

GPER1 Activation Exerts Anti-Tumor Activity in Multiple Myeloma


 , , , , , ,
, , , , , , 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Gene Expression Profiling Analysis
2.2. Cell Lines and Primary Samples
2.3. Drugs
2.4. Cell Viability and Proliferation Assays
2.5. Apoptosis Assays
2.6. Lentivirus Production, Transduction and Transfection of MM Cells
2.7. Reverse Transcription and Quantitative Real-Time Amplification (q-RT-PCR)
2.8. Western Blotting (WB)
2.9. Chromatin Immunoprecipitation (ChIP)
2.10. In Vivo Studies
2.11. Statistical Analysis
3. Results
3.1. GPER mRNA Is Down-Regulated in MM and Associates with Worse Overall Survival
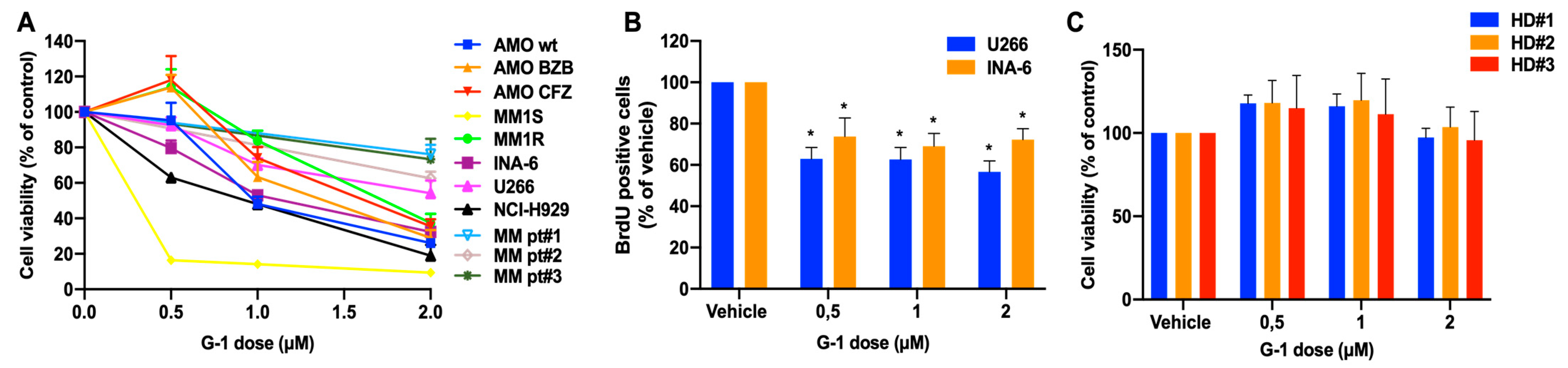
3.2. In Vitro Anti-Tumor Activity of the GPER1 Agonist G-1 in MM
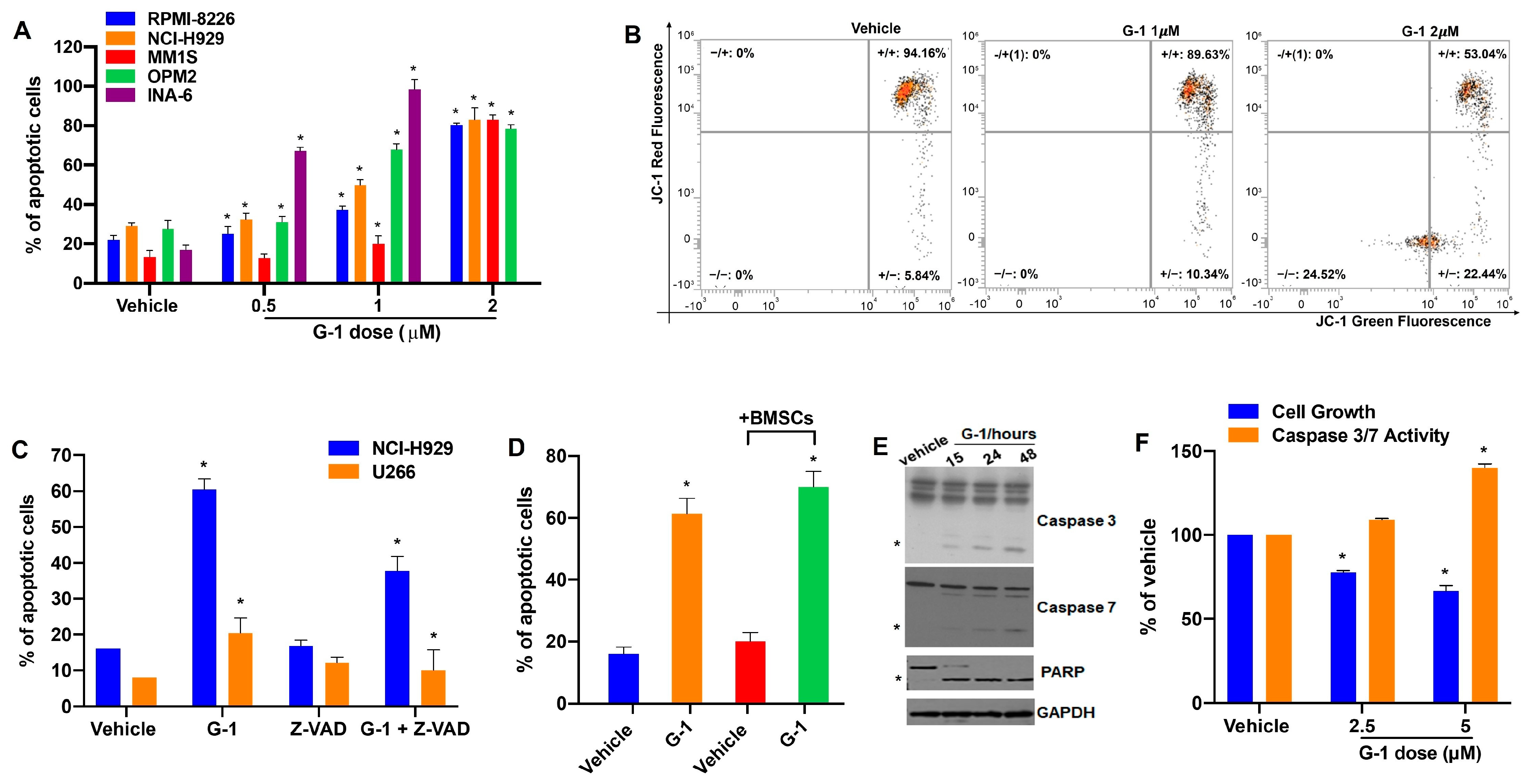
3.3. G-1 Is Pro-Apoptotic in MM
3.4. G-1 Upregulates the miR-29b Network in MM Cells
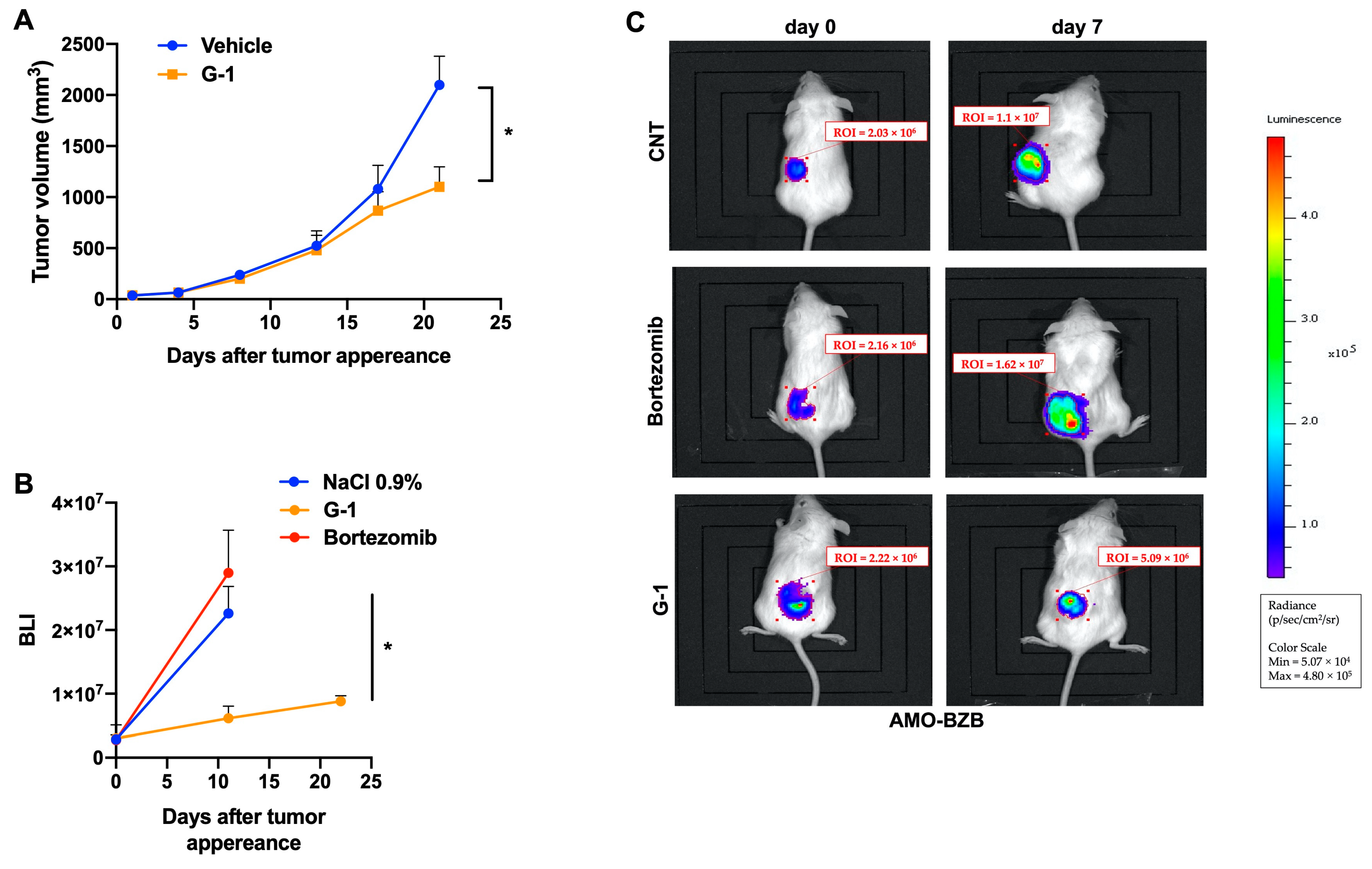
3.5. G-1 Exerts Anti-MM Activity In Vivo
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Hideshima, T.; Anderson, K.C. Signaling Pathway Mediating Myeloma Cell Growth and Survival. Cancers 2021, 13, 216. [Google Scholar] [CrossRef] [PubMed]
- Morelli, E.; Gullà, A.; Rocca, R.; Federico, C.; Raimondi, L.; Malvestiti, S.; Agosti, V.; Rossi, M.; Costa, G.; Giavaresi, G.; et al. The Non-Coding RNA Landscape of Plasma Cell Dyscrasias. Cancers 2020, 12, 320. [Google Scholar] [CrossRef]
- Taiana, E.; Gallo Cantafio, M.E.; Favasuli, V.K.; Bandini, C.; Viglietto, G.; Piva, R.; Neri, A.; Amodio, N. Genomic Instability in Multiple Myeloma: A “Non-Coding RNA” Perspective. Cancers 2021, 13, 2127. [Google Scholar] [CrossRef] [PubMed]
- Paradzik, T.; Bandini, C.; Mereu, E.; Labrador, M.; Taiana, E.; Amodio, N.; Neri, A.; Piva, R. The Landscape of Signaling Pathways and Proteasome Inhibitors Combinations in Multiple Myeloma. Cancers 2021, 13, 1235. [Google Scholar] [CrossRef]
- Ambrosio, F.A.; Costa, G.; Gallo Cantafio, M.E.; Torcasio, R.; Trapasso, F.; Alcaro, S.; Viglietto, G.; Amodio, N. Natural Agents as Novel Potential Source of Proteasome Inhibitors with Anti-Tumor Activity: Focus on Multiple Myeloma. Molecules 2023, 28, 1438. [Google Scholar] [CrossRef] [PubMed]
