
The Alternative TrkAIII Splice Variant, a Targetable Oncogenic Participant in Human Cutaneous Malignant Melanoma

 , ,
, ,  , , , , , , and
, , , , , , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Patients and CMM Tissues
2.2. Antibodies, Reagents and Cell Lines
2.3. Transient A375 Transfections
2.4. In Vitro Incucyte Cytotoxicity Assays
2.5. RNA Extraction and Reverse Transcriptase Polymerase Chain Reaction (RT-PCR) Analysis
2.6. DNA Sequencing
2.7. Protein Extraction and Western Blotting
2.8. Indirect IF
2.9. Statistical Analysis
3. Results
3.1. Alternative TrkA mRNA Splicing in CMM Tissues
3.2. Indirect IF Analyses
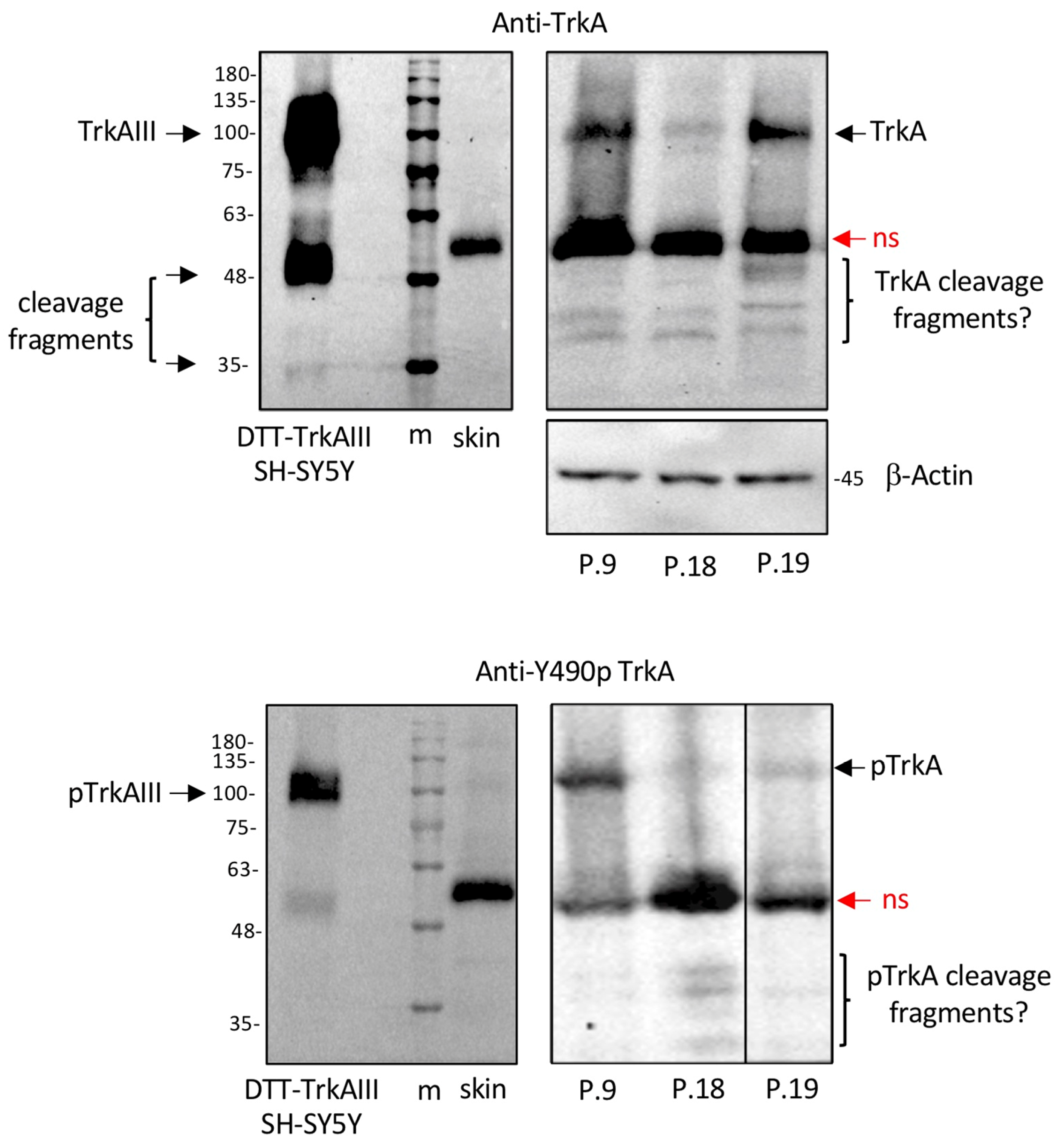
3.3. TrkA Isoform(s) Detected in CMM Extracts by Western Blotting
3.4. TrkAIII Expression in A375 Cells
3.5. Transient Fully Spliced TrkA and TrkAIII A375 Transfectants
3.6. TrkAIII Enhances A375 Resistance to DTT-Induced Death
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Davies, L.E.; Shalin, S.C.; Tackett, A. Current state of melanoma diagnosis and treatment. Cancer Biol. Ther. 2019, 20, 1366–1379. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garbe, C.; Amaral, T.; Peris, K.; Hauschild, A.; Arenberger, P.; Bastholt, L.; Bataille, V.; Del Marmol, V.; Dréno, B.; Fargnoli, M.C.; et al. European consensus-based interdisciplinary guideline for melanoma. Part 1: Diagnosis-Update 2019. Eur. J. Cancer 2020, 126, 141–158. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Welch, G.H.; Mazer, B.L.; Adamson, A.S. The rapid rise in cutaneous melanoma diagnoses. N. Eng. J. Med. 2021, 384, 72–74. [Google Scholar] [CrossRef] [PubMed]
- Dentro, S.C.; Leshchiner, I.; Haase, K.; Tarabichi, M.; Wintersinger, J.; Deshwar, A.G.; Yu, K.; Rubanova, Y.; Macintyre, G.; Demeulemeester, J.; et al. Characterizing genetic intra-tumor heterogeneity across 2658 human cancer genomes. Cell 2021, 184, 2239–2254. [Google Scholar] [CrossRef]
- Yaar, M.; Eller, M.S.; DiBenedetto, P.; Reenstra, W.R.; Zhai, S.; McQuaid, T.; Archambault, M.; Gilchrest, B.A. The trk family of receptors mediates nerve growth factor and neurotrophin-3 effects in melanocytes. J. Clin. Investig. 1994, 94, 1550–1562. [Google Scholar] [CrossRef] [PubMed]
- Truzzi, F.; Marconi, A.; Lotti, R.; Dallaglio, K.; French, L.E.; Hempstead, B.L.; Picelli, C. Neurotrophins and their receptors stimulate melanoma cell proliferation and migration. J. Invest Dermatol. 2008, 128, 2031–2040. [Google Scholar] [CrossRef] [Green Version]
