
Novel Insights into the Role of Kras in Myeloid Differentiation: Engaging with Wnt/β-Catenin Signaling

Abstract
:1. Introduction
2. Distinguishing the Functions of Kras from Those of Other Members of the Ras Family
3. Wnt Signaling Pathways
3.1. Wnt/β-Catenin Signaling Pathway
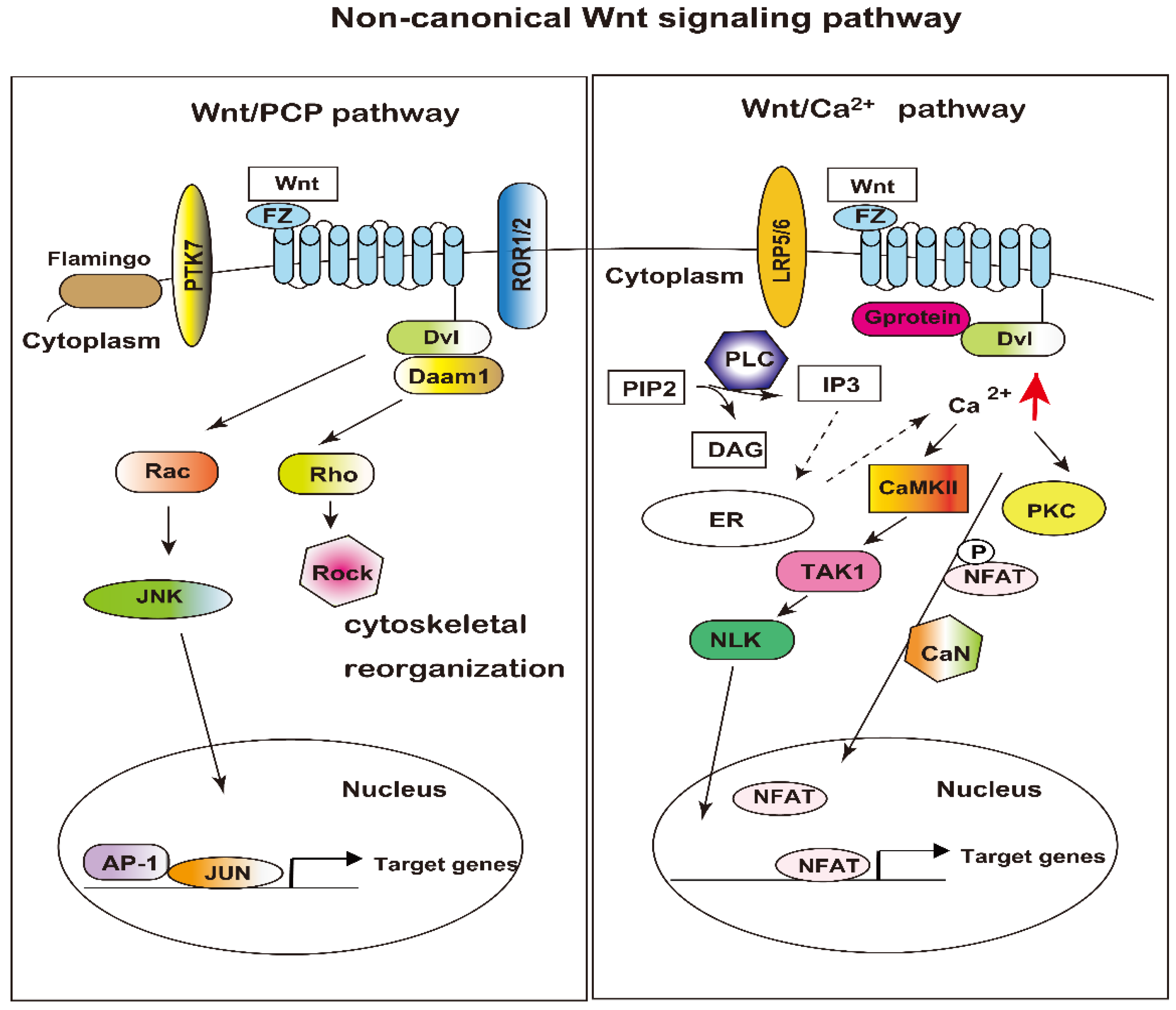
3.2. Non-Canonical Wnt Signaling Pathways
3.2.1. Planar Cell Polarity (Wnt/PCP) Pathway
3.2.2. Wnt Calcium (Wnt/Ca2+) Pathway
4. Wnt/β-Catenin Signaling in Normal Hematopoiesis and AML
5. Non-Canonical Wnt Signaling Pathways in AML
6. The Role of GSK3 in Hematopoietic Stem Cells
7. CCAAT Enhancer-Binding Proteins (C/EBPs)
7.1. C/EBPα
7.2. C/EBPβ
7.3. C/EBPε
8. G-CSF and G-CSF Receptor
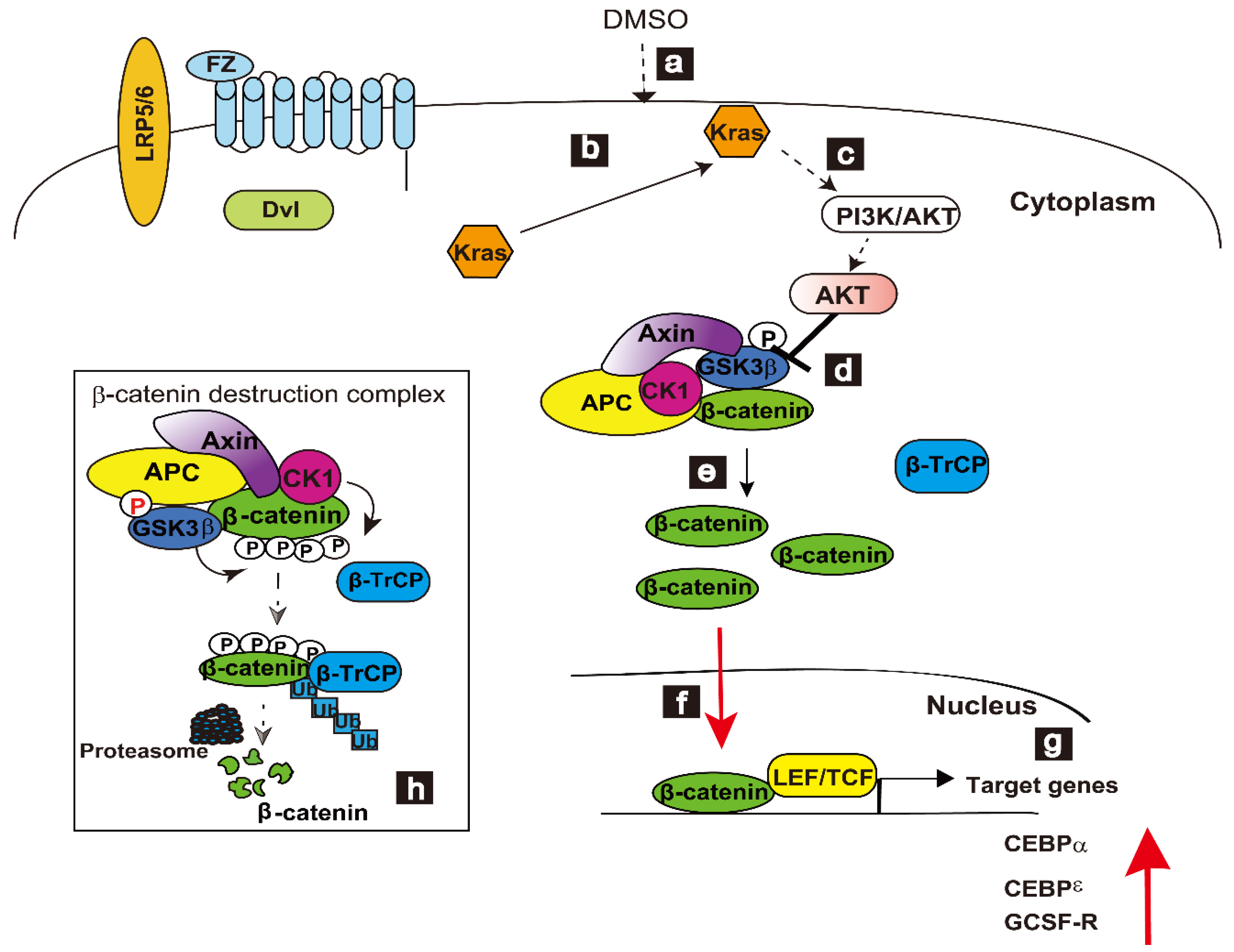
9. WT-Kras Engages with Wnt/β-Catenin Signaling Pathway in the DMSO-Induced Differentiation of HL-60 Cells
10. Level of β-Catenin Regulates DMSO-Induced Differentiation in HL-60 Cells
11. Applications to the Treatment of Patients with AML

{kind=link}
{kind=link}
{kind=link}
| Affected Target | Agent | Mechanism | Diseases | Clinical | References |
|---|---|---|---|---|---|
| CBP/β-catenin | PRI-724 | Inhibits interactions between CBP and β-catenin and prevents transcription of Wnt target genes | AML, CML | NCT01606579 (Phase I/II) completed, no results posted | [181] [182] |
| β-catenin | CWP232291 | Inhibits β-catenin transcriptional activity, leading to degradation of β-catenin and induction of apoptosis in leukemia cells | AML, CML | NCT01398462 (Phase I) completed, no results posted | [173] |
| TBL1/β-catenin | BC2059 (Tegavivint) | Inhibits β-catenin/transducin β-like 1 (TBL1) complex, degrades β-catenin and abrogates Wnt target gene expression | Refractory leukemia (AML) | NCT04874480 (Phase I) recruiting participants | [183] [184] |
| β-catenin S-phase-specific anti-metabolite drug | CWP232291+ AraC/cytarabine | Inhibit β-catenin transcriptional activity, induce apoptosis in leukemia cells | AML | NCT03055286 (Phase I/II) no results posted | |
| GSK3 | LY2090314 | Inhibits GSK3β, results consistent with Phase I trial results. Well tolerated, but no patients achieved CR or PR | AML | NCT01214603 (Phase II) | [100] |
| PAR receptor GSK3 | ATRA+LiCl | Inhibit phosphorylation of PU.1, enhance leukemia cells differentiation | AML, APL | NCT01820624 (Phase I) | [185] |
| FLT3 | Gilteritinib (ASP2215) | Inhibits FLT3 and AXL receptor, and inhibits cell growth | AML, relapsed or refractory FLT3-mutated AML | NCT03070093 (Phase I/II) completed (May 2021), approved for marketing NCT02421939, NCT03182244 (Phase III) | [171] |
| Cyclooxygenase-2 | Celecoxib +doxorubicin | Inhibit cell growth and induce apoptosis in leukemia cell lines | AML, primary AML blasts | NCT03878524 (Phase Ib) | [177] |
12. Perspective
13. Conclusions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
References
- Dalton, W.T., Jr.; Ahearn, M.J.; McCredie, K.B.; Freireich, E.J.; Stass, S.A.; Trujillo, J.M. HL-60 cell line was derived from a patient with FAB-M2 and not FAB-M3. Blood 1988, 71, 242–247. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Breitman, T.R.; Selonick, S.E.; Collins, S.E. Induction of differentiation of the human promyelocytic leukemia cell line (HL-60) by retinoic acid. Proc. Natl. Acad. Sci. USA 1980, 77, 2936–2940. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Collins, S.J. The HL-60 promyelocytic leukemia cell line: Proliferation, differentiation, and cellular oncogene expression. Blood 1987, 70, 1233–1244. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuliczkowski, K.; Darley, R.L.; Jacobs, A.; Padua, R.A.; Hoy, T.G. Upregulation of p21 RAS levels in HL-60 cells during differentiation induction with DMSO, all-trans-retinoic acid and TPA. Leuk. Res. 1995, 19, 291–296. [Google Scholar] [CrossRef] [PubMed]
- Neubauer, A.; Dodge, R.K.; George, S.L.; Davey, F.R.; Silver, R.T.; Schiffer, C.A.; Mayer, R.J.; Ball, E.D.; Wurster-Hill, D.; Bloomfield, C.D.; et al. Prognostic importance of mutations in the ras proto-oncogenes in de novo acute myeloid leukemia. Blood 1994, 83, 1603–1611. [Google Scholar] [CrossRef] [Green Version]
- Vetter, I.R.; Wittinghofer, A. The guanine nucleotide-binding switch in three dimensions. Science 2001, 294, 1299–1304. [Google Scholar] [CrossRef] [Green Version]
- Downward, J. Targeting RAS signalling pathways in cancer therapy. Nat. Rev. Cancer 2003, 3, 11–22. [Google Scholar] [CrossRef]
- Sunaga, N.; Imai, H.; Shimizu, K.; Shames, D.S.; Kakegawa, S.; Girard, L.; Sato, M.; Kaira, K.; Ishizuka, T.; Gazdar, A.F.; et al. Oncogenic KRAS-induced interleukin-8 overexpression promotes cell growth and migration and contributes to aggressive phenotypes of non-small cell lung cancer. Int. J. Cancer 2012, 130, 1733–1744. [Google Scholar] [CrossRef] [Green Version]
- Padavano, J.; Henkhaus, R.S.; Chen, H.; Skovan, B.A.; Cui, H.; Ignatenko, N.A. Mutant K-RAS Promotes Invasion and Metastasis in Pancreatic Cancer Through GTPase Signaling Pathways. Cancer Growth Metastasis 2015, 8, 95–113. [Google Scholar] [CrossRef] [Green Version]
- Esteban, L.M.; Vicario-Abejon, C.; Fernandez-Salguero, P.; Fernandez-Medarde, A.; Swaminathan, N.; Yienger, K.; Lopez, E.; Malumbres, M.; McKay, R.; Ward, J.M.; et al. Targeted genomic disruption of H-ras and N-ras, individually or in combination, reveals the dispensability of both loci for mouse growth and development. Mol. Cell Biol. 2001, 21, 1444–1452. [Google Scholar] [CrossRef]
- Damnernsawad, A.; Kong, G.; Wen, Z.; Liu, Y.; Rajagopalan, A.; You, X.; Wang, J.; Zhou, Y.; Ranheim, E.A.; Luo, H.R.; et al. Kras is required for adult hematopoiesis. Stem Cells 2016, 34, 1859–1871. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.; Zheng, Y.; You, X.; Yu, M.; Fu, G.; Su, X.; Zhou, F.; Zhu, W.; Wu, Z.; Zhang, J.; et al. Kras Is Critical for B Cell Lymphopoiesis. J. Immunol. 2016, 196, 1678–1685. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Z.; Wang, Y.; Vikis, H.G.; Johnson, L.; Liu, G.; Li, J.; Anderson, M.W.; Sills, R.C.; Hong, H.L.; Devereux, T.R.; et al. Wildtype Kras2 can inhibit lung carcinogenesis in mice. Nat. Genet. 2001, 29, 25–33. [Google Scholar] [CrossRef]
