
Secreted Metabolome of ALS-Related hSOD1(G93A) Primary Cultures of Myocytes and Implications for Myogenesis

 , , , , and
, , , , and Highlights
- The characterization of hSOD1(G93A) and hSOD1(WT) myocyte cultures by using liquid chromatography coupled with high-resolution mass spectrometry reveals a profound alteration in the abundance of a group of secreted molecules, including metabolites related to the pathways of amino acids and glycerolipid and pyrimidine metabolism.
- The secretome of healthy hSOD1(WT) myocytes allows the rescue of hSOD1(G93A) primary cultures of myocytes in defective myogenesis
Abstract
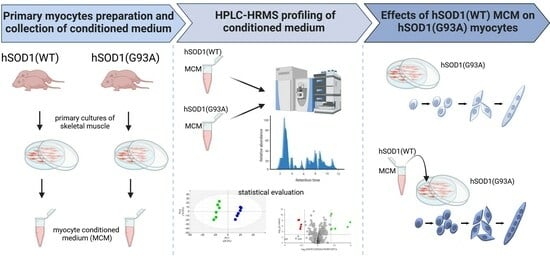
:
1. Introduction
2. Materials and Methods
2.1. Mouse Models
2.2. Primary Myocyte Cultures and Myocyte-Conditioned Medium (MCM) Preparation
2.3. Phenotypic Characterization of Myotubes
2.4. Immunocytochemistry
2.5. Western Blot Analysis
2.6. Metabolomic Analysis of the Secretome of Primary Cultures of Myocytes
2.7. Metabolomics Data Analysis
2.8. Statistical Analysis
3. Results
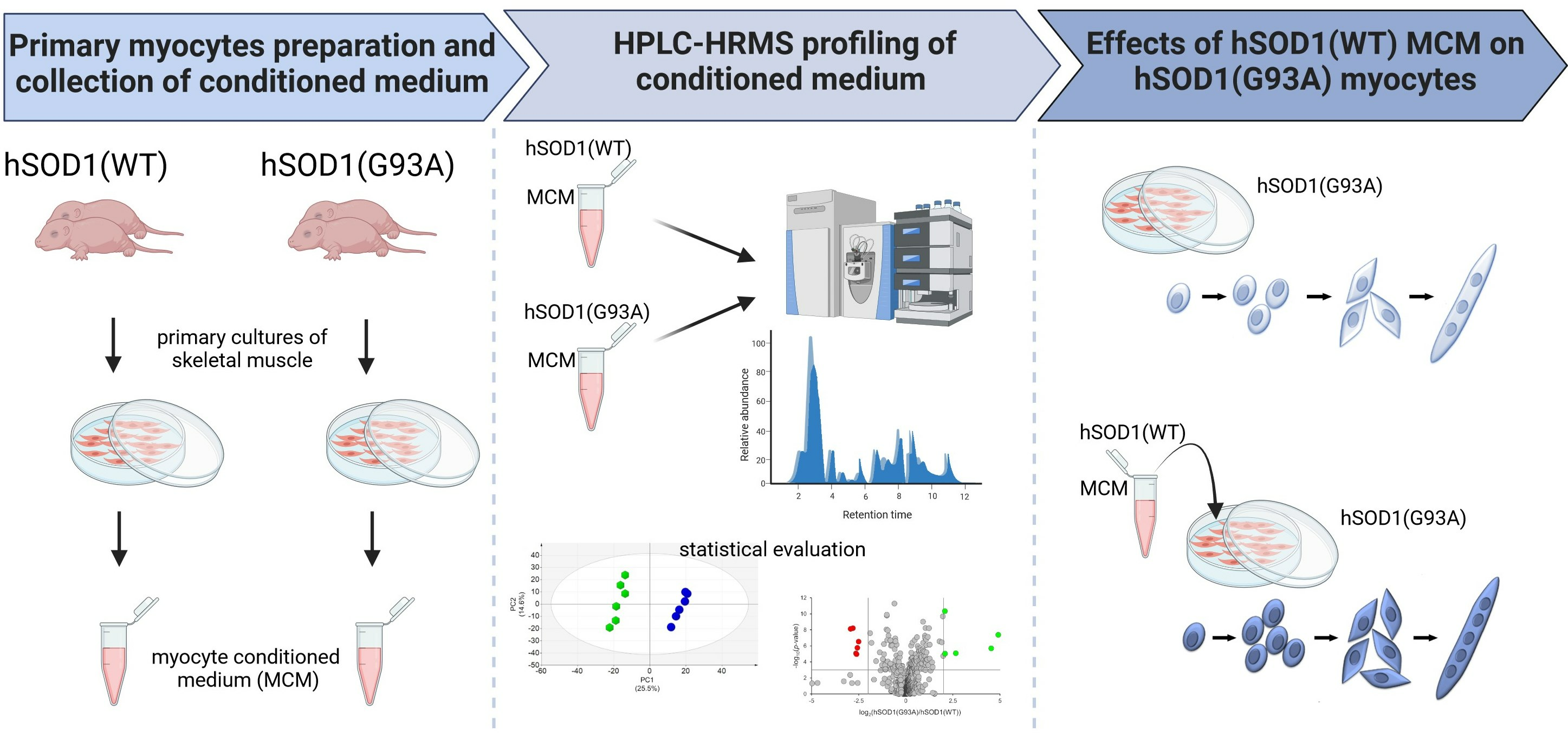
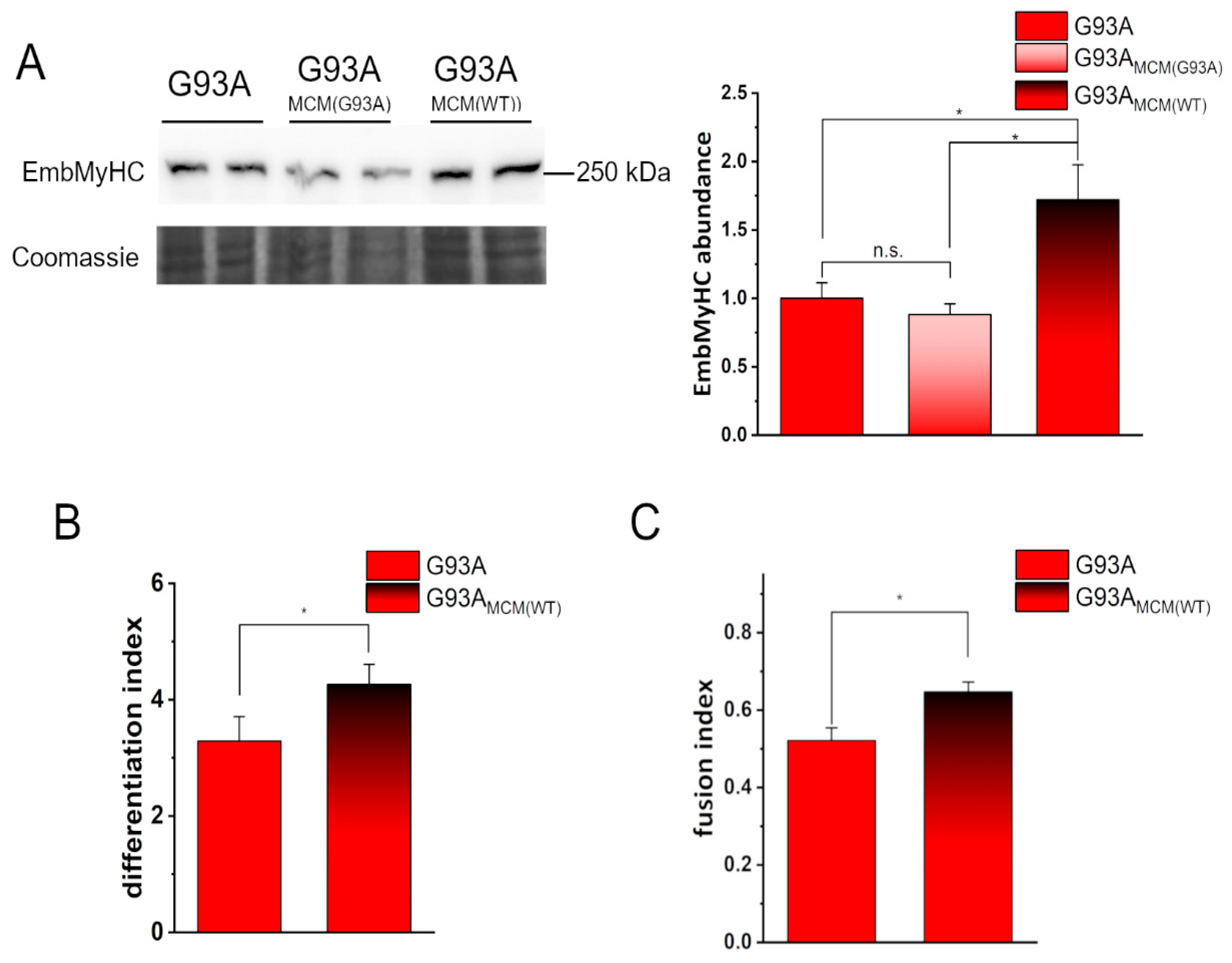
3.1. Impaired Myogenesis in hSOD1(G93A) Myocytes Compared to That of hSOD1(WT) Primary Cells
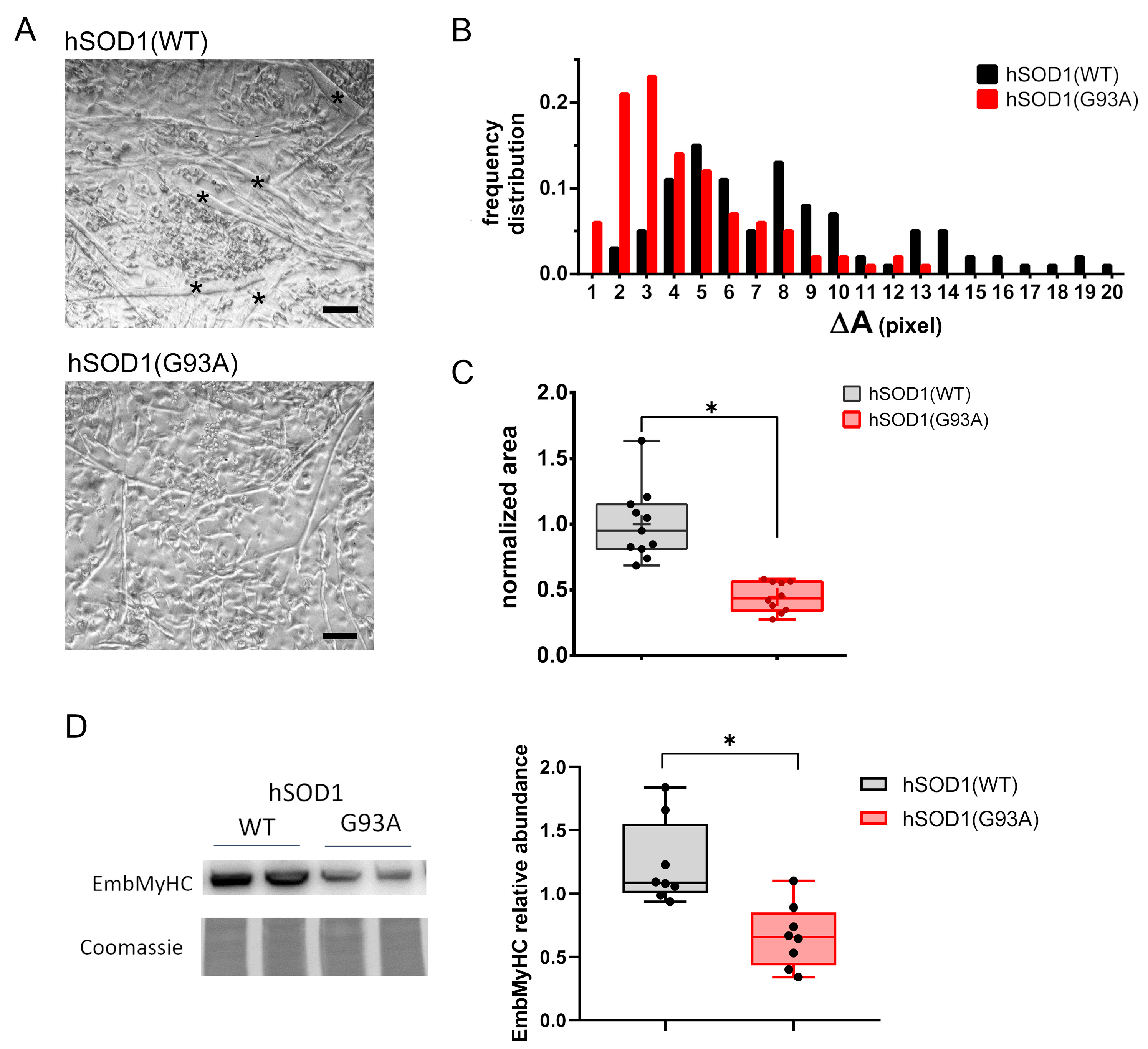
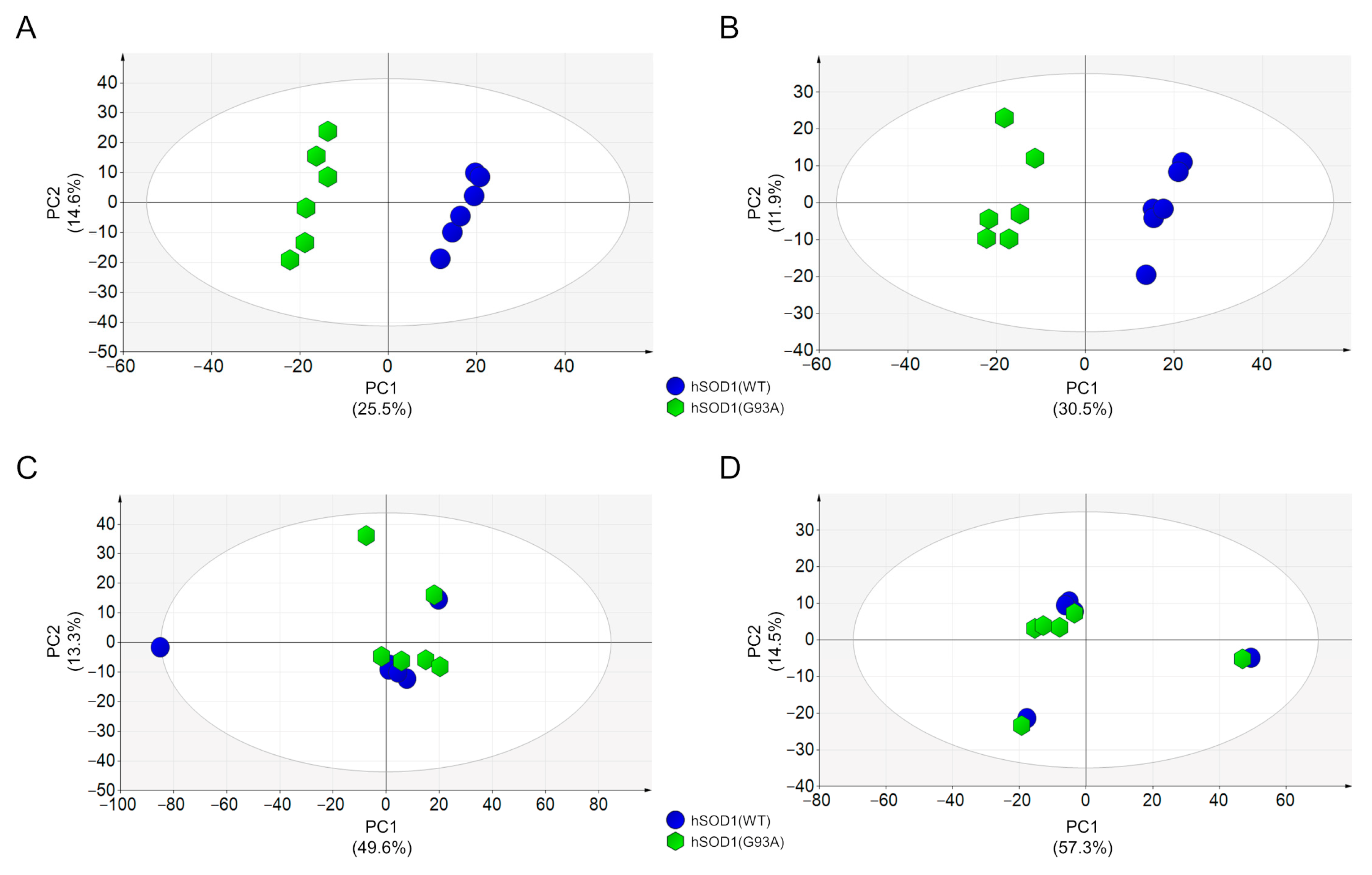
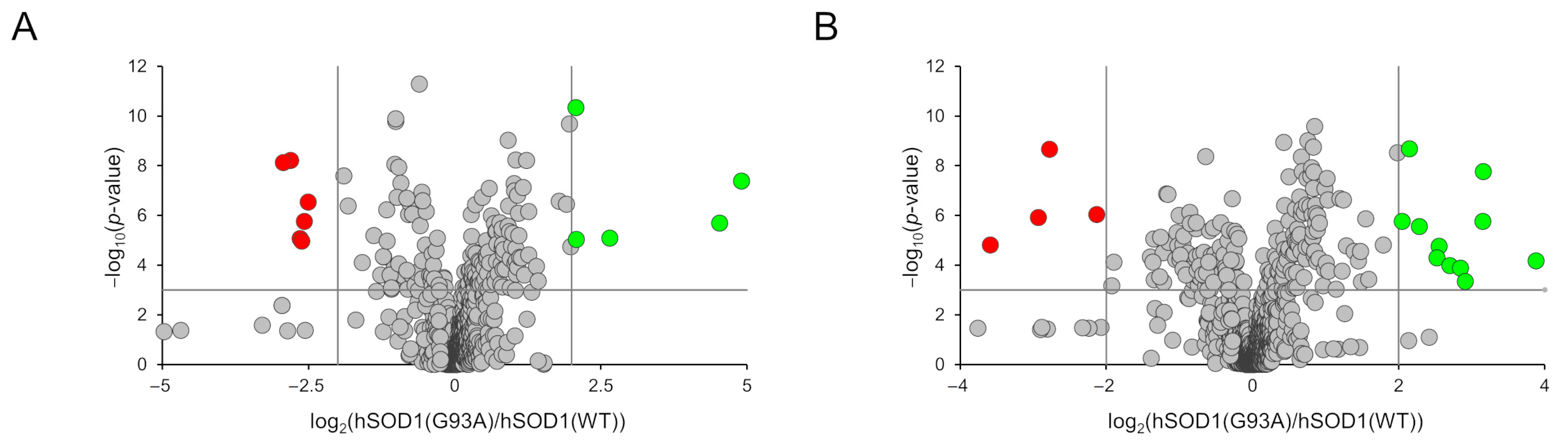
3.2. MCM Analysis by Using Liquid Chromatography–High Resolution Mass Spectrometry (LC–HRMS)
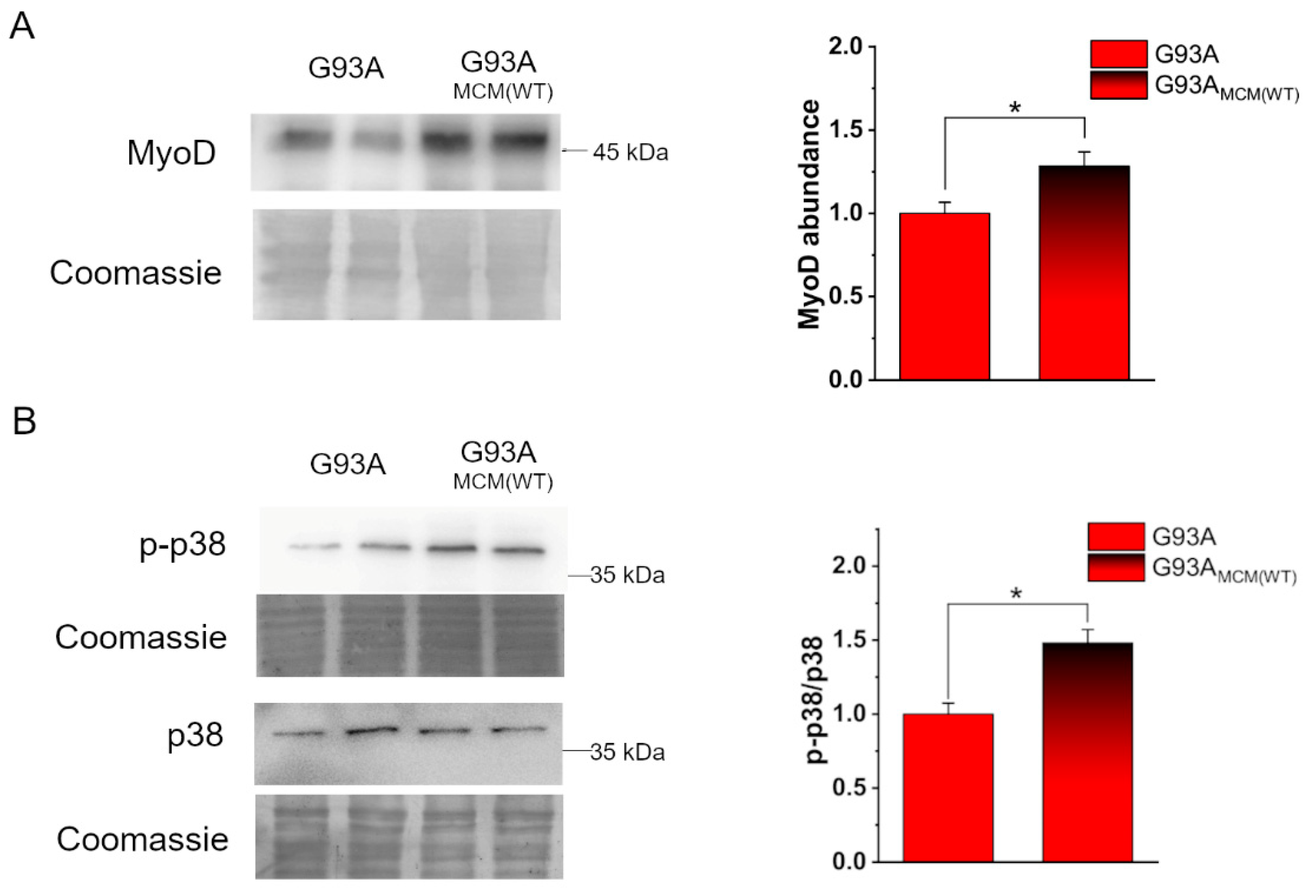
3.3. MCM from hSOD1(WT) Myocytes Rescued the Myogenic Differentiation of hSOD1(G93A) Cells
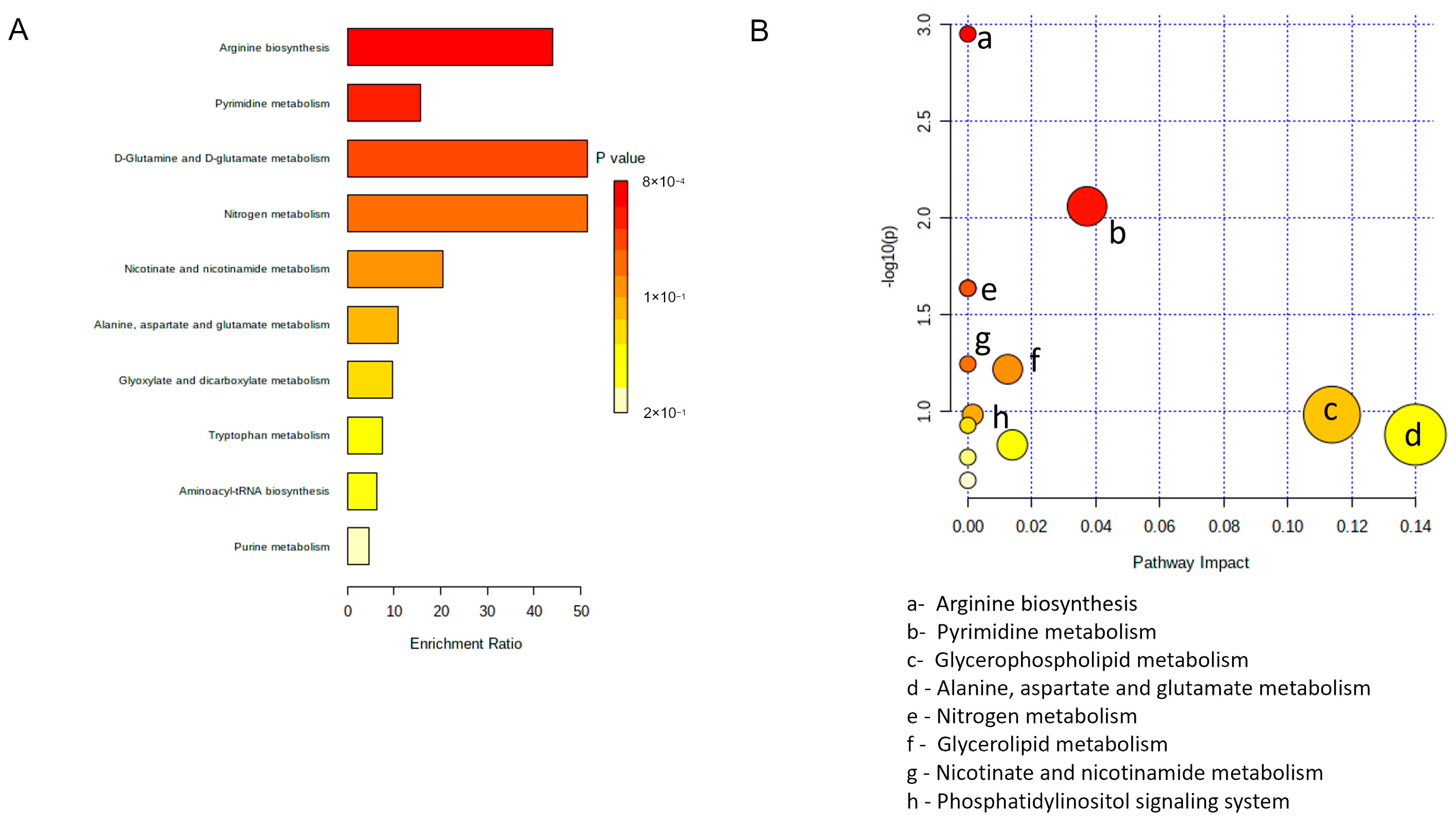
