed]
- Sun, H.; Ma, X. α5-nAChR modulates nicotine-induced cell migration and invasion in A549 lung cancer cells. Exp. Toxicol. Pathol. 2015, 67, 477–482. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Jia, Y.; Zhang, Y.; Zhou, D.; Sun, H.; Ma, X. α5-nAChR contributes to epithelial-mesenchymal transition and metastasis by regulating Jab1/Csn5 signalling in lung cancer. J. Cell Mol. Med. 2020, 24, 2497–2506. [Google Scholar] [CrossRef]
- Wang, F.; Gerzanich, V.; Wells, G.B.; Anand, R.; Peng, X.; Keyser, K.; Lindstrom, J. Assembly of human neuronal nicotinic receptor alpha5 subunits with alpha3, beta2, and beta4 subunits. J. Biol. Chem. 1996, 271, 17656–17665. [Google Scholar] [CrossRef] [Green Version]
- Le Novère, N.; Corringer, P.J.; Changeux, J.P. The diversity of subunit composition in nAChRs: Evolutionary origins, physiologic and pharmacologic consequences. J. Neurobiol. 2002, 53, 447–456. [Google Scholar] [CrossRef]
- Maskos, U. The nicotinic receptor alpha5 coding polymorphism rs16969968 as a major target in disease: Functional dissection and remaining challenges. J. Neurochem. 2020, 154, 241–250. [Google Scholar] [CrossRef] [Green Version]
- Scholze, P.; Huck, S. The α5 Nicotinic Acetylcholine Receptor Subunit Differentially Modulates α4β2* and α3β4* Receptors. Front. Synaptic. Neurosci. 2020, 12, 607959. [Google Scholar] [CrossRef]
- Ray, C.; Soderblom, E.J.; Bai, Y.; Carroll, F.I.; Caron, M.G.; Barak, L.S. Probing the Allosteric Role of α5 Subunit of α3β4α5 Nicotinic Acetylcholine Receptors by Functionally Selective Modulators and Ligands. ACS Chem. Biol. 2017, 12, 702–714. [Google Scholar] [CrossRef] [Green Version]
- Bailey, C.D.; Alves, N.C.; Nashmi, R.; De Biasi, M.; Lambe, E.K. Nicotinic α5 subunits drive developmental changes in the activation and morphology of prefrontal cortex layer VI neurons. Biol. Psychiatry 2012, 71, 120–128. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gotti, C.; Zoli, M.; Clementi, F. Brain nicotinic acetylcholine receptors: Native subtypes and their relevance. Trends Pharmacol. Sci. 2006, 27, 482–491. [Google Scholar] [CrossRef]
- Fowler, C.D.; Lu, Q.; Johnson, P.M.; Marks, M.J.; Kenny, P.J. Habenular α5 nicotinic receptor subunit signalling controls nicotine intake. Nature 2011, 471, 597–601. [Google Scholar] [CrossRef] [Green Version]
- Besson, M.; Guiducci, S.; Granon, S.; Guilloux, J.P.; Guiard, B.; Repérant, C.; Faure, P.; Pons, S.; Cannazza, G.; Zoli, M.; et al. Alterations in alpha5* nicotinic acetylcholine receptors result in midbrain- and hippocampus-dependent behavioural and neural impairments. Psychopharmacology 2016, 233, 3297–3314. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.C.; Cruchaga, C.; Saccone, N.L.; Bertelsen, S.; Liu, P.; Budde, J.P.; Duan, W.; Fox, L.; Grucza, R.A.; Kern, J.; et al. Risk for nicotine dependence and lung cancer is conferred by mRNA expression levels and amino acid change in CHRNA5. Hum. Mol. Genet. 2009, 18, 3125–3135. [Google Scholar] [CrossRef] [PubMed]
- Young, R.P.; Hopkins, R.J.; Hay, B.A.; Epton, M.J.; Black, P.N.; Gamble, G.D. Lung cancer gene associated with COPD: Triple whammy or possible confounding effect? Eur. Respir. J. 2008, 32, 1158–1164. [Google Scholar] [CrossRef]