- Pasini, L.; Re, A.; Tebaldi, T.; Ricci, G.; Boi, S.; Adami, V.; Barbareschi, M.; Quattrone, A. TrkA is amplified in malignant melanoma patients and induces an anti-proliferative response in cell lines. BMC Cancer 2015, 15, 777. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.J.; Kim, N.D.K.; Lee, S.H.; Kim, S.T.; Park, S.H.; Park, J.O.; Park, Y.S.; Lim, H.Y.; Kang, W.K.; Park, W.Y.; et al. NTRK gene amplification in patients with metastatic cancer. Precision Future Med. 2017, 1, 129–137. [Google Scholar] [CrossRef] [Green Version]
- Florennes, V.A.; Maelandsmo, G.; Holm, R.; Reich, R.; Lazarovici, P.; Davidson, B. Expression of activated TrkA protein in melanocytic tumors. Anatom. Pathol. 2004, 122, 412–420. [Google Scholar] [CrossRef]
- Moreira Antunes, L.C.; Cartell, A.; Brunetto de Farias, C.; Bakos, R.M.; Roesier, R.; Schwarzmann, G. Tropomyosin-related kinase receptor and neurotrophin expression in cutaneous melanoma is associated with poor prognosis and decreased survival. Oncology 2019, 97, 26–37. [Google Scholar] [CrossRef]
- Shalin, S. A review of kinase fusions in melanocytic tumors. Lab. Investig. 2017, 97, 158–165. [Google Scholar] [CrossRef] [Green Version]
- Forschner, A.; Forchhammer, S.; Bonzheim, I. NTRK gene fusions in melanoma: Detection, prevalence and potential therapeutic implications. J. Dtsch. Dermatol. Ges. 2020, 18, 1387–1392. [Google Scholar] [CrossRef] [PubMed]
- Marsland, M.; Dowdell, A.; Jiang, C.C.; Wilmott, J.S.; Scolyer, R.A.; Zhang, X.D.; Hondermarck, H.; Faulkner, S. Expression of NGF/ProNGF and their receptors TrkA, P75NTR and sortilin in melanoma. Int. J. Mol. Sci. 2022, 23, 4260. [Google Scholar] [CrossRef]
- Tacconelli, A.; Farina, A.R.; Cappabianca, L.; DeSantis, G.; Tessitore, A.; Vetuschi, A.; Sferra, R.; Rucci, N.; Argenti, B.; Screpanti, I.; et al. TrkA alternative splicing: A regulated tumor-promoting switch in human neuroblastoma. Cancer Cell 2004, 6, 347–360. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Farina, A.R.; Di Ianni, N.; Cappabianca, L.; Ruggeri, P.; Gneo, L.; Pellegrini, C.; Fargnoli, M.-C.; Mackay, A.R. The oncogenic neurotrophin receptor tropomyosin-related kinase variant, TrkAIII. J. Clin. Exp. Cancer Res. 2018, 37, 119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schramm, A.; Schowe, B.; Fielitz, K.; Heilmann, M.; Martin, M.; Marschall, T.; Koster, J.; Vandesompele, J.; Vermeulen, J.; de Preter, K.; et al. Exon-level expression analysis identify MYCN and NTRK1 as major determinants of alternative exon usage and robustly predict neuroblastoma outcome. Br. J. Cancer 2012, 107, 1409–1417. [Google Scholar] [CrossRef]
- Ruggeri, P.; Farina, A.R.; Cappabianca, L.; Di Ianni, N.; Ragone, M.; Merolle, S.; Gulino, A.; Mackay, A.R. Neurotrophin and neurotrophin receptor involvement in human neuroblastoma. In Neuroblastoma; Shimada, H., Ed.; Intech Open: London, UK, 2013. [Google Scholar]