3.4. Analysis and Annotation of Altered Metabolites in hSOD1(G93A) MCM
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- DeJesus-Hernandez, M.; Mackenzie, I.R.; Boeve, B.F.; Boxer, A.L.; Baker, M.; Rutherford, N.J.; Nicholson, A.M.; Finch, N.A.; Flynn, H.; Adamson, J.; et al. Expanded GGGGCC Hexanucleotide Repeat in Noncoding Region of C9ORF72 Causes Chromosome 9p-Linked FTD and ALS. Neuron 2011, 72, 245–256. [Google Scholar] [CrossRef]
- Renton, A.E.; Majounie, E.; Waite, A.; Simón-Sánchez, J.; Rollinson, S.; Gibbs, J.R.; Schymick, J.C.; Laaksovirta, H.; van Swieten, J.C.; Myllykangas, L.; et al. A Hexanucleotide Repeat Expansion in C9ORF72 Is the Cause of Chromosome 9p21-Linked ALS-FTD. Neuron 2011, 72, 257–268. [Google Scholar] [CrossRef] [PubMed]
- Bruijn, L.I.; Becher, M.W.; Lee, M.K.; Anderson, K.L.; Jenkins, N.A.; Copeland, N.G.; Sisodia, S.S.; Rothstein, J.D.; Borchelt, D.R.; Price, D.L.; et al. ALS-Linked SOD1 Mutant G85R Mediates Damage to Astrocytes and Promotes Rapidly Progressive Disease with SOD1-Containing Inclusions. Neuron 1997, 18, 327–338. [Google Scholar] [CrossRef] [PubMed]
- Rosen, D.R.; Siddique, T.; Patterson, D.; Figlewicz, D.A.; Sapp, P.; Hentati, A.; Donaldson, D.; Goto, J.; O’Regan, J.P.; Deng, H.X.; et al. Mutations in Cu/Zn Superoxide Dismutase Gene Are Associated with Familial Amyotrophic Lateral Sclerosis. Nature 1993, 362, 59–62. [Google Scholar] [CrossRef] [PubMed]
- Gurney, M.E.; Pu, H.; Chiu, A.Y.; Dal Canto, M.C.; Polchow, C.Y.; Alexander, D.D.; Caliendo, J.; Hentati, A.; Kwon, Y.W.; Deng, H.-X.X.; et al. Motor Neuron Degeneration in Mice That Express a Human Cu,Zn Superoxide Dismutase Mutation. Science 1994, 264, 1772–1775. [Google Scholar] [CrossRef] [PubMed]