- Barton, M.; Filardo, E.J.; Lolait, S.J.; Thomas, P.; Maggiolini, M.; Prossnitz, E.R. Twenty years of the G protein-coupled estrogen receptor GPER: Historical and personal perspectives. J. Steroid Biochem. Mol. Biol. 2018, 176, 4–15. [Google Scholar] [CrossRef] [PubMed]
- Thomas, P.; Pang, Y.; Filardo, E.J.; Dong, J. Identity of an Estrogen Membrane Receptor Coupled to a G Protein in Human Breast Cancer Cells. Endocrinology 2005, 146, 624–632. [Google Scholar] [CrossRef]
- Gonzalez de Valdivia, E.; Broselid, S.; Kahn, R.; Olde, B.; Leeb-Lundberg, L.M.F. G protein-coupled estrogen receptor 1 (GPER1)/GPR30 increases ERK1/2 activity through PDZ motif-dependent and -independent mechanisms. J. Biol. Chem. 2017, 292, 9932–9943. [Google Scholar] [CrossRef]
- Vrtačnik, P.; Ostanek, B.; Mencej-Bedrač, S.; Marc, J. The many faces of estrogen signaling. Biochem. Medica 2014, 24, 329–342. [Google Scholar] [CrossRef]
- Prossnitz, E.R.; Barton, M. The G-protein-coupled estrogen receptor GPER in health and disease. Nat. Rev. Endocrinol. 2011, 7, 715–726. [Google Scholar] [CrossRef]
- Filardo, E.J.; Graeber, C.T.; Quinn, J.A.; Resnick, M.B.; Giri, D.; DeLellis, R.A.; Steinhoff, M.M.; Sabo, E. Distribution of GPR30, a Seven Membrane–Spanning Estrogen Receptor, in Primary Breast Cancer and its Association with Clinicopathologic Determinants of Tumor Progression. Clin. Cancer Res. 2006, 12, 6359–6366. [Google Scholar] [CrossRef]
- Smith, H.O.; Arias-Pulido, H.; Kuo, D.Y.; Howard, T.; Qualls, C.R.; Lee, S.-J.; Verschraegen, C.F.; Hathaway, H.J.; Joste, N.E.; Prossnitz, E.R. GPR30 predicts poor survival for ovarian cancer. Gynecol. Oncol. 2009, 114, 465–471. [Google Scholar] [CrossRef]
- Santolla, M.F.; Vivacqua, A.; Lappano, R.; Rigiracciolo, D.C.; Cirillo, F.; Galli, G.R.; Talia, M.; Brunetti, G.; Miglietta, A.M.; Belfiore, A.; et al. GPER Mediates a Feedforward FGF2/FGFR1 Paracrine Activation Coupling CAFs to Cancer Cells toward Breast Tumor Progression. Cells 2019, 8, 223. [Google Scholar] [CrossRef] [PubMed]