- Cappabianca, L.; Guadagni, S.; Maccarone, R.; Sebastiano, M.; Chiominto, A.; Farina, A.R.; Mackay, A.R. A pilot study of alternative TrkAIII splicing in Merkel cell carcinoma: A potential oncogenic mechanism and novel therapeutic target. J. Exp. Clin. Cancer Res. 2019, 38, 424. [Google Scholar] [CrossRef]
- Guadagni, S.; Farina, A.R.; Cappabianca, L.; Sebastiano, M.; Maccarone, R.; Zelli, V.; Clementi, M.; Bruera, G.; Ricevuto, E.; Fiorentini, G.; et al. Multidisciplinary treatment, including locoregional chemotherapy, for Merkel-Polyomavirus positive Merkel cell carcinomas: Perspectives for patients exhibiting oncogenic alternative D exon 6-7 TrkAIII splicing of neurotrophin receptor tropomyosin-related kinase A. Int. J. Mol. Sci. 2020, 21, 8222. [Google Scholar]
- Arevalo, J.C.; Conde, B.; Hempstead, B.L.; Chao, M.V.; Martin-Zanca, D.; Perez, P. TrkA immunoglobulin-like ligand binding domains inhibit spontaneous activation of the receptor. Mol. Cell. Biol. 2000, 20, 5908–5916. [Google Scholar] [CrossRef] [Green Version]
- Watson, F.L.; Porcionatto, M.A.; Bhattacharyya, A.; Stiles, C.D.; Segal, R.A. TrkA glycosylation regulates receptor localization and activity. J. Neurobiol. 1999, 39, 323–336. [Google Scholar] [CrossRef]
- Ruggeri, P.; Farina, A.R.; Di Ianni, N.; Cappabianca, L.; Ragone, M.; Ianni, G.; Gulino, A.; Mackay, A.R. The TrkAIII oncoprotein inhibits mitochondrial free radical ROS-induced death of SH-SY5Y neuroblastoma cells by augmenting SOD2 expression and activity at the mitochondria, within the context of a tumor stem-cell like phenotype. PLoS ONE 2014, 9, e94568. [Google Scholar] [CrossRef] [PubMed]
- Farina, A.R.; Cappabianca, L.; Gneo, L.; Ruggeri, P.; Mackay, A.R. TrkAIII signals endoplasmic reticulum stress to the mitochondria in neuroblastoma cells, resulting in glycolytic metabolic adaptation. Oncotarget 2017, 9, 8368–8390. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luberg, K.; Park, R.; Aleksejeva, E.; Timmusk, T. Novel transcripts reveal a complex structure of the human TRKA gene and imply the presence of multiple protein isoforms. BMC Neurosci. 2015, 16, 78. [Google Scholar] [CrossRef] [Green Version]
- Cappabianca, L.; Sebastiano, M.; Ruggieri, M.; Sbaffone, M.; Zelli, V.; Farina, A.R.; Mackay, A.R. Doxorubicin-induced TrkAIII activation: A selection mechanism for resistant dormant neuroblastoma cells. Int. J. Mol. Sci. 2022, 23, 10895. [Google Scholar] [CrossRef] [PubMed]
- Farina, A.R.; Cappabianca, L.; Sebastiano, M.; Zelli, V.; Guadagni, S.; Mackay, A.R. Hypoxia-induced alternative splicing: The 11th hallmark of cancer. J. Clin. Exp. Cancer Res. 2020, 39, 110. [Google Scholar] [CrossRef]