- Dal Canto, M.C.; Gurney, M.E. Development of Central Nervous System Pathology in a Murine Transgenic Model of Human Amyotrophic Lateral Sclerosis. Am. J. Pathol. 1994, 145, 1271–1279. [Google Scholar]
- Chio, A.; Traynor, B.J.; Lombardo, F.; Fimognari, M.; Calvo, A.; Ghiglione, P.; Mutani, R.; Restagno, G. Prevalence of SOD1 Mutations in the Italian ALS Population. Neurology 2008, 70, 533–537. [Google Scholar] [CrossRef]
- Nardo, G.; Trolese, M.C.; Tortarolo, M.; Vallarola, A.; Freschi, M.; Pasetto, L.; Bonetto, V.; Bendotti, C. New Insights on the Mechanisms of Disease Course Variability in ALS from Mutant SOD1 Mouse Models. Brain Pathol. 2016, 26, 237–247. [Google Scholar] [CrossRef] [PubMed]
- Da Cruz, S.; Cleveland, D.W. Understanding the Role of TDP-43 and FUS/TLS in ALS and Beyond. Curr. Opin. Neurobiol. 2011, 21, 904–919. [Google Scholar] [CrossRef]
- Sun, Z.; Diaz, Z.; Fang, X.; Hart, M.P.; Chesi, A.; Shorter, J.; Gitler, A.D. Molecular Determinants and Genetic Modifiers of Aggregation and Toxicity for the Als Disease Protein Fus/Tls. PLoS Biol. 2011, 9, e1000614. [Google Scholar] [CrossRef]
- Birsa, N.; Bentham, M.P.; Fratta, P. Cytoplasmic Functions of TDP-43 and FUS and Their Role in ALS. Semin. Cell Dev. Biol. 2020, 99, 193–201. [Google Scholar] [CrossRef] [PubMed]
- Buratti, E. Chapter One—Functional Significance of TDP-43 Mutations in Disease. In Advances in Genetics; Friedmann, T., Dunlap, J.C., Goodwin, S.F.B.T.-A., Eds.; Academic Press: Cambridge, MA, USA, 2015; Volume 91, pp. 1–53. ISBN 0065-2660. [Google Scholar]