- Saccone, N.L.; Culverhouse, R.C.; Schwantes-An, T.H.; Cannon, D.S.; Chen, X.; Cichon, S.; Giegling, I.; Han, S.; Han, Y.; Keskitalo-Vuokko, K.; et al. Multiple independent loci at chromosome 15q25.1 affect smoking quantity: A meta-analysis and comparison with lung cancer and COPD. PLoS Genet. 2010, 6, e1001053. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pillai, S.G.; Ge, D.; Zhu, G.; Kong, X.; Shianna, K.V.; Need, A.C.; Feng, S.; Hersh, C.P.; Bakke, P.; Gulsvik, A.; et al. A genome-wide association study in chronic obstructive pulmonary disease (COPD): Identification of two major susceptibility loci. PLoS Genet. 2009, 5, e1000421. [Google Scholar] [CrossRef] [Green Version]
- Wilk, J.B.; Shrine, N.R.; Loehr, L.R.; Loehr, L.R.; Zhao, J.H.; Manichaikul, A.; Lopez, L.M.; Smith, A.V.; Heckbert, S.R.; Smolonska, J.; et al. Genome-wide association studies identify CHRNA5/3 and HTR4 in the development of airflow obstruction. Am. J. Respir. Crit. Care Med. 2012, 186, 622–632. [Google Scholar] [CrossRef] [Green Version]
- Fischer, H.; Liu, D.M.; Lee, A.; Harries, J.C.; Adams, D.J. Selective modulation of neuronal nicotinic acetylcholine receptor channel subunits by Go-protein subunits. J. Neurosci. 2005, 25, 3571–3577. [Google Scholar] [CrossRef] [Green Version]
- De Nunzio, C.; Andriole, G.L.; Thompson, I.M., Jr.; Freedland, S.J. Smoking and Prostate Cancer: A Systematic Review. Eur. Urol. Focus. 2015, 1, 28–38. [Google Scholar] [CrossRef] [PubMed]
- Huncharek, M.; Haddock, K.S.; Reid, R.; Kupelnick, B. Smoking as a risk factor for prostate cancer: A meta-analysis of 24 prospective cohort studies. Am. J. Public Health 2010, 100, 693–701. [Google Scholar] [CrossRef] [PubMed]
- Qi, J.C.; Xue, W.Y.; Zhang, Y.P.; Qu, C.B.; Lu, B.S.; Yin, Y.W.; Liu, K.L.; Wang, D.B.; Li, W.; Zhao, Z.M. Cholinergic α5 nicotinic receptor is involved in the proliferation and invasion of human prostate cancer cells. Oncol. Rep. 2020, 43, 159–168. [Google Scholar] [CrossRef] [Green Version]
- Schuller, H.M. Is cancer triggered by altered signalling of nicotinic acetylcholine receptors? Nat. Rev. Cancer 2009, 9, 195–205. [Google Scholar] [CrossRef]
- Tyagi, A.; Sharma, S.; Wu, K.; Xing, F.; Liu, Y.; Zhao, D.; Deshpande, R.P.; D’Agostino, R.B., Jr.; Watabe, K. Nicotine promotes breast cancer metastasis by stimulating N2 neutrophils and generating pre-metastatic niche in lung. Nat. Commun. 2021, 12, 474. [Google Scholar] [CrossRef]
- Wei, P.L.; Kuo, L.J.; Huang, M.T.; Ting, W.C.; Ho, Y.S.; Wang, W.; An, J.; Chang, Y.J. Nicotine enhances colon cancer cell migration by induction of fibronectin. Ann. Surg. Oncol. 2011, 18, 1782–1790. [Google Scholar] [CrossRef]
- Wong, H.P.; Yu, L.; Lam, E.K.; Tai, E.K.; Wu, W.K.; Cho, C.H. Nicotine promotes cell proliferation via alpha7-nicotinic acetylcholine receptor and catecholamine-synthesizing enzymes-mediated pathway in human colon adenocarcinoma HT-29 cells. Toxicol. Appl. Pharmacol. 2007, 221, 261–267. [Google Scholar] [CrossRef]
- Borgström, A.; Hauert, B.; Kappel, S.; Zoni, E.; Kiener, M.; Stokłosa, P.; Baur, R.; Spahn, M.; Kruithof-de Julio, M.; Peinelt, C. Small Molecular Inhibitors Block TRPM4 Currents in Prostate Cancer Cells, with Limited Impact on Cancer Hallmark Functions. J. Mol. Biol. 2021, 433, 166665. [Google Scholar] [CrossRef] [PubMed]
- Stokłosa, P.; Borgström, A.; Hauert, B.; Baur, R.; Peinelt, C. Investigation of Novel Small Molecular TRPM4 Inhibitors in Colorectal Cancer Cells. Cancers 2021, 13, 5400. [Google Scholar] [CrossRef] [PubMed]
- Human Protein Atlas. Available online: https://www.proteinatlas.org (accessed on 1 May 2023).