- Xiao, W.; Loscalzo, J. Metabolic responses to reductive stress. Antiox. Redox Signal. 2020, 32, 1330–1347. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.; Tew, K.D. Reductive stress in cancer. Adv. Cancer Res. 2021, 152, 383–413. [Google Scholar]
- Oakes, S.A. Endoplasmic reticulum stress signaling in cancer cells. Am. J. Pathol. 2020, 190, 934–946. [Google Scholar] [CrossRef] [Green Version]
- Sykes, E.K.; Mactier, S.; Christopherson, R.I. Melanoma and the unfolded protein response. Cancers 2016, 8, 30. [Google Scholar] [CrossRef] [Green Version]
- Eigner, K.; Filik, Y.; Mark, F.; Schutz, B.; Klambauer, G.; Moriggl, R.; Hengstschlager, M.; Stangl, H.; Mikula, M.; Rohrl, C. The unfolded protein response impacts melanoma progression by enhancing FGF expression and can be antagonized by a chemical chaperone. Sci. Rep. 2017, 7, 17498. [Google Scholar] [CrossRef] [Green Version]
- Tavernier, Q.; Legras, A.; Didelot, A.; Normand, C.; Gibault, L.; Badoual, C.; Le Pimpec-Barthes, F.; Puig, P.L.; Blons, H.; Pallet, N. High expression of spliced X-box binding protein 1 in lung tumors is associated with cancer aggressiveness and epithelial-to-mesenchymal transition. Sci. Rep. 2020, 10, 10188. [Google Scholar] [CrossRef] [PubMed]
- Carew, N.T.; Nelson, A.M.; Liang, Z.; Smith, S.A.; Milcarek, C. Linking endoplasmic reticular stress and alternative splicing. Int. J. Mol. Sci. 2018, 19, 3919. [Google Scholar] [CrossRef] [PubMed]
- Krebs, J.; Groenendyk, J.; Michalak, M. Ca2+-signaling, alternative splicing and endoplasmic reticulum stress-response. Neurochem. Res. 2011, 36, 1198–1211. [Google Scholar] [CrossRef] [PubMed]
- Pinto, P.; Giorgi, C.; Siviero, R.; Zecchini, E.; Rizzuto, R. Calcium and apoptosis: ER-mitochonria Ca2+ transfer in the control of apoptosis. Oncogene 2008, 27, 6407–6418. [Google Scholar] [CrossRef] [Green Version]
- Xie, J. Control of alternative pre-mRNA splicing by Ca++ signals. Biochim. Biophys. Acta. 2008, 1779, 438–452. [Google Scholar] [CrossRef] [Green Version]
- Shabbir, M.; Stuart, R. Lestaurtinib, a multitargeted tyrosine kinase inhibitor: From bench to bedside. Expert Opin. Investig. Drugs 2010, 19, 427–436. [Google Scholar] [CrossRef]
- Iyer, R.; Wehrmann, L.; Golden, R.L.; Naraparaju, K.; Croucher, J.L.; MacFarland, S.P.; Cuan, P.; Kolla, V.; Wei, G.; Cam, L.; et al. Entrectinib is a potent inhibitor of Trk-driven neuroblastomas in a xenograft mouse model. Cancer Lett. 2016, 372, 179–186. [Google Scholar] [CrossRef] [Green Version]
- Litchfield, K.; Reading, J.L.; Lim, E.L.; Xu, H.; Liu, P.; Al-Bakir, M.; Wong, Y.N.S.; Rowan, A.; Funt, S.A.; Merghoub, T.; et al. Escape from nonsense-mediated decay associates with anti-tumor immunogenicity. Nature Commun. 2020, 11, 3800. [Google Scholar] [CrossRef]