- Ratti, A.; Buratti, E. Physiological Functions and Pathobiology of TDP-43 and FUS/TLS Proteins. J. Neurochem. 2016, 138, 95–111. [Google Scholar] [CrossRef] [PubMed]
- Buratti, E.; Baralle, F.E. The Molecular Links between TDP-43 Dysfunction and Neurodegeneration. In Advances in Genetics; Academic Press: Cambridge, MA, USA, 2009; Volume 66, pp. 1–34. [Google Scholar] [CrossRef]
- Wu, H.; Xiong, W.C.; Mei, L. To Build a Synapse: Signaling Pathways in Neuromuscular Junction Assembly. Development 2010, 137, 1017–1033. [Google Scholar] [CrossRef]
- Li, L.; Xiong, W.C.; Mei, L. Neuromuscular Junction Formation, Aging, and Disorders. Annu. Rev. Physiol. 2018, 80, 159–188. [Google Scholar] [CrossRef] [PubMed]
- Verma, S.; Khurana, S.; Vats, A.; Sahu, B.; Ganguly, N.K.; Chakraborti, P.; Gourie-Devi, M.; Taneja, V. Neuromuscular Junction Dysfunction in Amyotrophic Lateral Sclerosis. Mol. Neurobiol. 2022, 59, 1502–1527. [Google Scholar] [CrossRef] [PubMed]
- Taylor, J.P.; Brown, R.H.J.; Cleveland, D.W. Decoding ALS: From Genes to Mechanism. Nature 2016, 539, 197–206. [Google Scholar] [CrossRef] [PubMed]
- Boillée, S.; Yamanaka, K.; Lobsiger, C.S.; Copeland, N.G.; Jenkins, N.A.; Kassiotis, G.; Kollias, G.; Cleveland, D.W. Onset and Progression in Inherited ALS Determined by Motor Neurons and Microglia. Science 2006, 312, 1389–1392. [Google Scholar] [CrossRef]
- Yamanaka, K.; Boillee, S.; Roberts, E.A.; Garcia, M.L.; McAlonis-Downes, M.; Mikse, O.R.; Cleveland, D.W.; Goldstein, L.S.B. Mutant SOD1 in Cell Types Other than Motor Neurons and Oligodendrocytes Accelerates Onset of Disease in ALS Mice. Proc. Natl. Acad. Sci. USA 2008, 105, 7594–7599. [Google Scholar] [CrossRef]