- Staffas, A.; Karlsson, C.; Persson, M.; Palmqvist, L.; Bergo, M.O. Wild-type KRAS inhibits oncogenic KRAS-induced T-ALL in mice. Leukemia 2015, 29, 1032–1040. [Google Scholar] [CrossRef]
- Kong, G.; Chang, Y.I.; Damnernsawad, A.; You, X.; Du, J.; Ranheim, E.A.; Lee, W.; Ryu, M.J.; Zhou, Y.; Xing, Y.; et al. Loss of wild-type Kras promotes activation of all Ras isoforms in oncogenic Kras-induced leukemogenesis. Leukemia 2016, 30, 1542–1551. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Wynshaw-Boris, A. The canonical Wnt pathway in early mammalian embryogenesis and stem cell maintenance/differentiation. Curr. Opin. Genet. Dev. 2004, 14, 533–539. [Google Scholar] [CrossRef]
- Cadigan, K.M.; Liu, Y.I. Wnt signaling: Complexity at the surface. J. Cell Sci. 2006, 119, 395–402. [Google Scholar] [CrossRef] [Green Version]
- Clevers, H. Wnt/beta-catenin signaling in development and disease. Cell 2006, 127, 469–480. [Google Scholar] [CrossRef] [Green Version]
- Chien, A.J.; Conrad, W.H.; Moon, R.T. A Wnt survival guide: From flies to human disease. J. Investig. Dermatol. 2009, 129, 1614–1627. [Google Scholar] [CrossRef] [Green Version]
- Staal, F.J.; Sen, J.M. The canonical Wnt signaling pathway plays an important role in lymphopoiesis and hematopoiesis. Eur. J. Immunol. 2008, 38, 1788–1794. [Google Scholar] [CrossRef]
- Malhotra, S.; Kincade, P.W. Wnt-related molecules and signaling pathway equilibrium in hematopoiesis. Cell Stem Cell 2009, 4, 27–36. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Staal, F.J.; Luis, T.C. Wnt signaling in hematopoiesis: Crucial factors for self-renewal, proliferation, and cell fate decisions. J. Cell Biochem. 2010, 109, 844–849. [Google Scholar] [CrossRef] [PubMed]
- Reya, T.; O’Riordan, M.; Okamura, R.; Devaney, E.; Willert, K.; Nusse, R.; Grosschedl, R. Wnt signaling regulates B lymphocyte proliferation through a LEF-1 dependent mechanism. Immunity 2000, 13, 15–24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fodde, R.; Brabletz, T. Wnt/beta-catenin signaling in cancer stemness and malignant behavior. Curr. Opin. Cell Biol. 2007, 19, 150–158. [Google Scholar] [CrossRef] [PubMed]
- Staal, F.J.; Chhatta, A.; Mikkers, H. Caught in a Wnt storm: Complexities of Wnt signaling in hematopoiesis. Exp. Hematol. 2016, 44, 451–457. [Google Scholar] [CrossRef]
- Gruszka, A.M.; Valli, D.; Alcalay, M. Wnt Signalling in Acute Myeloid Leukaemia. Cells 2019, 8, 1403. [Google Scholar] [CrossRef] [Green Version]
- Frenquelli, M.; Tonon, G. WNT Signaling in Hematological Malignancies. Front. Oncol. 2020, 10, 615190. [Google Scholar] [CrossRef]
- Scholl, C.; Gilliland, D.G.; Fröhling, S. Deregulation of signaling pathways in acute myeloid leukemia. Semin. Oncol. 2008, 35, 336–345. [Google Scholar] [CrossRef]
- Gandillet, A.; Park, S.; Lassailly, F.; Griessinger, E.; Vargaftig, J.; Filby, A.; Lister, T.A.; Bonnet, D. Heterogeneous sensitivity of human acute myeloid leukemia to beta-catenin down-modulation. Leukemia 2011, 25, 770–780. [Google Scholar] [CrossRef] [Green Version]
- Yokoyama, N.; Kim, Y.J.; Hirabayashi, Y.; Tabe, Y.; Takamori, K.; Ogawa, H.; Iwabuchi, K. Kras promotes myeloid differentiation through Wnt/beta-catenin signaling. FASEB Bioadv. 2019, 1, 435–449. [Google Scholar] [CrossRef]
- Ostrem, J.M.; Peters, U.; Sos, M.L.; Wells, J.A.; Shokat, K.M. K-Ras(G12C) inhibitors allosterically control GTP affinity and effector interactions. Nature 2013, 503, 548–551. [Google Scholar] [CrossRef] [Green Version]
- Hall, A. G proteins and small GTPases: Distant relatives keep in touch. Science 1998, 280, 2074–2075. [Google Scholar] [CrossRef]
- Ayllón, V.; Rebollo, A. Ras-induced cellular events (review). Mol. Membr. Biol. 2000, 17, 65–73. [Google Scholar]
- Simanshu, D.K.; Nissley, D.V.; McCormick, F. RAS Proteins and Their Regulators in Human Disease. Cell 2017, 170, 17–33. [Google Scholar] [CrossRef] [Green Version]
- Campbell, P.M.; Der, C.J. Oncogenic Ras and its role in tumor cell invasion and metastasis. Semin. Cancer Biol. 2004, 14, 105–114. [Google Scholar] [CrossRef]
- Fotiadou, P.P.; Takahashi, C.; Rajabi, H.N.; Ewen, M.E. Wild-type NRas and KRas perform distinct functions during transformation. Mol. Cell Biol. 2007, 27, 6742–6755. [Google Scholar] [CrossRef] [Green Version]
- Johnson, L.; Greenbaum, D.; Cichowski, K.; Mercer, K.; Murphy, E.; Schmitt, E.; Bronson, R.T.; Umanoff, H.; Edelmann, W.; Kucherlapati, R.; et al. K-ras is an essential gene in the mouse with partial functional overlap with N-ras. Genes Dev. 1997, 11, 2468–2481. [Google Scholar] [CrossRef] [Green Version]
- Castellano, E.; Downward, J. RAS Interaction with PI3K: More Than Just Another Effector Pathway. Genes Cancer 2011, 2, 261–274. [Google Scholar] [CrossRef] [Green Version]
- Yen, A.; Roberson, M.S.; Varvayanis, S.; Lee, A.T. Retinoic acid induced mitogen-activated protein (MAP)/extracellular signal-regulated kinase (ERK) kinase-dependent MAP kinase activation needed to elicit HL-60 cell differentiation and growth arrest. Cancer Res. 1998, 58, 3163–3172. [Google Scholar]
- Willumsen, B.M.; Christensen, A.; Hubbert, N.L.; Papageorge, A.G.; Lowy, D.R. The p21 ras C-terminus is required for transformation and membrane association. Nature 1984, 310, 583–586. [Google Scholar] [CrossRef]
- Wright, L.P.; Philips, M.R. Thematic review series: Lipid posttranslational modifications. CAAX modification and membrane targeting of Ras. J. Lipid Res. 2006, 47, 883–891. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsai, F.D.; Lopes, M.S.; Zhou, M.; Court, H.; Ponce, O.; Fiordalisi, J.J.; Gierut, J.J.; Cox, A.D.; Haigis, K.M.; Philips, M.R. K-Ras4A splice variant is widely expressed in cancer and uses a hybrid membrane-targeting motif. Proc. Natl. Acad. Sci. USA 2015, 112, 779–784. [Google Scholar] [CrossRef] [PubMed]
- Jackson, J.H.; Cochrane, C.G.; Bourne, J.R.; Solski, P.A.; Buss, J.E.; Der, C.J. Farnesol modification of Kirsten-ras exon 4B protein is essential for transformation. Proc. Natl. Acad. Sci. USA 1990, 87, 3042–3046. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jackson, J.H.; Li, J.W.; Buss, J.E.; Der, C.J.; Cochrane, C.G. Polylysine domain of K-ras 4B protein is crucial for malignant transformation. Proc. Natl. Acad. Sci. USA 1994, 91, 12730–12734. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, H.; Liu, P.; Zhang, R.; Wu, M.; Li, D.; Zhao, X.; Zhang, C.; Jiao, B.; Chen, B.; Chen, Z.; et al. Roles of palmitoylation and the KIKK membrane-targeting motif in leukemogenesis by oncogenic KRAS4A. J. Hematol. Oncol. 2015, 8, 132. [Google Scholar] [CrossRef] [Green Version]