- Cancer Genom Atlas (TCGA). Available online: https://www.cancer.gov/about-nci/organiza-tion/ccg/research/structural-genomics/tcga (accessed on 1 May 2023).
- Siegel, R.L.; Miller, K.D.; Wagle, N.S.; Jemal, A. Cancer statistics, 2023. CA Cancer J. Clin. 2023, 73, 17–48. [Google Scholar] [CrossRef] [PubMed]
- Fu, Y.; Ci, H.; Du, W.; Dong, Q.; Jia, H. CHRNA5 Contributes to Hepatocellular Carcinoma Progression by Regulating YAP Activity. Pharmaceutics 2022, 14, 275. [Google Scholar] [CrossRef] [PubMed]
- Jia, Y.; Sun, H.; Wu, H.; Zhang, H.; Zhang, X.; Xiao, D.; Ma, X.; Wang, Y. Nicotine Inhibits Cisplatin-Induced Apoptosis via Regulating α5-nAChR/AKT Signaling in Human Gastric Cancer Cells. PLoS ONE 2016, 11, e0149120. [Google Scholar] [CrossRef] [PubMed]
- Pucci, S.; Fasoli, F.; Moretti, M.; Benfante, R.; Di Lascio, S.; Viani, P.; Daga, A.; Gordon, T.J.; McIntosh, M.; Zoli, M.; et al. Choline and nicotine increase glioblastoma cell proliferation by binding and activating α7- and α9- containing nicotinic receptors. Pharmacol. Res. 2021, 163, 105336. [Google Scholar] [CrossRef] [PubMed]
- Cedillo, J.L.; Bordas, A.; Arnalich, F.; Esteban-Rodríguez, I.; Martín-Sánchez, C.; Extremera, M.; Atienza, G.; Rios, J.J.; Arribas, R.L.; Montiel, C. Anti-tumoral activity of the human-specific duplicated form of α7-nicotinic receptor subunit in tobacco-induced lung cancer progression. Lung Cancer 2019, 128, 134–144. [Google Scholar] [CrossRef]
- Maldifassi, M.C.; Momboisse, F.; Guerra, M.J.; Vielma, A.H.; Maripillán, J.; Báez-Matus, X.; Flores-Muñoz, C.; Cádiz, B.; Schmachtenberg, O.; Martínez, A.D.; et al. The interplay between α7 nicotinic acetylcholine receptors, pannexin-1 channels and P2X7 receptors elicit exocytosis in chromaffin cells. J. Neurochem. 2021, 157, 1789–1808. [Google Scholar] [CrossRef]
- Kappel, S.; Stokłosa, P.; Hauert, B.; Ross-Kaschitza, D.; Borgström, A.; Baur, R.; Galván, J.A.; Zlobec, I.; Peinelt, C. TRPM4 is highly expressed in human colorectal tumor buds and contributes to proliferation, cell cycle, and invasion of colorectal cancer cells. Mol. Oncol. 2019, 13, 2393–2405. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Albuquerque, E.X.; Pereira, E.F.; Alkondon, M.; Rogers, S.W. Mammalian nicotinic acetylcholine receptors: From structure to function. Physiol. Rev. 2009, 89, 73–120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dinicola, S.; Masiello, M.G.; Proietti, S.; Coluccia, P.; Fabrizi, G.; Catizone, A.; Ricci, G.; de Toma, G.; Bizzarri, M.; Cucina, A. Nicotine increases colon cancer cell migration and invasion through epithelial to mesenchymal transition (EMT): COX-2 involvement. J. Cell Physiol. 2018, 233, 4935–4948. [Google Scholar] [CrossRef]