- Yamanaka, K.; Komine, O. The Multi-Dimensional Roles of Astrocytes in ALS. Neurosci. Res. 2018, 126, 31–38. [Google Scholar] [CrossRef]
- Nijssen, J.; Comley, L.H.; Hedlund, E. Motor Neuron Vulnerability and Resistance in Amyotrophic Lateral Sclerosis. Acta Neuropathol. 2017, 133, 863–885. [Google Scholar] [CrossRef]
- Martineau, É.; Di Polo, A.; Vande Velde, C.; Robitaille, R. Dynamic Neuromuscular Remodeling Precedes Motor-Unit Loss in a Mouse Model of ALS. Elife 2018, 7, e41973. [Google Scholar] [CrossRef] [PubMed]
- Son, Y.J.; Thompson, W.J. Schwann Cell Processes Guide Regeneration of Peripheral Axons. Neuron 1995, 14, 125–132. [Google Scholar] [CrossRef] [PubMed]
- Lépine, S.; Castellanos-Montiel, M.J.; Durcan, T.M. TDP-43 Dysregulation and Neuromuscular Junction Disruption in Amyotrophic Lateral Sclerosis. Transl. Neurodegener. 2022, 11, 56. [Google Scholar] [CrossRef] [PubMed]
- Jensen, L.; Jørgensen, L.H.; Bech, R.D.; Frandsen, U.; Schrøder, H.D. Skeletal Muscle Remodelling as a Function of Disease Progression in Amyotrophic Lateral Sclerosis. Biomed. Res. Int. 2016, 2016, 5930621. [Google Scholar] [CrossRef] [PubMed]
- Tsitkanou, S.; Lindsay, A.; Della Gatta, P. The Role of Skeletal Muscle in Amyotrophic Lateral Sclerosis: A ‘Dying-Back’ or ‘Dying-Forward’ Phenomenon? J. Physiol. 2019, 597, 5527–5528. [Google Scholar] [CrossRef] [PubMed]
- Loeffler, J.P.; Picchiarelli, G.; Dupuis, L.; Gonzalez De Aguilar, J.L. The Role of Skeletal Muscle in Amyotrophic Lateral Sclerosis. Brain Pathol. 2016, 26, 227–236. [Google Scholar] [CrossRef] [PubMed]