- Aran, V. K-RAS4A: Lead or Supporting Role in Cancer Biology? Front. Mol. Biosci. 2021, 8, 729830. [Google Scholar] [CrossRef]
- Okudela, K.; Hayashi, H.; Ito, T.; Yazawa, T.; Suzuki, T.; Nakane, Y.; Sato, H.; Ishi, H.; KeQin, X.; Masuda, A.; et al. K-ras gene mutation enhances motility of immortalized airway cells and lung adenocarcinoma cells via Akt activation: Possible contribution to non-invasive expansion of lung adenocarcinoma. Am. J. Pathol. 2004, 164, 91–100. [Google Scholar] [CrossRef]
- Liu, H.; Liang, Z.; Zhou, C.; Zeng, Z.; Wang, F.; Hu, T.; He, X.; Wu, X.; Wu, X.; Lan, P. Mutant KRAS triggers functional reprogramming of tumor-associated macrophages in colorectal cancer. Signal Transduct. Target. Ther. 2021, 6, 144. [Google Scholar] [CrossRef]
- Zdanov, S.; Mandapathil, M.; Abu Eid, R.; Adamson-Fadeyi, S.; Wilson, W.; Qian, J.; Carnie, A.; Tarasova, N.; Mkrtichyan, M.; Berzofsky, J.A.; et al. Mutant KRAS Conversion of Conventional T Cells into Regulatory T Cells. Cancer Immunol. Res. 2016, 4, 354–365. [Google Scholar] [CrossRef] [Green Version]
- Liao, W.; Overman, M.J.; Boutin, A.T.; Shang, X.; Zhao, D.; Dey, P.; Li, J.; Wang, G.; Lan, Z.; Li, J.; et al. KRAS-IRF2 Axis Drives Immune Suppression and Immune Therapy Resistance in Colorectal Cancer. Cancer Cell 2019, 35, 559–572. [Google Scholar] [CrossRef] [Green Version]
- Nusse, R.; Clevers, H. Wnt/β-Catenin Signaling, Disease, and Emerging Therapeutic Modalities. Cell 2017, 169, 985–999. [Google Scholar] [CrossRef] [Green Version]
- Kikuchi, A.; Kishida, S.; Yamamoto, H. Regulation of Wnt signaling by protein-protein interaction and post-translational modifications. Exp. Mol. Med. 2006, 38, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Trejo-Solis, C.; Escamilla-Ramirez, A.; Jimenez-Farfan, D.; Castillo-Rodriguez, R.A.; Flores-Najera, A.; Cruz-Salgado, A. Crosstalk of the Wnt/β-Catenin Signaling Pathway in the Induction of Apoptosis on Cancer Cells. Pharmaceuticals 2021, 14, 871. [Google Scholar] [CrossRef]
- Staal, F.J.; Luis, T.C.; Tiemessen, M.M. WNT signalling in the immune system: WNT is spreading its wings. Nat. Rev. Immunol. 2008, 8, 581–593. [Google Scholar] [CrossRef]
- Reya, T.; Duncan, A.W.; Ailles, L.; Domen, J.; Scherer, D.C.; Willert, K.; Hintz, L.; Nusse, R.; Weissman, I.L. A role for Wnt signalling in self-renewal of haematopoietic stem cells. Nature 2003, 423, 409–414. [Google Scholar] [CrossRef]
- Ranes, M.; Zaleska, M.; Sakalas, S.; Knight, R.; Guettler, S. Reconstitution of the destruction complex defines roles of AXIN polymers and APC in β-catenin capture, phosphorylation, and ubiquitylation. Mol. Cell 2021, 81, 3246–3261. [Google Scholar] [CrossRef]
- Stamos, J.L.; Weis, W.I. The β-catenin destruction complex. Cold Spring Harb. Perspect. Biol. 2013, 5, a007898. [Google Scholar] [CrossRef]
- Yost, C.; Torres, M.; Miller, J.R.; Huang, E.; Kimelman, D.; Moon, R.T. The axis-inducing activity, stability, and subcellular distribution of beta-catenin is regulated in Xenopus embryos by glycogen synthase kinase 3. Genes Dev. 1996, 10, 1443–1454. [Google Scholar] [CrossRef] [Green Version]
- Liu, C.; Li, Y.; Semenov, M.; Han, C.; Baeg, G.H.; Tan, Y.; Zhang, Z.; Lin, X.; He, X. Control of beta-catenin phosphorylation/degradation by a dual-kinase mechanism. Cell 2002, 108, 837–847. [Google Scholar] [CrossRef] [Green Version]
- Metcalfe, C.; Bienz, M. Inhibition of GSK3 by Wnt signalling–two contrasting models. J. Cell Sci. 2011, 124, 3537–3544. [Google Scholar] [CrossRef] [Green Version]
- Gammons, M.V.; Renko, M.; Johnson, C.M.; Rutherford, T.J.; Bienz, M. Wnt Signalosome Assembly by DEP Domain Swapping of Dishevelled. Mol. Cell 2016, 64, 92–104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gammons, M.; Bienz, M. Multiprotein complexes governing Wnt signal transduction. Curr. Opin. Cell Biol. 2018, 51, 42–49. [Google Scholar] [CrossRef] [PubMed]
- Yokoyama, N.; Golebiewska, U.; Wang, H.Y.; Malbon, C.C. Wnt-dependent assembly of supermolecular Dishevelled-3-based complexes. J. Cell Sci. 2010, 123, 3693–3702. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Colozza, G.; Koo, B.K. Wnt/β-catenin signaling: Structure, assembly and endocytosis of the signalosome. Dev. Growth Differ. 2021, 63, 199–218. [Google Scholar] [CrossRef] [PubMed]
- Barker, N.; Hurlstone, A.; Musisi, H.; Miles, A.; Bienz, M.; Clevers, H. The chromatin remodelling factor Brg-1 interacts with β-catenin to promote target gene activation. EMBO J. 2001, 20, 4935–4943. [Google Scholar] [CrossRef] [Green Version]
- Daulat, A.M.; Borg, J.P. Wnt/Planar cell polarity signaling: New opportunities for cancer treatment. Trends Cancer 2017, 3, 113–125. [Google Scholar] [CrossRef] [Green Version]
- De, A. Wnt/Ca2+ signaling pathway: A brief overview. Acta Biochim. Biophys Sin 2011, 43, 745–756. [Google Scholar] [CrossRef]
- Danek, P.; Kardosova, M.; Janeckova, L.; Karkoulia, E.; Vanickova, K.; Fabisik, M.; Lozano-Asencio, C.; Benoukraf, T.; Tirado-Magallanes, R.; Zhou, Q. β-Catenin-TCF/LEF signaling promotes steady-state and emergency granulopoiesis via G-CSF receptor upregulation. Blood 2020, 136, 2574–2587. [Google Scholar] [CrossRef]
- Zhang, D.E.; Zhang, P.; Wang, N.D.; Hetherington, C.J.; Darlington, G.J.; Tenen, D.G. Absence of granulocyte colony-stimulating factor signaling and neutrophil development in CCAAT enhancer binding protein alpha-deficient mice. Proc. Natl. Acad. Sci. USA 1997, 94, 569–574. [Google Scholar] [CrossRef] [Green Version]
- Boettcher, S. Wnt to the rescue! A new role in granulopoiesis. Blood 2020, 136, 2487–2489. [Google Scholar] [CrossRef]
- Heidel, F.H.; Arreba-Tutusaus, P.; Armstrong, S.A.; Fischer, T. Evolutionarily conserved signaling pathways: Acting in the shadows of acute myelogenous leukemia’s genetic diversity. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2015, 21, 240–248. [Google Scholar] [CrossRef] [Green Version]