- Tammimäki, A.; Herder, P.; Li, P.; Esch, C.; Laughlin, J.R.; Akk, G.; Stitzel, J.A. Impact of human D398N single nucleotide polymorphism on intracellular calcium response mediated by α3β4α5 nicotinic acetylcholine receptors. Neuropharmacology 2012, 63, 1002–1011. [Google Scholar] [CrossRef] [Green Version]
- Breum, A.W.; Falk, S.; Svendsen, C.S.A.; Nicolaisen, T.S.; Mathiesen, C.V.; Maskos, U.; Clemmensen, C. Divergent Roles of α5 and β4 Nicotinic Receptor Subunits in Food Reward and Nicotine-induced Weight Loss in Male Mice. Endocrinology 2022, 163, bqac079. [Google Scholar] [CrossRef]
- Forget, B.; Scholze, P.; Langa, F.; Morel, C.; Pons, S.; Mondoloni, S.; Besson, M.; Durand-de Cuttoli, R.; Hay, A.; Tricoire, L.; et al. A Human Polymorphism in CHRNA5 Is Linked to Relapse to Nicotine Seeking in Transgenic Rats. Curr. Biol. 2018, 28, 3244–3253.e7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Exley, R.; McIntosh, J.M.; Marks, M.J.; Maskos, U.; Cragg, S.J. Striatal α5 nicotinic receptor subunit regulates dopamine transmission in dorsal striatum. J. Neurosci. 2012, 32, 2352–2356. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Improgo, M.R.; Scofield, M.D.; Tapper, A.R.; Gardner, P.D. The nicotinic acetylcholine receptor CHRNA5/A3/B4 gene cluster: Dual role in nicotine addiction and lung cancer. Prog. Neurobiol. 2010, 92, 212–226. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hollenhorst, M.I.; Krasteva-Christ, G. Nicotinic Acetylcholine Receptors in the Respiratory Tract. Molecules 2021, 26, 6097. [Google Scholar] [CrossRef] [PubMed]
- Shehwana, H.; Keskus, A.G.; Ozdemir, S.E.; Acikgöz, A.A.; Biyik-Sit, R.; Cagnan, I.; Gunes, D.; Jahja, E.; Cingir-Koker, S.; Olmezer, G.; et al. CHRNA5 belongs to the secondary estrogen signalling network exhibiting prognostic significance in breast cancer. Cell Oncol. 2021, 44, 453–472. [Google Scholar] [CrossRef]
- Jackson, K.J.; Marks, M.J.; Vann, R.E.; Chen, X.; Gamage, T.F.; Warner, J.A.; Damaj, M.I. Role of alpha5 nicotinic acetylcholine receptors in pharmacological and behavioural effects of nicotine in mice. J. Pharmacol. Exp. Ther. 2010, 334, 137–146. [Google Scholar] [CrossRef] [Green Version]
- Brown, R.W.; Collins, A.C.; Lindstrom, J.M.; Whiteaker, P. Nicotinic alpha5 subunit deletion locally reduces high-affinity agonist activation without altering nicotinic receptor numbers. J. Neurochem. 2007, 103, 204–215. [Google Scholar] [CrossRef]
- Strathdee, G. Epigenetic versus genetic alterations in the inactivation of E-cadherin. Semin. Cancer Biol. 2002, 12, 373–379. [Google Scholar] [CrossRef]