- Fischer, L.R.; Culver, D.G.; Tennant, P.; Davis, A.A.; Wang, M.; Castellano-Sanchez, A.; Khan, J.; Polak, M.A.; Glass, J.D. Amyotrophic Lateral Sclerosis Is a Distal Axonopathy: Evidence in Mice and Man. Exp. Neurol. 2004, 185, 232–240. [Google Scholar] [CrossRef]
- Tsitkanou, S.; Della Gatta, P.A.; Russell, A.P. Skeletal Muscle Satellite Cells, Mitochondria, and MicroRNAs: Their Involvement in the Pathogenesis of ALS. Front. Physiol. 2016, 7, 403. [Google Scholar] [CrossRef]
- Peggion, C.; Scalcon, V.; Massimino, M.L.; Nies, K.; Lopreiato, R.; Rigobello, M.P.; Bertoli, A. SOD1 in ALS: Taking Stock in Pathogenic Mechanisms and the Role of Glial and Muscle Cells. Antioxidants 2022, 11, 614. [Google Scholar] [CrossRef]
- Pradat, P.F.; Barani, A.; Wanschitz, J.; Dubourg, O.; Lombès, A.; Bigot, A.; Mouly, V.; Bruneteau, G.; Salachas, F.; Lenglet, T.; et al. Abnormalities of Satellite Cells Function in Amyotrophic Lateral Sclerosis. Amyotroph. Lateral Scler. 2011, 12, 264–271. [Google Scholar] [CrossRef]
- Scaramozza, A.; Marchese, V.; Papa, V.; Salaroli, R.; Sorarù, G.; Angelini, C.; Cenacchi, G. Skeletal Muscle Satellite Cells in Amyotrophic Lateral Sclerosis. Ultrastruct. Pathol. 2014, 38, 295–302. [Google Scholar] [CrossRef]
- Dobrowolny, G.; Aucello, M.; Musarò, A. Muscle Atrophy Induced by SOD1G93A Expression Does Not Involve the Activation of Caspase in the Absence of Denervation. Skelet. Muscle 2011, 1, 3. [Google Scholar] [CrossRef] [PubMed]