- Goentoro, L.; Kirschner, M.W. Evidence that fold-change, and not absolute level, of beta-catenin dictates Wnt signaling. Mol. Cell 2009, 36, 872–884. [Google Scholar] [CrossRef]
- Luis, T.C.; Ichii, M.; Brugman, M.H.; Kincade, P.; Staal, F.J. Wnt signaling strength regulates normal hematopoiesis and its deregulation is involved in leukemia development. Leukemia 2012, 26, 414–421. [Google Scholar] [CrossRef] [Green Version]
- Luis, T.C.; Naber, B.A.; Roozen, P.P.; Brugman, M.H.; de Haas, E.F.; Ghazvini, M.; Fibbe, W.E.; van Dongen, J.J.; Fodde, R.; Staal, F.J. Canonical wnt signaling regulates hematopoiesis in a dosage-dependent fashion. Cell Stem Cell 2011, 9, 345–356. [Google Scholar] [CrossRef] [Green Version]
- Zhao, C.; Blum, J.; Chen, A.; Kwon, H.Y.; Jung, S.H.; Cook, J.M.; Lagoo, A.; Reya, T. Loss of beta-catenin impairs the renewal of normal and CML stem cells in vivo. Cancer Cell 2007, 12, 528–541. [Google Scholar] [CrossRef] [Green Version]
- Fleming, H.E.; Janzen, V.; Lo Celso, C.; Guo, J.; Leahy, K.M.; Kronenberg, H.M.; Scadden, D.T. Wnt signaling in the niche enforces hematopoietic stem cell quiescence and is necessary to preserve self-renewal in vivo. Cell Stem Cell 2008, 2, 274–283. [Google Scholar] [CrossRef] [Green Version]
- Luis, T.C.; Naber, B.A.; Fibbe, W.E.; van Dongen, J.J.; Staal, F.J. Wnt3a nonredundantly controls hematopoietic stem cell function and its deficiency results in complete absence of canonical Wnt signaling. Blood 2010, 116, 496–497. [Google Scholar] [CrossRef]
- Li, J.; Mizukami, Y.; Zhang, X.; Jo, W.S.; Chung, D.C. Oncogenic K-ras stimulates Wnt signaling in colon cancer through inhibition of GSK-3beta. Gastroenterology 2005, 128, 1907–1918. [Google Scholar] [CrossRef]
- Lemieux, E.; Cagnol, S.; Beaudry, K.; Carrier, J.; Rivard, N. Oncogenic KRAS signalling promotes the Wnt/beta-catenin pathway through LRP6 in colorectal cancer. Oncogene 2015, 34, 4914–4927. [Google Scholar] [CrossRef]
- Liang, H.; Chen, Q.; Coles, A.H.; Anderson, S.J.; Pihan, G.; Bradley, A.; Gerstein, R.; Jurecic, R.; Jones, S.N. Wnt5a inhibits B cell proliferation and functions as a tumor suppressor in hematopoietic tissue. Cancer Cell 2003, 4, 349–360. [Google Scholar] [CrossRef] [Green Version]
- Heidel, F.H.; Bullinger, L.; Arreba-Tutusaus, P.; Wang, Z.; Gaebel, J.; Hirt, C.; Niederwieser, D.; Lane, S.W.; Döhner, K.; Vasioukhin, V.; et al. The cell fate determinant Llgl1 influences HSC fitness and prognosis in AML. J. Exp. Med. 2013, 210, 15–22. [Google Scholar] [CrossRef] [PubMed]
- Beekman, R.; Valkhof, M.G.; Sanders, M.A.; van Strien, P.M.; Haanstra, J.R.; Broeders, L.; Geertsma-Kleinekoort, W.M.; Veerman, A.J.; Valk, P.J.; Verhaak, R.G.; et al. Sequential gain of mutations in severe congenital neutropenia progressing to acute myeloid leukemia. Blood 2012, 119, 5071–5077. [Google Scholar] [CrossRef] [PubMed]
- Prebet, T.; Lhoumeau, A.C.; Arnoulet, C.; Aulas, A.; Marchetto, S.; Audebert, S.; Puppo, F.; Chabannon, C.; Sainty, D.; Santoni, M.J. The cell polarity PTK7 receptor acts as a modulator of the chemotherapeutic response in acute myeloid leukemia and impairs clinical outcome. Blood 2010, 116, 2315–2323. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lhoumeau, A.C.; Arcangeli, M.L.; De Grandis, M.; Giordano, M.; Orsoni, J.C.; Lembo, F.; Bardin, F.; Marchetto, S.; Aurrand-Lions, S.; Borg, J.P. Ptk7-deficient mice have decreased hematopoietic stem cell pools as a result of deregulated proliferation and migration. J. Immunol. 2016, 196, 4367–4377. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zang, S.; Liu, N.; Wang, H.; Wald, D.N.; Shao, N.; Zhang, J.; Ma, D.; Ji, C.; Tse, W. Wnt signaling is involved in 6-benzylthioinosine-induced AML cell differentiation. BMC Cancer 2014, 14, 886. [Google Scholar] [CrossRef] [Green Version]
- McCubrey, J.A.; Steelman, L.S.; Bertrand, F.E.; Davis, N.M.; Sokolosky, M.; Abrams, S.L.; Montalto, G.; D’Assoro, A.B.; Libra, M.; Nicoletti, F.; et al. GSK-3 as potential target for therapeutic intervention in cancer. Oncotarget 2014, 5, 2881–2911. [Google Scholar] [CrossRef] [Green Version]
- Huang, J.; Zhang, Y.; Bersenev, A.; O’Brien, W.T.; Tong, W.; Emerson, S.G.; Klein, P.S. Pivotal role for glycogen synthase kinase-3 in hematopoietic stem cell homeostasis in mice. J. Clin. Investig. 2009, 119, 3519–3529. [Google Scholar] [CrossRef]
- Wang, Y.; Krivtsov, A.V.; Sinha, A.U.; North, T.E.; Goessling, W.; Feng, Z.; Zon, L.I.; Armstrong, S.A. The Wnt/beta-catenin pathway is required for the development of leukemia stem cells in AML. Science 2010, 327, 1650–1653. [Google Scholar] [CrossRef] [Green Version]
- Cohen, P.; Frame, S. The renaissance of GSK3. Nat. Rev. Mol. Cell Biol. 2001, 2, 769–776. [Google Scholar] [CrossRef]
- Kaidanovich-Beilin, O.; Woodgett, J.R. GSK-3: Functional Insights from Cell Biology and Animal Models. Front. Mol. Neurosci. 2011, 4, 40. [Google Scholar] [CrossRef] [Green Version]
- Martelli, A.M.; Evangelisti, C.; Paganelli, F.; Chiarini, F.; McCubrey, J.A. GSK-3: A multifaceted player in acute leukemias. Leukemia 2021, 35, 1829–1842. [Google Scholar] [CrossRef]
- Guezguez, B.; Almakadi, M.; Benoit, Y.D.; Shapovalova, Z.; Rahmig, S.; Fiebig-Comyn, A.; Casado, F.L.; Tanasijevic, B.; Bresolin, S.; Masetti, R.; et al. GSK3 deficiencies in hematopoietic stem cells initiate pre-neoplastic state that is predictive of clinical outcomes of human acute leukemia. Cancer Cell 2016, 29, 61–74. [Google Scholar] [CrossRef]
- Wang, Z.; Smith, K.S.; Murphy, M.; Piloto, O.; Somervaille, T.C.; Cleary, M.L. Glycogen synthase kinase 3 in MLL leukaemia maintenance and targeted therapy. Nature 2008, 455, 1205–1209. [Google Scholar] [CrossRef] [Green Version]
- Hozumi, M. Differentiation therapy of leukemia: Achievements, limitations and future prospects. Int. J. Hematol. 1998, 68, 107–129. [Google Scholar] [CrossRef]
- Meng-Er, H.; Yu-Chen, Y.; Shu-Rong, C.; Jin-Ren, C.; Jia-Xiang, L.; Lin, Z.; Zhen-Yi, W. Use of all-trans retinoic acid in the treatment of acute promyelocytic leukemia. Blood 1988, 72, 567–572. [Google Scholar]
- Gupta, K.; Gulen, F.; Sun, L.; Aguilera, R.; Chakrabarti, A.; Kiselar, J.; Agarwal, M.K.; Wald, D.N. GSK3 is a regulator of RAR-mediated differentiation. Leukemia 2012, 26, 1277–1285. [Google Scholar] [CrossRef] [Green Version]