- Talia, M.; De Francesco, E.; Rigiracciolo, D.; Muoio, M.; Muglia, L.; Belfiore, A.; Maggiolini, M.; Sims, A.; Lappano, R. The G Protein-Coupled Estrogen Receptor (GPER) Expression Correlates with Pro-Metastatic Pathways in ER-Negative Breast Cancer: A Bioinformatics Analysis. Cells 2020, 9, 622. [Google Scholar] [CrossRef]
- Hirtz, A.; Lebourdais, N.; Rech, F.; Bailly, Y.; Vaginay, A.; Smaïl-Tabbone, M.; Dubois-Pot-Schneider, H.; Dumond, H. GPER Agonist G-1 Disrupts Tubulin Dynamics and Potentiates Temozolomide to Impair Glioblastoma Cell Proliferation. Cells 2021, 10, 3438. [Google Scholar] [CrossRef]
- Chimento, A.; Casaburi, I.; Bartucci, M.; Patrizii, M.; Dattilo, R.; Avena, P.; Andò, S.; Pezzi, V.; Sirianni, R. Selective GPER activation decreases proliferation and activates apoptosis in tumor Leydig cells. Cell Death Dis. 2013, 4, e747. [Google Scholar] [CrossRef] [PubMed]
- Ambrosini, G.; Natale, C.A.; Musi, E.; Garyantes, T.; Schwartz, G.K. The GPER Agonist LNS8801 Induces Mitotic Arrest and Apoptosis in Uveal Melanoma Cells. Cancer Res. Commun. 2023, 3, 540–547. [Google Scholar] [CrossRef] [PubMed]
- Pepermans, R.A.; Sharma, G.; Prossnitz, E.R. G Protein-Coupled Estrogen Receptor in Cancer and Stromal Cells: Functions and Novel Therapeutic Perspectives. Cells 2021, 10, 672. [Google Scholar] [CrossRef] [PubMed]
- Ren, J.; Tao, Y.; Peng, M.; Xiao, Q.; Jing, Y.; Huang, J.; Yang, J.; Lin, C.; Sun, M.; Lei, L.; et al. Targeted activation of GPER enhances the efficacy of venetoclax by boosting leukemic pyroptosis and CD8+ T cell immune function in acute myeloid leukemia. Cell Death Dis. 2022, 13, 915. [Google Scholar] [CrossRef]
- Torres-López, L.; Olivas-Aguirre, M.; Villatoro-Gómez, K.; Dobrovinskaya, O. The G-Protein–Coupled Estrogen Receptor Agonist G-1 Inhibits Proliferation and Causes Apoptosis in Leukemia Cell Lines of T Lineage. Front. Cell Dev. Biol. 2022, 10, 811479. [Google Scholar] [CrossRef]