- Dobrowolny, G.; Bernardini, C.; Martini, M.; Baranzini, M.; Barba, M.; Musarò, A. Muscle Expression of SOD1G93A Modulates MicroRNA and MRNA Transcription Pattern Associated with the Myelination Process in the Spinal Cord of Transgenic Mice. Front. Cell Neurosci. 2015, 9, 463. [Google Scholar] [CrossRef] [PubMed]
- Dobrowolny, G.; Giacinti, C.; Pelosi, L.; Nicoletti, C.; Winn, N.; Barberi, L.; Molinaro, M.; Rosenthal, N.; Musarò, A. Muscle Expression of a Local Igf-1 Isoform Protects Motor Neurons in an ALS Mouse Model. J. Cell Biol. 2005, 168, 193–199. [Google Scholar] [CrossRef] [PubMed]
- Dobrowolny, G.; Aucello, M.; Rizzuto, E.; Beccafico, S.; Mammucari, C.; Bonconpagni, S.; Belia, S.; Wannenes, F.; Nicoletti, C.; Del Prete, Z.; et al. Skeletal Muscle Is a Primary Target of SOD1G93A-Mediated Toxicity. Cell Metab. 2008, 8, 425–436. [Google Scholar] [CrossRef]
- Martin, L.J.; Wong, M. Skeletal Muscle-Restricted Expression of Human SOD1 in Transgenic Mice Causes a Fatal ALS-Like Syndrome. Front. Neurol. 2020, 11, 592851. [Google Scholar] [CrossRef]
- Manzano, R.; Toivonen, J.M.; Calvo, A.C.; Oliván, S.; Zaragoza, P.; Rodellar, C.; Montarras, D.; Osta, R. Altered in Vitro Proliferation of Mouse SOD1-G93A Skeletal Muscle Satellite Cells. Neurodegener. Dis. 2013, 11, 153–164. [Google Scholar] [CrossRef]
- Martini, M.; Dobrowolny, G.; Aucello, M.; Musarò, A. Postmitotic Expression of SOD1G93A Gene Affects the Identity of Myogenic Cells and Inhibits Myoblasts Differentiation. Mediat. Inflamm. 2015, 2015, 537853. [Google Scholar] [CrossRef]
- Das, D.K.; Graham, Z.A.; Cardozo, C.P. Myokines in Skeletal Muscle Physiology and Metabolism: Recent Advances and Future Perspectives. Acta Physiol. 2020, 228, e13367. [Google Scholar] [CrossRef]