- Gupta, K.; Stefan, T.; Ignatz-Hoover, J.; Moreton, S.; Parizher, G.; Saunthararajah, Y.; Wald, D.N. GSK-3 Inhibition Sensitizes Acute Myeloid Leukemia Cells to 1,25D-Mediated Differentiation. Cancer Res. 2016, 76, 2743–2753. [Google Scholar] [CrossRef] [Green Version]
- Holmes, T.; O’Brien, T.A.; Knight, R.; Lindeman, R.; Shen, S.; Song, E.; Symonds, G.; Dolnikov, A. Glycogen synthase kinase-3beta inhibition preserves hematopoietic stem cell activity and inhibits leukemic cell growth. Stem Cells 2008, 26, 1288–1297. [Google Scholar] [CrossRef] [Green Version]
- Song, E.Y.; Palladinetti, P.; Klamer, G.; Ko, K.H.; Lindeman, R.; O’Brien, T.A.; Dolnikov, A. Glycogen synthase kinase–3β inhibitors suppress leukemia cell growth. Exp. Hematol. 2010, 38, 908–921. [Google Scholar] [CrossRef]
- Rizzieri, D.A.; Cooley, S.; Odenike, O.; Moonan, L.; Chow, K.H.; Jackson, K.; Wang, X.; Brail, L.; Borthakur, G. An open-label phase 2 study of glycogen synthase kinase-3 inhibitor LY2090314 in patients with acute leukemia. Leuk. Lymphoma 2016, 57, 1800–1806. [Google Scholar] [CrossRef]
- Li, C.W.; Lim, S.O.; Xia, W.; Lee, H.H.; Chan, L.C.; Kuo, C.W.; Khoo, K.H.; Chang, S.S.; Cha, J.H.; Kim, T.; et al. Glycosylation and stabilization of programmed death ligand-1 suppresses T-cell activity. Nat. Commun. 2016, 7, 12632. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, F.; Yang, L.; Xiao, M.; Zhang, Z.; Shen, J.; Anuchapreeda, S.; Tima, S.; Chiampanichayakul, S.; Xiao, Z. PD-L1 regulates cell proliferation and apoptosis in acute myeloid leukemia by activating PI3K-AKT signaling pathway. Sci. Rep. 2022, 12, 11444. [Google Scholar] [CrossRef]
- Dimou, A.; Syrigos, K.N. The Role of GSK3β in T lymphocytes in the tumor microenvironment. Front. Oncol. 2020, 10, 1221. [Google Scholar] [CrossRef] [PubMed]
- Dong, Y.; Han, Y.; Huang, Y.; Jiang, S.; Huang, Z.; Chen, R.; Yu, Z.; Yu, K.; Zhang, S. PD-L1 is expressed and promotes the expansion of regulatory T Cells in acute myeloid leukemia. Front. Immunol. 2020, 11, 1710. [Google Scholar] [CrossRef] [PubMed]
- Ramji, D.P.; Foka, P. CCAAT/enhancer-binding proteins: Structure, function, and regulation. Biochem. J. 2002, 365, 561–575. [Google Scholar] [CrossRef] [Green Version]
- Akagi, T.; Saitoh, T.; O’Kelly, J.; Akira, S.; Gombart, A.F.; Koeffler, H.P. Impaired response to GM-CSF and G-CSF, and enhanced apoptosis in C/EBPbeta-deficient hematopoietic cells. Blood 2008, 111, 2999–3004. [Google Scholar] [CrossRef] [Green Version]
- Tolomeo, M.; Grimaudo, S. The “Janus” role of C/EBPs family members in cancer progression. Int. J. Mol. Sci. 2020, 21, 4308. [Google Scholar] [CrossRef]
- Cloutier, A.; Guindi, C.; Larivee, P.; Dubois, C.M.; Amrani, A.; McDonald, P.P. Inflammatory cytokine production by human neutrophils involves C/EBP transcription factors. J. Immunol. 2009, 182, 563–571. [Google Scholar] [CrossRef] [Green Version]
- Nerlov, C. The C/EBP family of transcription factors: A paradigm for interaction between gene expression and proliferation control. Trends Cell Biol. 2007, 17, 318–324. [Google Scholar] [CrossRef]
- Radomska, H.S.; Huettner, C.S.; Zhang, P.; Cheng, T.; Scadden, D.T.; Tenen, D.G. CCAAT/enhancer binding protein alpha is a regulatory switch sufficient for induction of granulocytic development from bipotential myeloid progenitors. Mol. Cell Biol. 1998, 18, 4301–4314. [Google Scholar] [CrossRef] [Green Version]
- Skokowa, J.; Cario, G.; Uenalan, M.; Schambach, A.; Germeshausen, M.; Battmer, K.; Zeidler, C.; Lehmann, U.; Eder, M.; Baum, C.; et al. LEF-1 is crucial for neutrophil granulocytopoiesis and its expression is severely reduced in congenital neutropenia. Nat. Med. 2006, 12, 1191–1197. [Google Scholar] [CrossRef]
- Smith, L.T.; Hohaus, S.; Gonzalez, D.A.; Dziennis, S.E.; Tenen, D.G. PU.1 (Spi-1) and C/EBP alpha regulate the granulocyte colony-stimulating factor receptor promoter in myeloid cells. Blood 1996, 88, 1234–1247. [Google Scholar] [CrossRef] [Green Version]
- Zhang, P.; Iwama, A.; Datta, M.W.; Darlington, G.J.; Link, D.C.; Tenen, D.G. Upregulation of interleukin 6 and granulocyte colony-stimulating factor receptors by transcription factor CCAAT enhancer binding protein alpha (C/EBP alpha) is critical for granulopoiesis. J. Exp. Med. 1998, 188, 1173–1184. [Google Scholar] [CrossRef]
- Skokowa, J.; Klimiankou, M.; Klimenkova, O.; Lan, D.; Gupta, K.; Hussein, K.; Carrizosa, E.; Kusnetsova, I.; Li, Z.; Sustmann, C.; et al. Interactions among HCLS1, HAX1 and LEF-1 proteins are essential for G-CSF-triggered granulopoiesis. Nat. Med. 2012, 18, 1550–1559. [Google Scholar] [CrossRef] [Green Version]
- Tanaka, T.; Akira, S.; Yoshida, K.; Umemoto, M.; Yoneda, Y.; Shirafuji, N.; Fujiwara, H.; Suematsu, S.; Yoshida, N.; Kishimoto, T. Targeted disruption of the NF-IL6 gene discloses its essential role in bacteria killing and tumor cytotoxicity by macrophages. Cell 1995, 80, 353–361. [Google Scholar] [CrossRef] [Green Version]
- Collins, S.J.; Ulmer, J.; Purton, L.E.; Darlington, G. Multipotent hematopoietic cell lines derived from C/EBPalpha(-/-) knockout mice display granulocyte macrophage-colony-stimulating factor, granulocyte- colony-stimulating factor, and retinoic acid-induced granulocytic differentiation. Blood 2001, 98, 2382–2388. [Google Scholar] [CrossRef] [Green Version]
- Hirai, H.; Zhang, P.; Dayaram, T.; Hetherington, C.J.; Mizuno, S.; Imanishi, J.; Akashi, K.; Tenen, D.G. C/EBPbeta is required for ‘emergency’ granulopoiesis. Nat. Immunol. 2006, 7, 732–739. [Google Scholar] [CrossRef]
- Akira, S.; Isshiki, H.; Sugita, T.; Tanabe, O.; Kinoshita, S.; Nishio, Y.; Nakajima, T.; Hirano, T.; Kishimoto, T. A nuclear factor for IL-6 expression (NF-IL6) is a member of a C/EBP family. EMBO J. 1990, 9, 1897–1906. [Google Scholar] [CrossRef]
- Descombes, P.; Chojkier, M.; Lichtsteiner, S.; Falvey, E.; Schibler, U. LAP, a novel member of the C/EBP gene family, encodes a liver-enriched transcriptional activator protein. Genes Dev. 1990, 4, 1541–1551. [Google Scholar] [CrossRef] [Green Version]
- Poli, V.; Mancini, F.P.; Cortese, R. IL-6DBP, a nuclear protein involved in interleukin-6 signal transduction, defines a new family of leucine zipper proteins related to C/EBP. Cell 1990, 63, 643–653. [Google Scholar] [CrossRef]