- Bracke, M.E.; Van Roy, F.M.; Mareel, M.M. The E-cadherin/catenin complex in invasion and metastasis. Curr. Top Microbiol. Immunol. 1996, 213, 123–161. [Google Scholar] [CrossRef]
- Kang, Y.; Massagué, J. Epithelial-mesenchymal transitions: Twist in development and metastasis. Cell 2004, 118, 277–279. [Google Scholar] [CrossRef] [Green Version]
- Bukholm, I.K.; Nesland, J.M.; Børresen-Dale, A.L. Re-expression of E-cadherin, alpha-catenin and beta-catenin, but not of gamma-catenin, in metastatic tissue from breast cancer patients. J. Pathol. 2000, 190, 15–19. [Google Scholar] [CrossRef]
- Lou, Y.; Preobrazhenska, O.; auf dem Keller, U.; Sutcliffe, M.; Barclay, L.; McDonald, P.C.; Roskelley, C.; Overall, C.M.; Dedhar, S. Epithelial-mesenchymal transition (EMT) is not sufficient for spontaneous murine breast cancer metastasis. Dev. Dyn. 2008, 237, 2755–2768. [Google Scholar] [CrossRef] [PubMed]
- Hollestelle, A.; Peeters, J.K.; Smid, M.; Timmermans, M.; Verhoog, L.C.; Westenend, P.J.; Heine, A.A.; Chan, A.; Sieuwerts, A.M.; Wiemer, E.A.; et al. Loss of E-cadherin is not a necessity for epithelial to mesenchymal transition in human breast cancer. Breast Cancer Res. Treat. 2013, 138, 47–57. [Google Scholar] [CrossRef] [PubMed]
- Querzoli, P.; Coradini, D.; Pedriali, M.; Boracchi, P.; Ambrogi, F.; Raimondi, E.; La Sorda, R.; Lattanzio, R.; Rinaldi, R.; Lunardi, M.; et al. An immunohistochemically positive E-cadherin status is not always predictive for a good prognosis in human breast cancer. Br. J. Cancer 2010, 103, 1835–1839. [Google Scholar] [CrossRef] [Green Version]
- Wang, M.L.; Hsu, Y.F.; Liu, C.H.; Kuo, C.H.; Chen, Y.C.; Yeh, Y.C.; Ho, H.L.; Wu, Y.C.; Chou, T.Y.; Wu, C.W. Low-Dose Nicotine Activates EGFR Signaling via α5-nAChR and Promotes Lung Adenocarcinoma Progression. Int. J. Mol. Sci. 2020, 21, 6829. [Google Scholar] [CrossRef]
- Oshikawa, J.; Toya, Y.; Fujita, T.; Egawa, M.; Kawabe, J.; Umemura, S.; Ishikawa, Y. Nicotinic acetylcholine receptor alpha 7 regulates cAMP signal within lipid rafts. Am. J. Physiol. Cell Physiol. 2003, 285, C567–C574. [Google Scholar] [CrossRef] [Green Version]
- Cheng, Q.; Yakel, J.L. Activation of α7 nicotinic acetylcholine receptors increases intracellular cAMP levels via activation of AC1 in hippocampal neurons. Neuropharmacology 2015, 95, 405–414. [Google Scholar] [CrossRef] [Green Version]
- Zhu, S.; Huang, S.; Xia, G.; Wu, J.; Shen, Y.; Wang, Y.; Ostrom, R.S.; Du, A.; Shen, C.; Xu, C. Anti-inflammatory effects of α7-nicotinic ACh receptors are exerted through interactions with adenylyl cyclase-6. Br. J. Pharmacol. 2021, 178, 2324–2338. [Google Scholar] [CrossRef]