- Wang, D.; Zavadil, J.; Martin, L.; Parisi, F.; Friedman, E.; Levy, D.; Harding, H.; Ron, D.; Gardner, L.B. Inhibition of nonsense-mediated RNA decay by the tumor microenvironment promotes tumorigenesis. Mol. Cell. Biol. 2011, 17, 3670–3680. [Google Scholar] [CrossRef] [Green Version]
- Shuang, Q.; Jiao, Z.; Lu, G.; Yao, B.; Wang, T.; Rong, W.; Xu, J.; Fan, T.; Sun, X.; Yang, R.; et al. PD-L1 lncRNA splice isoform promotes lung adenocarcinoma progression via enhancing c-Myc activity. Genome Biol. 2021, 22, 104. [Google Scholar]
- Kim, W.K.; Yun, S.; Kwon, Y.; You, K.T.; Shin, N.; Kim, J.; Kim, H. mRNAs containing NMD-competent premature termination codons are stabilized and translated under UPF1 depletion. Sci. Reports 2017, 7, 15833. [Google Scholar] [CrossRef] [Green Version]
- Oren, Y.S.; McClure, M.L.; Rowe, S.M.; Sorscher, E.J.; Bester, A.C.; Manor, M.; Kerem, E.; Rivlin, J.; Zahdeh, F.; Mann, M.; et al. The unfolded protein response affects readthrough of premature termination stop codons. EMBO Mol. Med. 2014, 6, 685–701. [Google Scholar] [CrossRef] [Green Version]
- Yoshida, H. Unconventional splicing of XBP-1 mRNA in the unfolded protein response. Antioxid. Redox Signal. 2007, 9, 2323–2333. [Google Scholar] [CrossRef]
- Li, H.; Korennykh, A.V.; Behrman, S.L.; Walter, P. Mammalian endoplasmic reticulum stress sensor IRE1 signals by dynamic clustering. Proc. Natl. Acad. Sci. USA 2010, 107, 16113–16118. [Google Scholar] [CrossRef] [Green Version]
- DuRose, J.B.; Tam, A.B.; Niwa, M. Intrinsic capacities of molecular sensors of the unfolded protein response to sense alternate forms of endoplasmic reticulum stress. Mol Biol Cell 2006, 17, 3095–3107. [Google Scholar] [CrossRef] [Green Version]
- Farina, A.R.; Cappabianca, L.; Ruggeri, P.; Gneo, L.; Maccarone, R.; Mackay, A.R. Retrograde TrkAIII transport from the ERGIC to ER: A re-localisation mechanism for oncogenic activity. Oncotarget 2015, 6, 35636–35651. [Google Scholar] [CrossRef] [Green Version]
- Seiler, M.; Peng, S.; Agrawal, A.A.; Palacino, J.; Teng, T.; Zhu, P.; Smith, P.G. Somatic mutational landscape of splicing factor genes and their functional consequences across 33 cancer types. Cell Rep. 2018, 23, 282–296. [Google Scholar] [CrossRef] [Green Version]
- Zhao, F.; Li, P.; Chen, S.R.; Louis, C.F.; Fruen, B.R. Dantrolene inhibition of ryanodine receptor Ca2+ release channels. Molecular mechanism and isoform selectivity. J. Biol. Chem. 2001, 276, 13810–13816. [Google Scholar] [CrossRef] [Green Version]
- Lebeau, P.F.; Platko, K.; Hyun Byun, J.; Austin, R.C. Calcium as a reliable marker for the quantitative assessment of endoplasmic reticulum stress in live cells. J. Biol. Chem. 2021, 296, 100779. [Google Scholar] [CrossRef]