- Ford, A.M.; Bennett, C.A.; Healy, L.E.; Towatari, M.; Greaves, M.F.; Enver, T. Regulation of the myeloperoxidase enhancer binding proteins Pu1, C-EBP alpha, -beta, and -delta during granulocyte-lineage specification. Proc. Natl. Acad. Sci. USA 1996, 93, 10838–10843. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oelgeschläger, M.; Nuchprayoon, I.; Lüscher, B.; Friedman, A.D. C/EBP, c-Myb, and PU.1 cooperate to regulate the neutrophil elastase promoter. Mol. Cell Biol. 1996, 16, 4717–4725. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hirai, H.; Yokota, A.; Tamura, A.; Sato, A.; Maekawa, T. Non-steady-state hematopoiesis regulated by the C/EBPbeta transcription factor. Cancer Sci. 2015, 106, 797–802. [Google Scholar] [CrossRef] [PubMed]
- Morosetti, R.; Park, D.J.; Chumakov, A.M.; Grillier, I.; Shiohara, M.; Gombart, A.F.; Nakamaki, T.; Weinberg, K.; Koeffler, H.P. A novel, myeloid transcription factor, C/EBP epsilon, is upregulated during granulocytic, but not monocytic, differentiation. Blood 1997, 90, 2591–2600. [Google Scholar] [CrossRef] [PubMed]
- Yamanaka, R.; Barlow, C.; Lekstrom-Himes, J.; Castilla, L.H.; Liu, P.P.; Eckhaus, M.; Decker, T.; Wynshaw-Boris, A.; Xanthopoulos, K.G. Impaired granulopoiesis, myelodysplasia, and early lethality in CCAAT/enhancer binding protein epsilon-deficient mice. Proc. Natl. Acad. Sci. USA 1997, 94, 13187–13192. [Google Scholar] [CrossRef] [Green Version]
- Lekstrom-Himes, J.A. The role of C/EBP(epsilon) in the terminal stages of granulocyte differentiation. Stem Cells 2001, 19, 125–133. [Google Scholar] [CrossRef]
- Mollinedo, F.; Lopez-Perez, R.; Gajate, C. Differential gene expression patterns coupled to commitment and acquisition of phenotypic hallmarks during neutrophil differentiation of human leukaemia HL-60 cells. Gene 2008, 419, 16–26. [Google Scholar] [CrossRef]
- Nakajima, H.; Ihle, J.N. Granulocyte colony-stimulating factor regulates myeloid differentiation through CCAAT/enhancer-binding protein epsilon. Blood 2001, 98, 897–905. [Google Scholar] [CrossRef] [Green Version]
- Dührsen, U.; Villeval, J.L.; Boyd, J.; Kannourakis, G.; Morstyn, G.; Metcalf, D. Effects of recombinant human granulocyte colony-stimulating factor on hematopoietic progenitor cells in cancer patients. Blood 1988, 72, 2074–2081. [Google Scholar] [CrossRef] [Green Version]
- Mehta, H.M.; Malandra, M.; Corey, S.J. G-CSF and GM-CSF in Neutropenia. J. Immunol. 2015, 195, 1341–1349. [Google Scholar] [CrossRef] [Green Version]
- Cheers, C.; Haigh, A.M.; Kelso, A.; Metcalf, D.; Stanley, E.R.; Young, A.M. Production of colony-stimulating factors (CSFs) during infection: Separate determinations of macrophage-, granulocyte-, granulocyte-macrophage-, and multi-CSFs. Infect. Immun. 1988, 56, 247–251. [Google Scholar] [CrossRef] [Green Version]
- Kawakami, M.; Tsutsumi, H.; Kumakawa, T.; Abe, H.; Hirai, M.; Kurosawa, S.; Mori, M.; Fukushima, M. Levels of serum granulocyte colony-stimulating factor in patients with infections. Blood 1990, 76, 1962–1964. [Google Scholar] [CrossRef] [Green Version]
- Hartung, T.; Doecke, W.D.; Bundschuh, D.; Foote, M.A.; Gantner, F.; Hermann, C.; Lenz, A.; Milwee, S.; Rich, B.; Simon, B.; et al. Effect of filgrastim treatment on inflammatory cytokines and lymphocyte functions. Clin. Pharmacol. Ther. 1999, 66, 415–424. [Google Scholar] [CrossRef]
- Dahl, R.; Walsh, J.C.; Lancki, D.; Laslo, P.; Iyer, S.R.; Singh, H.; Simon, M.C. Regulation of macrophage and neutrophil cell fates by the PU.1:C/EBPalpha ratio and granulocyte colony-stimulating factor. Nat. Immunol. 2003, 4, 1029–1036. [Google Scholar] [CrossRef]
- Sheng, Y.; Ju, W.; Huang, Y.; Li, J.; Ozer, H.; Qiao, X.; Qian, Z. Activation of wnt/β-catenin signaling blocks monocyte-macrophage differentiation through antagonizing PU.1-targeted gene transcription. Leukemia 2016, 30, 2106–2109. [Google Scholar] [CrossRef] [Green Version]
- Morris, K.T.; Khan, H.; Ahmad, A.; Weston, L.L.; Nofchissey, R.A.; Pinchuk, I.V.; Beswick, E.J. G-CSF and G-CSFR are highly expressed in human gastric and colon cancers and promote carcinoma cell proliferation and migration. Br. J. Cancer 2014, 110, 1211–1220. [Google Scholar] [CrossRef] [Green Version]
- El Fakih, R.; Rasheed, W.; Hawsawi, Y.; Alsermani, M.; Hassanein, M. Targeting FLT3 mutations in acute myeloid leukemia. Cells 2018, 7, 4. [Google Scholar] [CrossRef] [Green Version]
- Kotb, A.; El Fakih, R.; Hanbali, A.; Hawsawi, Y.; Alfraih, F.; Hashmi, S.; Aljurf, M. Philadelphia-like acute lymphoblastic leukemia: Diagnostic dilemma and management perspectives. Exp. Hematol. 2018, 67, 1–9. [Google Scholar] [CrossRef]
- Basu, S.; Hodgson, G.; Katz, M.; Dunn, A.R. Evaluation of role of G-CSF in the production, survival, and release of neutrophils from bone marrow into circulation. Blood 2002, 100, 854–861. [Google Scholar] [CrossRef] [Green Version]
- Theyab, A.; Algahtani, M.; Alsharif, K.F.; Hawsawi, Y.M.; Alghamdi, A.; Alghamdi, A.; Akinwale, J. New insight into the mechanism of granulocyte colony-stimulating factor (G-CSF) that induces the mobilization of neutrophils. Hematology 2021, 26, 628–636. [Google Scholar] [CrossRef]
- Dwivedi, P.; Greis, K.D. Granulocyte colony-stimulating factor receptor signaling in severe congenital neutropenia, chronic neutrophilic leukemia, and related malignancies. Exp. Hematol. 2017, 46, 9–20. [Google Scholar] [CrossRef] [Green Version]
- Ward, A.C.; Loeb, D.M.; Soede-Bobok, A.A.; Touw, I.P.; Friedman, A.D. Regulation of granulopoiesis by transcription factors and cytokine signals. Leukemia 2000, 14, 973–990. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fukunaga, R.; Ishizaka-Ikeda, E.; Nagata, S. Growth and differentiation signals mediated by different regions in the cytoplasmic domain of granulocyte colony-stimulating factor receptor. Cell 1993, 74, 1079–1087. [Google Scholar] [CrossRef] [PubMed]
- Akbarzadeh, S.; Ward, A.C.; McPhee, D.O.; Alexander, W.S.; Lieschke, G.J.; Layton, J.E. Tyrosine residues of the granulocyte colony-stimulating factor receptor transmit proliferation and differentiation signals in murine bone marrow cells. Blood 2002, 99, 879–887. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hermans, M.H.; van de Geijn, G.J.; Antonissen, C.; Gits, J.; van Leeuwen, D.; Ward, A.C.; Touw, I.P. Signaling mechanisms coupled to tyrosines in the granulocyte colony-stimulating factor receptor orchestrate G-CSF-induced expansion of myeloid progenitor cells. Blood 2003, 101, 2584–2590. [Google Scholar] [CrossRef]