- King, J.R.; Nordman, J.C.; Bridges, S.P.; Lin, M.K.; Kabbani, N. Identification and Characterization of a G Protein-binding Cluster in α7 Nicotinic Acetylcholine Receptors. J. Biol. Chem. 2015, 290, 20060–20070. [Google Scholar] [CrossRef] [Green Version]
- King, J.R.; Gillevet, T.C.; Kabbani, N. A G-protein-coupled α7 nicotinic receptor regulates signaling and TNF-α release in microglia. FEBS Open Bio. 2017, 7, 1350–1361. [Google Scholar] [CrossRef]
- Nordman, J.C.; Muldoon, P.; Clark, S.; Damaj, M.I.; Kabbani, N. The α4 nicotinic receptor promotes CD4+ T-cell proliferation and a helper T-cell immune response. Mol. Pharmacol. 2014, 85, 50–61. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chrestia, J.F.; Bruzzone, A.; Esandi, M.D.C.; Bouzat, C. Tyrosine phosphorylation differentially fine-tunes ionotropic and metabotropic responses of human α7 nicotinic acetylcholine receptor. Cell. Mol. Life. Sci. 2021, 78, 5381–5395. [Google Scholar] [CrossRef] [PubMed]
- Kabbani, N.; Nordman, J.C.; Corgiat, B.A.; Veltri, D.P.; Shehu, A.; Seymour, V.A.; Adams, D.J. Are nicotinic acetylcholine receptors coupled to G proteins? Bioessays 2013, 35, 1025–1034. [Google Scholar] [CrossRef] [PubMed]
- Razani-Boroujerdi, S.; Boyd, R.T.; Dávila-García, M.I.; Nandi, J.S.; Mishra, N.C.; Singh, S.P.; Pena-Philippides, J.C.; Langley, R.; Sopori, M.L. T cells express alpha7-nicotinic acetylcholine receptor subunits that require a functional TCR and leukocyte-specific protein tyrosine kinase for nicotine-induced Ca2+ response. J. Immunol. 2007, 179, 2889–2898. [Google Scholar] [CrossRef]
- Skok, M.V. To channel or not to channel? Functioning of nicotinic acetylcholine receptors in leukocytes. J. Leukoc. Biol. 2009, 86, 1–3. [Google Scholar] [CrossRef]
- Chernyavsky, A.; Chen, Y.; Wang, P.H.; Grando, S.A. Pemphigus vulgaris antibodies target the mitochondrial nicotinic acetylcholine receptors that protect keratinocytes from apoptolysis. Int. Immunopharmacol. 2015, 29, 76–80. [Google Scholar] [CrossRef]
- Gergalova, G.; Lykhmus, O.; Kalashnyk, O.; Koval, L.; Chernyshov, V.; Kryukova, E.; Tsetlin, V.; Komisarenko, S.; Skok, M. Mitochondria express α7 nicotinic acetylcholine receptors to regulate Ca2+ accumulation and cytochrome c release: Study on isolated mitochondria. PLoS ONE 2012, 7, e31361. [Google Scholar] [CrossRef]
- Skok, M.; Gergalova, G.; Lykhmus, O.; Kalashnyk, O.; Koval, L.; Uspenska, K. Nicotinic acetylcholine receptors in mitochondria: Subunit composition, function and signaling. Neurotransmitter 2016, 3, e1290. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Protein | Gene Name | Probe ID |
|---|---|---|
| α3 subunit * | CHRNA3 | Hs01088199_m1 |
| α4 subunit * | CHRNA4 | Hs00181247_m1 |
| α5 subunit * | CHRNA5 | Hs00181248_m1 |