- Deniaud, A.; el dien Sharaf, O.; Maillier, E.; Poncet, D.; Kroemer, G.; Lemaire, C.; Brenner, C. Endoplasmic reticulum stress induces calcium-dependent permeability transition, mitochondrial outer membrane permeabilization and apoptosis. Oncogene 2008, 27, 285–299. [Google Scholar] [CrossRef] [Green Version]
- Farina, A.R.; Tacconelli, A.; Cappabianca, L.; Cea, G.; Chioda, A.; Romanelli, A.; Pensato, S.; Pedone, C.; Gulino, A.; Mackay, A.R. The neuroblastoma tumour suppressor TrkAI and its oncogenic alternative TrkAIII splice variant exhibit geldanamycin-sensitive interactions with Hsp90 in human neuroblastoma cells. Oncogene 2009, 28, 4075–4095. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sweeney, J.G.; Liang, J.; Antonopoulos, A.; Giovannone, N.; Kang, S.; Mondala, T.S.; Head, S.R.; King, S.L.; Tani, Y.; Brackett, D.; et al. Loss of GCNT2/I-branched glycans enhances melanoma growth and survival. Nat. Commun. 2018, 9, 3368. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Müller, J.; Krijgsman, O.; Tsoi, J.; Robert, L.; Hugo, W.; Song, C.; Kong, X.; Possik, P.A.; Cornelissen-Steijger, P.D.M.; Geukes Foppen, M.H.; et al. Low MITF/AXL ratio predicts early resistance to multiple targeted drugs in melanoma. Nat. Commun. 2014, 5, 5712. [Google Scholar] [CrossRef] [PubMed]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| PRIMARY CMMs. | |||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Patient | P.1 | P.2 | P.3 | P.4 | P.5 | P.6 | P.7 | P.8 | P.28a FFPE | P.29a FFPE | P.30a FFPE | NS | |
| TrkA relative to 18S rRNA | High | Mod/High | High | High | High | Mod/ Low | Low | High | ND | ND | ND | High | |
| TrkA exon 1-8 Alt. splicing | Yes | Yes | No | Yes | Yes | No | Yes | No | ND | ND | ND | No | |
| TrkAIII exon 1–8 RT-PCR | ++ | + | - | + | - | - | - | - | ND | ND | ND | - | |
| TrkAIII exon 5–8 RT-PCR | +++ | + | ± | - | - | - | - | ND | ND | ND | ND | - | |
| TrkAIII exon 5–8 RT-PCR FFPE | + | + | ± | ± | - | - | - | ND | + | + | + | - | |
| TrkA IF | ++± | +++ | + | +++ | ++ | ++ | - | ND | +++ | +++ | +++ | +++ | |
| pTrkA IF | ++ | +++ | - | +± | - | ± | - | ND | +++ | ++ | +++ | - | |
| Xbp1 splicing | Yes | Yes | Yes | Yes | ND | ND | ND | ND | Yes | Yes | Yes | No | |
| METASTATIC CMMs | |||||||||||||
| Patient | P.9 | P.10 | P.11 | P.12 | P.13 | P.14 | P.15 | P.16 | P.17 | P.18 | P.19 | P.20 | |
| TrkA relative to 18S rRNA | Mod/ High | High | Mod/ Low | Mod/ Low | Mod/ High | Mod/ Low | Low | Mod/ High | Mod/ High | High | High | High | |
| TrkA exon 1-8 Alt splicing | Yes | Yes | Yes | Yes | Yes | Yes | Yes | Yes | Yes | Yes | Yes | Yes | |