- Kendrick, T.S.; Bogoyevitch, M.A. Activation of mitogen-activated protein kinase pathways by the granulocyte colony-stimulating factor receptor: Mechanisms and functional consequences. Front. Biosci. 2007, 12, 591–607. [Google Scholar] [CrossRef] [Green Version]
- Manz, M.G.; Boettcher, S. Emergency granulopoiesis. Nat. Rev. Immunol. 2014, 14, 302–314. [Google Scholar] [CrossRef]
- Maun, N.A.; Gaines, P.; Khanna-Gupta, A.; Zibello, T.; Enriquez, L.; Goldberg, L.; Berliner, N. G-CSF signaling can differentiate promyelocytes expressing a defective retinoic acid receptor: Evidence for divergent pathways regulating neutrophil differentiation. Blood 2004, 103, 1693–1701. [Google Scholar] [CrossRef] [Green Version]
- Wang, Q.F.; Friedman, A.D. CCAAT/enhancer-binding proteins are required for granulopoiesis independent of their induction of the granulocyte colony-stimulating factor receptor. Blood 2002, 99, 2776–2785. [Google Scholar] [CrossRef] [Green Version]
- Ip, S.H.; Cooper, R.A. Decreased membrane fluidity during differentiation of human promyelocytic leukemia cells in culture. Blood 1980, 56, 227–232. [Google Scholar] [CrossRef] [Green Version]
- Lee, Y.; Pincus, P.A.; Hyeon, C. Effects of dimethyl sulfoxide on surface water near phospholipid bilayers. Biophys. J. 2016, 111, 2481–2491. [Google Scholar] [CrossRef] [Green Version]
- Zylber-Katz, E.; Glazer, R.I. Phospholipid- and Ca2+-dependent protein kinase activity and protein phosphorylation patterns in the differentiation of human promyelocytic leukemia cell line HL-60. Cancer Res. 1985, 45, 5159–5164. [Google Scholar]
- Fontana, J.A.; Reppucci, A.; Durham, J.P.; Miranda, D. Correlation between the induction of leukemic cell differentiation by various retinoids and modulation of protein kinases. Cancer Res. 1986, 46, 2468–2473. [Google Scholar]
- Chung, E.J.; Hwang, S.H.; Nguyen, P.; Lee, S.; Kim, J.S.; Kim, J.W.; Henkart, P.A.; Bottaro, D.P.; Soon, L.; Bonvini, P.; et al. Regulation of leukemic cell adhesion, proliferation, and survival by beta-catenin. Blood 2002, 100, 982–990. [Google Scholar] [CrossRef]
- Willert, K.; Jones, K.A. Wnt signaling: Is the party in the nucleus? Genes Dev. 2006, 20, 1394–1404. [Google Scholar] [CrossRef]
- Ying, Q.-L.; Wray, J.; Nichols, J.; Batlle-Morera, L.; Doble, B.; Woodgett, J.; Cohen, P.; Smith, A. The ground state of embryonic stem cell self-renewal. Nature 2008, 453, 519. [Google Scholar] [CrossRef] [Green Version]
- Cross, D.A.; Alessi, D.R.; Cohen, P.; Andjelkovich, M.; Hemmings, B.A. Inhibition of glycogen synthase kinase-3 by insulin mediated by protein kinase B. Nature 1995, 378, 785–789. [Google Scholar] [CrossRef]
- Liu, L.; Zhu, S.; Gong, Z.; Low, B.C. K-ras/PI3K-Akt signaling is essential for zebrafish hematopoiesis and angiogenesis. PloS ONE 2008, 3, e2850. [Google Scholar] [CrossRef] [Green Version]
- Mologni, L.; Brussolo, S.; Ceccon, M.; Gambacorti-Passerini, C. Synergistic effects of combined Wnt/KRAS inhibition in colorectal cancer cells. PloS ONE 2012, 7, e51449. [Google Scholar] [CrossRef] [Green Version]
- Wagstaff, M.; Coke, B.; Hodgkiss, G.R.; Morgan, R.G. Targeting β-catenin in acute myeloid leukaemia: Past, present, and future perspectives. Biosci. Rep. 2022, 42, 4. [Google Scholar] [CrossRef]
- Perry, J.M.; He, X.C.; Sugimura, R.; Grindley, J.C.; Haug, J.S.; Ding, S.; Li, L. Cooperation between both Wnt/{beta}-catenin and PTEN/PI3K/Akt signaling promotes primitive hematopoietic stem cell self-renewal and expansion. Genes Dev. 2011, 25, 1928–1942. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ming, M.; Wang, S.; Wu, W.; Senyuk, V.; Le Beau, M.M.; Nucifora, G.; Qian, Z. Activation of Wnt/β-catenin protein signaling induces mitochondria-mediated apoptosis in hematopoietic progenitor cells. J. Biol. Chem. 2012, 287, 22683–22690. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kato, Y.; Naiki, Y.; Komatsu, T.; Takahashi, K.; Nakamura, J.; Koide, N. A Wnt Pathway Activator Induces Apoptosis and Cell Death in Mouse Monocytic Leukemia Cells. Oncol. Res. 2017, 25, 479–483. [Google Scholar] [CrossRef]
- Su, N.; Wang, P.; Li, Y. Role of Wnt/β-catenin pathway in inducing autophagy and apoptosis in multiple myeloma cells. Oncol. Lett. 2016, 12, 4623–4629. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, L.; Guo, C.; Wang, F.; Fu, L. KRAS mutation: From undruggable to druggable in cancer. Signal Transduct. Target. Ther. 2021, 6, 386. [Google Scholar] [CrossRef]
- Moore, A.R.; Rosenberg, S.C.; McCormick, F.; Malek, S. RAS-targeted therapies: Is the undruggable drugged? Nat. Rev. Drug Discov. 2020, 19, 533–552. [Google Scholar] [CrossRef]
- Remsberg, J.R.; Suciu, R.M.; Zambetti, N.A.; Hanigan, T.W.; Firestone, A.J.; Inguva, A.; Long, A.; Ngo, N.; Lum, K.M.; Henry, C.L.; et al. ABHD17 regulation of plasma membrane palmitoylation and N-Ras-dependent cancer growth. Nat. Chem. Biol. 2021, 17, 856–864. [Google Scholar] [CrossRef]
- Thomé, C.H.; Ferreira, G.A.; Pereira-Martins, D.A.; dos Santos, G.A.; Ortiz, C.A.; de Souza, L.E.B.; Sobral, L.M.; Silva, C.L.A.; Scheucher, P.S.; Gil, C.D.; et al. NTAL is associated with treatment outcome, cell proliferation and differentiation in acute promyelocytic leukemia. Sci. Rep. 2020, 10, 1–12. [Google Scholar] [CrossRef]
- Lv, K.; Ren, J.G.; Han, X.; Gui, J.; Gong, C.; Tong, W. Depalmitoylation rewires FLT3-ITD signaling and exacerbates leukemia progression. Blood 2021, 138, 2244–2255. [Google Scholar] [CrossRef]
- Parcells, B.W.; Ikeda, A.K.; Simms-Waldrip, T.; Moore, T.B.; Sakamoto, K.M. FMS-like tyrosine kinase 3 in normal hematopoiesis and acute myeloid leukemia. Stem Cells 2006, 24, 1174–1184. [Google Scholar] [CrossRef]
- Levis, M.; Perl, A.E. Gilteritinib: Potent targeting of FLT3 mutations in AML. Blood Adv. 2020, 4, 1178–1191. [Google Scholar] [CrossRef] [Green Version]
- Zhou, B.; Hao, Q.; Liang, Y.; Kong, E. Protein palmitoylation in cancer: Molecular functions and therapeutic potential. Mol. Oncol. 2022, 17, 3–26. [Google Scholar] [CrossRef]