- Zhou, L.; Yu, T.; Yang, F.; Han, J.; Zuo, B.; Huang, L.; Bai, X.; Jiang, M.; Wu, D.; Chen, S.; et al. G Protein-Coupled Estrogen Receptor Agonist G-1 Inhibits Mantle Cell Lymphoma Growth in Preclinical Models. Front. Oncol. 2021, 11, 668617. [Google Scholar] [CrossRef]
- Morelli, E.; Hunter, Z.R.; Fulciniti, M.; Gullà, A.; Perrotta, I.D.; Zuccalà, V.; Federico, C.; Juli, G.; Manzoni, M.; Ronchetti, D.; et al. Therapeutic activation of G protein-coupled estrogen receptor 1 in Waldenström Macroglobulinemia. Exp. Hematol. Oncol. 2022, 11, 54. [Google Scholar] [CrossRef]
- Cardona-Benavides, I.J.; de Ramón, C.; Gutiérrez, N.C. Genetic Abnormalities in Multiple Myeloma: Prognostic and Therapeutic Implications. Cells 2021, 10, 336. [Google Scholar] [CrossRef]
- Agnelli, L.; Mosca, L.; Fabris, S.; Lionetti, M.; Andronache, A.; Kwee, I.; Todoerti, K.; Verdelli, D.; Battaglia, C.; Bertoni, F.; et al. A SNP microarray and FISH-based procedure to detect allelic imbalances in multiple myeloma: An integrated genomics approach reveals a wide gene dosage effect. Genes Chromosomes Cancer 2009, 48, 603–614. [Google Scholar] [CrossRef]
- Chng, W.-J.; Huang, G.F.; Chung, T.H.; Ng, S.B.; Gonzalez-Paz, N.; Troska-Price, T.; Mulligan, G.; Chesi, M.; Bergsagel, P.L.; Fonseca, R. Clinical and biological implications of MYC activation: A common difference between MGUS and newly diagnosed multiple myeloma. Leukemia 2011, 25, 1026–1035. [Google Scholar] [CrossRef] [PubMed]
- Gutiérrez, N.C.; Ocio, E.M.; de Las Rivas, J.; Maiso, P.; Delgado, M.; Fermiñán, E.; Arcos, M.J.; Sánchez, M.L.; Hernández, J.M.; San Miguel, J.F. Gene expression profiling of B lymphocytes and plasma cells from Waldenström’s macroglobulinemia: Comparison with expression patterns of the same cell counterparts from chronic lymphocytic leukemia, multiple myeloma and normal individuals. Leukemia 2007, 21, 541–549. [Google Scholar] [CrossRef] [PubMed]
- Lombardi, L.; Poretti, G.; Mattioli, M.; Fabris, S.; Agnelli, L.; Bicciato, S.; Kwee, I.; Rinaldi, A.; Ronchetti, D.; Verdelli, D.; et al. Molecular characterization of human multiple myeloma cell lines by integrative genomics: Insights into the biology of the disease. Genes Chromosomes Cancer 2007, 46, 226–238. [Google Scholar] [CrossRef] [PubMed]