| α7 subunit * | CHRNA7 | Hs01063372_m1 |
| dupα7 subunit * | CHRFAM7A | Hs04189909_m1 |
| α9 subunit * | CHRNA9 | Hs00395558_m1 |
| β2 subunit * | CHRNB2 | Hs01114010_g1 |
| β4 subunit * | CHRNB4 | Hs00609523_m1 |
| CD47 | CD47 | Hs00179953_m1 |
| PDL-1 | CD274 | Hs00204257_m1 |
| Vimentin | VIM | Hs00958111_m1 |
| E-cadherin | CDH1 | Hs01023895_m1 |
| TATA-binding protein | TBP | Hs00427621_m1 |
| nAChR Subunit | MCF7 | A549 | PC9 | SW480 | SW620 | CACO2 | DU145 | PC3 | LNCAP |
|---|---|---|---|---|---|---|---|---|---|
| α3 | 0.024 ±0.002 | 0.057 ±0.014 | n.d. | 0.261 ±0.054 | 0.007 ±0.002 | n.d. | 0.002 ±0.0 | n.d. | 0.013 ±0.0 |
| α4 | n.d. | 0.005 ±0.001 | n.d. | 0.035 ±0.012 | n.d. | 0.004 ±0.002 | n.d. | n.d. | n.d. |
| α5 | 1.431 ±0.001 | 3.396 ±0.743 | 1.020 ±0.044 | 0.461 ±0.114 | 3.739 ±0.284 | 0.698 ±0.301 | 1.444 ±0.087 | 0.321 ±0.075 | 0.469 ±0.052 |
| α7 | 0.002 ±0.0 | 0.141 ±0.033 | n.d. | 0.010 ±0.004 | 0.007 ±0.002 | 0.011 ±0.005 | 0.013 ±0.002 | 0.001 ±0.0 | 0.098 ±0.021 |
| dup α7 | n.d. | 1.211 ±0.766 | 0.009 ±0.005 | 0.002 ±0.001 | 0.058 ±0.021 | 0.012 ±0.004 | 0.271 ±0.053 | 0.077 ±0.025 | 0.010 ±0.002 |
| α9 | n.d. | 0.001 ±0.0 | 0.001 ±0.0 | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. |
| β2 | 0.003 ±0.0 | 0.102 ±0.017 | 0.002 ±0.0 | 0.055 ±0.016 | 0.263 ±0.097 | 0.003 ±0.001 | 0.001 ±0.0 | 0.001 ±0.0 | 0.001 ±0.0 |
| β4 | 0.031 ±0.007 | 0.107 ±0.006 | 0.009 ±0.003 | 0.050 ±0.019 | 0.013 ±0.005 | 0.009 ±0.004 | 0.02 2 ± 0.0 | 0.001 ±0.0 | n.d. |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Papapostolou, I.; Ross-Kaschitza, D.; Bochen, F.; Peinelt, C.; Maldifassi, M.C. Contribution of the α5 nAChR Subunit and α5SNP to Nicotine-Induced Proliferation and Migration of Human Cancer Cells. Cells 2023, 12, 2000. https://doi.org/10.3390/cells12152000
Papapostolou I, Ross-Kaschitza D, Bochen F, Peinelt C, Maldifassi MC. Contribution of the α5 nAChR Subunit and α5SNP to Nicotine-Induced Proliferation and Migration of Human Cancer Cells. Cells. 2023; 12(15):2000. https://doi.org/10.3390/cells12152000
Chicago/Turabian StylePapapostolou, Irida, Daniela Ross-Kaschitza, Florian Bochen, Christine Peinelt, and Maria Constanza Maldifassi. 2023. "Contribution of the α5 nAChR Subunit and α5SNP to Nicotine-Induced Proliferation and Migration of Human Cancer Cells" Cells 12, no. 15: 2000. https://doi.org/10.3390/cells12152000
APA StylePapapostolou, I., Ross-Kaschitza, D., Bochen, F., Peinelt, C., & Maldifassi, M. C. (2023). Contribution of the α5 nAChR Subunit and α5SNP to Nicotine-Induced Proliferation and Migration of Human Cancer Cells. Cells, 12(15), 2000. https://doi.org/10.3390/cells12152000