- Lee, J.H.; Faderl, S.; Pagel, J.M.; Jung, C.W.; Yoon, S.S.; Pardanani, A.D.; Becker, P.S.; Lee, H.; Choi, J.; Lee, K.; et al. Phase 1 study of CWP232291 in patients with relapsed or refractory acute myeloid leukemia and myelodysplastic syndrome. Blood Adv. 2020, 4, 2032–2043. [Google Scholar] [CrossRef]
- Hu, S.; Ueda, M.; Stetson, L.; Ignatz-Hoover, J.; Moreton, S.; Chakrabarti, A.; Xia, Z.; Karan, G.; de Lima, M.; Agrawal, M.K.; et al. A Novel Glycogen Synthase Kinase-3 inhibitor optimized for acute myeloid leukemia differentiation activity. Mol. Cancer Ther. 2016, 15, 1485–1494. [Google Scholar] [CrossRef] [Green Version]
- Levis, M.; Allebach, J.; Tse, K.F.; Zheng, R.; Baldwin, B.R.; Smith, B.D.; Jones-Bolin, S.; Ruggeri, B.; Dionne, C.; Small, D. A FLT3-targeted tyrosine kinase inhibitor is cytotoxic to leukemia cells in vitro and in vivo. Blood 2002, 99, 3885–3891. [Google Scholar] [CrossRef]
- Tickenbrock, L.; Schwäble, J.; Wiedehage, M.; Steffen, B.; Sargin, B.; Choudhary, C.; Brandts, C.; Berdel, W.E.; Müller-Tidow, C.; Serve, H. Flt3 tandem duplication mutations cooperate with Wnt signaling in leukemic signal transduction. Blood 2005, 105, 3699–3706. [Google Scholar] [CrossRef]
- Chen, C.; Xu, W.; Wang, C.M. Combination of celecoxib and doxorubicin increases growth inhibition and apoptosis in acute myeloid leukemia cells. Leuk. Lymphoma 2013, 54, 2517–2522. [Google Scholar] [CrossRef]
- Ysebaert, L.; Chicanne, G.; Demur, C.; De Toni, F.; Prade-Houdellier, N.; Ruidavets, J.B.; Mansat-De Mas, V.; Rigal-Huguet, F.; Laurent, G.; Payrastre, B.; et al. Expression of beta-catenin by acute myeloid leukemia cells predicts enhanced clonogenic capacities and poor prognosis. Leukemia 2006, 20, 1211–1216. [Google Scholar] [CrossRef] [Green Version]
- Chen, C.C.; Gau, J.P.; You, J.Y.; Lee, K.D.; Yu, Y.B.; Lu, C.H.; Lin, J.T.; Lan, C.; Lo, W.H.; Liu, J.M.; et al. Prognostic significance of beta-catenin and topoisomerase IIalpha in de novo acute myeloid leukemia. Am. J. Hematol. 2009, 84, 87–92. [Google Scholar] [CrossRef]
- Morgan, R.G.; Pearn, L.; Liddiard, K.; Pumford, S.L.; Burnett, A.K.; Tonks, A.; Darley, R.L. γ-Catenin is overexpressed in acute myeloid leukemia and promotes the stabilization and nuclear localization of β-catenin. Leukemia 2013, 27, 336–343. [Google Scholar] [CrossRef]
- Zhou, H.; Mak, P.Y.; Mu, H.; Mak, D.H.; Zeng, Z.; Cortes, J.; Liu, Q.; Andreeff, M.; Carter, B.Z. Combined inhibition of β-catenin and Bcr-Abl synergistically targets tyrosine kinase inhibitor-resistant blast crisis chronic myeloid leukemia blasts and progenitors in vitro and in vivo. Leukemia 2017, 31, 2065–2074. [Google Scholar] [CrossRef] [PubMed]
- Jiang, X.; Mak, P.Y.; Mu, H.; Tao, W.; Mak, D.H.; Kornblau, S.; Zhang, Q.; Ruvolo, P.; Burks, J.K.; Zhang, W.; et al. Disruption of Wnt/β-catenin exerts antileukemia activity and synergizes with FLT3 inhibition in FLT3-mutant acute myeloid leukemia. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2018, 24, 2417–2429. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soldi, R.; Halder, T.G.; Sampson, S.; Vankayalapati, H.; Weston, A.; Thode, T.; Bhalla, K.N.; Ng, S.; del Villar, R.R.; Drenner, K.; et al. The Small Molecule BC-2059 inhibits Wingless/integrated (Wnt)-dependent gene transcription in cancer through disruption of the Transducin β-Like 1-β-Catenin protein complex. J. Pharmacol. Exp. Ther. 2021, 378, 77–86. [Google Scholar] [CrossRef] [PubMed]
- Savvidou, I.; Khong, T.; Cuddihy, A.; McLean, C.; Horrigan, S.; Spencer, A. β-Catenin Inhibitor BC2059 Is Efficacious as Monotherapy or in Combination with Proteasome Inhibitor Bortezomib in Multiple Myeloma. Mol. Cancer Ther. 2017, 16, 1765–1778. [Google Scholar] [CrossRef] [Green Version]
- Ueda, M.; Stefan, T.; Stetson, L.; Ignatz-Hoover, J.J.; Tomlinson, B.; Creger, R.J.; Cooper, B.; Lazarus, H.M.; de Lima, M.; Wald, D.N.; et al. Phase I Trial of Lithium and Tretinoin for Treatment of Relapsed and Refractory Non-promyelocytic Acute Myeloid Leukemia. Front. Oncol. 2020, 10, 327. [Google Scholar] [CrossRef]
- Madan, V.; Koeffler, H.P. Differentiation therapy of myeloid leukemia: Four decades of development. Haematologica 2021, 106, 26–38. [Google Scholar] [CrossRef]



| DMSO treatment | 0 day | 1 day | 3 days | 5 days |
| Kras translocation (plasma membrane) | 1.0 | 1.5 | 1.5 | 1.5 |
| Kras activity (plasma membrane) | 1.0 | 1.7 | 2.0 | 2.0 |
| pGSK3β/GSK3β ratio at Ser9 (cytoplasm) | 1.0 | 1.3 | 2.0 | 11.9 |
| GSK3β/actin (cytoplasm) | 1.0 | 1.0 | 1.0 | 1.0 |
| β-catenin (nucleus) | 1.0 | 3.2 | 4.3 | 5.6 |
| TCF4 (nucleus) | 1.0 | 1.3 | 1.8 | 4.6 |
| C/EBPα (nucleus) | 1.0 | 1.4 | 2.1 | 2.1 |
| C/EBPε (nucleus) | 1.0 | 1.1 | 2.5 | 2.1 |
| G-CSFR (nucleus) | 1.0 | 1.9 | 2.8 | 3.5 |
| Lef/Tcf-sensitive transcription | 1.0 | N. D | N. D | 2.5 |
| Target Protein | Agents | Mechanism | Diseases | References |
|---|---|---|---|---|
| Kras4A | 2-bromopalmitate (2-BP) | Inhibits translocation to the membrane | AML, CML | [45] |
| LAT2 | ABHD17A/B/C (ABD957, depalmitoylase inhibitor) | Palmitoylation of LAT2 increases leukemia cell proliferation | AML, APL | [167] [168] |
| FLT3-ITD | Gilteritinib+Palmostatin B (palm B, depalmitoylase inhibitor) | Increased palmitoylated FLT3-ITD inhibits leukemia cell growth | AML, primary AML blasts | [169] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yokoyama, N.; Nakayama, H.; Iwabuchi, K. Novel Insights into the Role of Kras in Myeloid Differentiation: Engaging with Wnt/β-Catenin Signaling. Cells 2023, 12, 322. https://doi.org/10.3390/cells12020322
Yokoyama N, Nakayama H, Iwabuchi K. Novel Insights into the Role of Kras in Myeloid Differentiation: Engaging with Wnt/β-Catenin Signaling. Cells. 2023; 12(2):322. https://doi.org/10.3390/cells12020322
Chicago/Turabian StyleYokoyama, Noriko, Hitoshi Nakayama, and Kazuhisa Iwabuchi. 2023. "Novel Insights into the Role of Kras in Myeloid Differentiation: Engaging with Wnt/β-Catenin Signaling" Cells 12, no. 2: 322. https://doi.org/10.3390/cells12020322
APA StyleYokoyama, N., Nakayama, H., & Iwabuchi, K. (2023). Novel Insights into the Role of Kras in Myeloid Differentiation: Engaging with Wnt/β-Catenin Signaling. Cells, 12(2), 322. https://doi.org/10.3390/cells12020322