- Ronchetti, D.; Lionetti, M.; Mosca, L.; Agnelli, L.; Andronache, A.; Fabris, S.; Deliliers, G.L.; Neri, A. An integrative genomic approach reveals coordinated expression of intronic miR-335, miR-342, and miR-561 with deregulated host genes in multiple myeloma. BMC Med. Genom. 2008, 1, 37. [Google Scholar] [CrossRef]
- Mulligan, G.; Mitsiades, C.; Bryant, B.; Zhan, F.; Chng, W.J.; Roels, S.; Koenig, E.; Fergus, A.; Huang, Y.; Richardson, P.; et al. Gene expression profiling and correlation with outcome in clinical trials of the proteasome inhibitor bortezomib. Blood 2007, 109, 3177–3188. [Google Scholar] [CrossRef]
- Todoerti, K.; Gallo Cantafio, M.E.; Oliverio, M.; Juli, G.; Rocca, C.; Citraro, R.; Tassone, P.; Procopio, A.; De Sarro, G.; Neri, A.; et al. Oleil Hydroxytyrosol (HTOL) Exerts Anti-Myeloma Activity by Antagonizing Key Survival Pathways in Malignant Plasma Cells. Int. J. Mol. Sci. 2021, 22, 11639. [Google Scholar] [CrossRef]
- Juli, G.; Oliverio, M.; Bellizzi, D.; Gallo Cantafio, M.E.; Grillone, K.; Passarino, G.; Colica, C.; Nardi, M.; Rossi, M.; Procopio, A.; et al. Anti-tumor Activity and Epigenetic Impact of the Polyphenol Oleacein in Multiple Myeloma. Cancers 2019, 11, 990. [Google Scholar] [CrossRef]
- Amodio, N.; Gallo Cantafio, M.; Botta, C.; Agosti, V.; Federico, C.; Caracciolo, D.; Ronchetti, D.; Rossi, M.; Driessen, C.; Neri, A.; et al. Replacement of miR-155 Elicits Tumor Suppressive Activity and Antagonizes Bortezomib Resistance in Multiple Myeloma. Cancers 2019, 11, 236. [Google Scholar] [CrossRef] [PubMed]
- Hirtz, A.; Bailly, Y.; Rech, F.; Pierson, J.; Dumond, H.; Dubois-Pot-Schneider, H. Molecular Characterization of the Dual Effect of the GPER Agonist G-1 in Glioblastoma. Int. J. Mol. Sci. 2022, 23, 14309. [Google Scholar] [CrossRef] [PubMed]
- Wei, W.; Chen, Z.-J.; Zhang, K.-S.; Yang, X.-L.; Wu, Y.-M.; Chen, X.-H.; Huang, H.-B.; Liu, H.-L.; Cai, S.-H.; Du, J.; et al. The activation of G protein-coupled receptor 30 (GPR30) inhibits proliferation of estrogen receptor-negative breast cancer cells in vitro and in vivo. Cell Death Dis. 2014, 5, e1428. [Google Scholar] [CrossRef]