| TrkAIII exon 1–8 RT-PCR | +++ | +++ | +++ | ++ | +++ | + | ± | +++ | +++ | + | + | ± | |
| TrkAIII exon 5–8 RT-PCR | ± | + | ++ | + | ++± | + | + | ND | ND | + | + | + | |
| TrkAIII exon 5–8 RT-PCR FFPE | + | + | + | + | + | + | + | ND | ND | + | + | - | |
| TrkA IF | ++± | +++ | ++± | ++± | ++ | ++± | ++ | ND | ND | ++± | ++± | ++ | |
| pTrkA IF | ++ | +++ | ++± | +± | ++ | ± | ++ | ND | ND | ++± | ++± | +± | |
| Xbp1 splicing | Yes | Yes | Yes | Yes | Yes | Yes | Yes | ND | ND | Yes | Yes | Yes | |
| Patient | P:21 | P.22 | P.23 | P.24 | P.25 | P.26 | P.27 | P.28b FFPE | P.29b FFPE | P.30b FFPE | |||
| TrkA relative to 18S rRNA | High | High | Low | Low | Mod/ High | No | No | ND | ND | ND | |||
| TrkA exon 1-8 Alt splicing | Yes | No | No | No | No | No | No | ND | ND | ND | |||
| TrkAIII exon 1–8 RT-PCR | - | - | - | - | - | - | - | ND | ND | ND | |||
| TrkAIII exon 5–8 RT-PCR | - | ± | - | - | ND | ± | - | ND | ND | ND | |||
| TrkAIII exon 5–8 RT-PCR FFPE | ± | - | + | - | ND | - | - | + | + | ± | |||
| TrkA IF | ++± | + | +++ | - | ND | - | - | +++ | +++ | +++ | |||
| pTrkA IF | ++± | ± | +++ | - | ND | - | - | +++ | ++ | +++ | |||
| Xbp1 splicing | Yes | Yes | Yes | Yes | ND | Yes | Yes | Yes | Yes | Yes | |||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cappabianca, L.; Zelli, V.; Pellegrini, C.; Sebastiano, M.; Maccarone, R.; Clementi, M.; Chiominto, A.; Ruggeri, P.; Cardelli, L.; Ruggieri, M.; et al. The Alternative TrkAIII Splice Variant, a Targetable Oncogenic Participant in Human Cutaneous Malignant Melanoma. Cells 2023, 12, 237. https://doi.org/10.3390/cells12020237
Cappabianca L, Zelli V, Pellegrini C, Sebastiano M, Maccarone R, Clementi M, Chiominto A, Ruggeri P, Cardelli L, Ruggieri M, et al. The Alternative TrkAIII Splice Variant, a Targetable Oncogenic Participant in Human Cutaneous Malignant Melanoma. Cells. 2023; 12(2):237. https://doi.org/10.3390/cells12020237
Chicago/Turabian StyleCappabianca, Lucia, Veronica Zelli, Cristina Pellegrini, Michela Sebastiano, Rita Maccarone, Marco Clementi, Alessandro Chiominto, Pierdomenico Ruggeri, Ludovica Cardelli, Marianna Ruggieri, and et al. 2023. "The Alternative TrkAIII Splice Variant, a Targetable Oncogenic Participant in Human Cutaneous Malignant Melanoma" Cells 12, no. 2: 237. https://doi.org/10.3390/cells12020237
APA StyleCappabianca, L., Zelli, V., Pellegrini, C., Sebastiano, M., Maccarone, R., Clementi, M., Chiominto, A., Ruggeri, P., Cardelli, L., Ruggieri, M., Sbaffone, M., Fargnoli, M. -C., Guadagni, S., Farina, A. R., & Mackay, A. R. (2023). The Alternative TrkAIII Splice Variant, a Targetable Oncogenic Participant in Human Cutaneous Malignant Melanoma. Cells, 12(2), 237. https://doi.org/10.3390/cells12020237