- Wang, C.; Lv, X.; He, C.; Hua, G.; Tsai, M.-Y.; Davis, J.S. The G-protein-coupled estrogen receptor agonist G-1 suppresses proliferation of ovarian cancer cells by blocking tubulin polymerization. Cell Death Dis. 2013, 4, e869. [Google Scholar] [CrossRef]
- McDermott, A.; Kim, K.; Kasper, S.; Ho, S.-M.; Leung, Y.-K. The androgen receptor inhibits transcription of GPER1 by preventing Sp1 and Sp3 from binding to the promoters in prostate cancer cells. Oncotarget 2022, 13, 46–60. [Google Scholar] [CrossRef] [PubMed]
- Fulciniti, M.; Amin, S.; Nanjappa, P.; Rodig, S.; Prabhala, R.; Li, C.; Minvielle, S.; Tai, Y.; Tassone, P.; Avet-Loiseau, H.; et al. Significant Biological Role of Sp1 Transactivation in Multiple Myeloma. Clin. Cancer Res. 2011, 17, 6500–6509. [Google Scholar] [CrossRef]
- Amodio, N.; Di Martino, M.T.; Foresta, U.; Leone, E.; Lionetti, M.; Leotta, M.; Gullà, A.M.; Pitari, M.R.; Conforti, F.; Rossi, M.; et al. miR-29b sensitizes multiple myeloma cells to bortezomib-induced apoptosis through the activation of a feedback loop with the transcription factor Sp1. Cell Death Dis. 2012, 3, e436. [Google Scholar] [CrossRef] [PubMed]
- Rae, J.M.; Johnson, M.D. What does an orphan G-protein-coupled receptor have to do with estrogen? Breast Cancer Res. 2005, 7, 243. [Google Scholar] [CrossRef] [PubMed]
- Gauduchon, J.; Gouilleux, F.; Maillard, S.; Marsaud, V.; Renoir, J.-M.; Sola, B. 4-Hydroxytamoxifen inhibits proliferation of multiple myeloma cells in vitro through down-regulation of c-Myc, up-regulation of p27Kip1, and modulation of Bcl-2 family members. Clin. Cancer Res. 2005, 11, 2345–2354. [Google Scholar] [CrossRef]
- Decaudin, D.; Etienne, M.-C.; de Cremoux, P.; Maciorowski, Z.; Vantelon, J.-M.; Voog, E.; Urien, S.; Tran-Perennou, C.; Renee, N.; Vielh, P.; et al. Multicenter Phase II Feasibility Trial of High-Dose Tamoxifen in Patients with Refractory or Relapsed Multiple Myeloma. JNCI J. Natl. Cancer Inst. 2004, 96, 636–637. [Google Scholar] [CrossRef] [PubMed]
- Xu, S.; Yu, S.; Dong, D.; Lee, L.T.O. G Protein-Coupled Estrogen Receptor: A Potential Therapeutic Target in Cancer. Front. Endocrinol. 2019, 10, 725. [Google Scholar] [CrossRef]
- He, Y.; Su, Y.; Zeng, J.; Chong, W.; Hu, X.; Zhang, Y.; Peng, X. Cancer-specific survival after diagnosis in men versus women: A pan-cancer analysis. MedComm 2022, 3, e145. [Google Scholar] [CrossRef]
- Lee, S.-J.; Kim, T.W.; Park, G.L.; Hwang, Y.S.; Cho, H.J.; Kim, J.-T.; Lee, H.G. G protein-coupled estrogen receptor-1 agonist induces chemotherapeutic effect via ER stress signaling in gastric cancer. BMB Rep. 2019, 52, 647–652. [Google Scholar] [CrossRef] [PubMed]
- Natale, C.A.; Li, J.; Pitarresi, J.R.; Norgard, R.J.; Dentchev, T.; Capell, B.C.; Seykora, J.T.; Stanger, B.Z.; Ridky, T.W. Pharmacologic Activation of the G Protein–Coupled Estrogen Receptor Inhibits Pancreatic Ductal Adenocarcinoma. Cell. Mol. Gastroenterol. Hepatol. 2020, 10, 868–880.e1. [Google Scholar] [CrossRef]
- Bologa, C.G.; Revankar, C.M.; Young, S.M.; Edwards, B.S.; Arterburn, J.B.; Kiselyov, A.S.; Parker, M.A.; Tkachenko, S.E.; Savchuck, N.P.; Sklar, L.A.; et al. Virtual and biomolecular screening converge on a selective agonist for GPR30. Nat. Chem. Biol. 2006, 2, 207–212. [Google Scholar] [CrossRef] [PubMed]
- Dennis, M.K.; Burai, R.; Ramesh, C.; Petrie, W.K.; Alcon, S.N.; Nayak, T.K.; Bologa, C.G.; Leitao, A.; Brailoiu, E.; Deliu, E.; et al. In vivo effects of a GPR30 antagonist. Nat. Chem. Biol. 2009, 5, 421–427. [Google Scholar] [CrossRef]
- Dennis, M.K.; Field, A.S.; Burai, R.; Ramesh, C.; Petrie, W.K.; Bologa, C.G.; Oprea, T.I.; Yamaguchi, Y.; Hayashi, S.-I.; Sklar, L.A.; et al. Identification of a GPER/GPR30 antagonist with improved estrogen receptor counterselectivity. J. Steroid Biochem. Mol. Biol. 2011, 127, 358–366. [Google Scholar] [CrossRef]
- Shen, Y.; Li, C.; Zhou, L.; Huang, J. G protein-coupled oestrogen receptor promotes cell growth of non-small cell lung cancer cells via YAP1/QKI/circNOTCH1/m6A methylated NOTCH1 signalling. J. Cell. Mol. Med. 2021, 25, 284–296. [Google Scholar] [CrossRef]
- Jacenik, D.; Beswick, E.J.; Krajewska, W.M.; Prossnitz, E.R. G protein-coupled estrogen receptor in colon function, immune regulation and carcinogenesis. World J. Gastroenterol. 2019, 25, 4092–4104. [Google Scholar] [CrossRef] [PubMed]
- Amodio, N.; Rossi, M.; Raimondi, L.; Pitari, M.R.; Botta, C.; Tagliaferri, P.; Tassone, P. miR-29s: A family of epi-miRNAs with therapeutic implications in hematologic malignancies. Oncotarget 2015, 6, 12837–12861. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Wu, L.-C.; Pang, J.; Santhanam, R.; Schwind, S.; Wu, Y.-Z.; Hickey, C.J.; Yu, J.; Becker, H.; Maharry, K.; et al. Sp1/NFkappaB/HDAC/miR-29b regulatory network in KIT-driven myeloid leukemia. Cancer Cell 2010, 17, 333–347. [Google Scholar] [CrossRef] [PubMed]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gallo Cantafio, M.E.; Torcasio, R.; Scionti, F.; Mesuraca, M.; Ronchetti, D.; Pistoni, M.; Bellizzi, D.; Passarino, G.; Morelli, E.; Neri, A.; et al. GPER1 Activation Exerts Anti-Tumor Activity in Multiple Myeloma. Cells 2023, 12, 2226. https://doi.org/10.3390/cells12182226
Gallo Cantafio ME, Torcasio R, Scionti F, Mesuraca M, Ronchetti D, Pistoni M, Bellizzi D, Passarino G, Morelli E, Neri A, et al. GPER1 Activation Exerts Anti-Tumor Activity in Multiple Myeloma. Cells. 2023; 12(18):2226. https://doi.org/10.3390/cells12182226
Chicago/Turabian StyleGallo Cantafio, Maria Eugenia, Roberta Torcasio, Francesca Scionti, Maria Mesuraca, Domenica Ronchetti, Mariaelena Pistoni, Dina Bellizzi, Giuseppe Passarino, Eugenio Morelli, Antonino Neri, and et al. 2023. "GPER1 Activation Exerts Anti-Tumor Activity in Multiple Myeloma" Cells 12, no. 18: 2226. https://doi.org/10.3390/cells12182226
APA StyleGallo Cantafio, M. E., Torcasio, R., Scionti, F., Mesuraca, M., Ronchetti, D., Pistoni, M., Bellizzi, D., Passarino, G., Morelli, E., Neri, A., Viglietto, G., & Amodio, N. (2023). GPER1 Activation Exerts Anti-Tumor Activity in Multiple Myeloma. Cells, 12(18), 2226. https://doi.org/10.3390/cells12182226