- Trovato, E.; Di Felice, V.; Barone, R. Extracellular Vesicles: Delivery Vehicles of Myokines. Front. Physiol. 2019, 10, 522. [Google Scholar] [CrossRef]
- Chikazawa, M.; Shimizu, M.; Yamauchi, Y.; Sato, R. Bridging Molecules Are Secreted from the Skeletal Muscle and Potentially Regulate Muscle Differentiation. Biochem. Biophys. Res. Commun. 2020, 522, 113–120. [Google Scholar] [CrossRef] [PubMed]
- Henningsen, J.; Rigbolt, K.T.G.; Blagoev, B.; Pedersen, B.K.; Kratchmarova, I. Dynamics of the Skeletal Muscle Secretome during Myoblast Differentiation. Mol. Cell. Proteom. 2010, 9, 2482–2496. [Google Scholar] [CrossRef] [PubMed]
- Pedersen, B.K.; Febbraio, M.A. Muscles, Exercise and Obesity: Skeletal Muscle as a Secretory Organ. Nat. Rev. Endocrinol. 2012, 8, 457–465. [Google Scholar] [CrossRef] [PubMed]
- Pedersen, B.K.; Febbraio, M.A. Muscle as an Endocrine Organ: Focus on Muscle-Derived Interleukin-6. Physiol. Rev. 2008, 88, 1379–1406. [Google Scholar] [CrossRef] [PubMed]
- Florin, A.; Lambert, C.; Sanchez, C.; Zappia, J.; Durieux, N.; Tieppo, A.M.; Mobasheri, A.; Henrotin, Y. The Secretome of Skeletal Muscle Cells: A Systematic Review. Osteoarthr. Cartil. Open 2020, 2, 100019. [Google Scholar] [CrossRef]
- Aguer, C.; Loro, E.; Di Raimondo, D. Editorial: The Role of the Muscle Secretome in Health and Disease. Front. Physiol. 2020, 11, 1101. [Google Scholar] [CrossRef]
- Lin, W.; Song, H.; Shen, J.; Wang, J.; Yang, Y.; Yang, Y.; Cao, J.; Xue, L.; Zhao, F.; Xiao, T.; et al. Functional Role of Skeletal Muscle-Derived Interleukin-6 and Its Effects on Lipid Metabolism. Front. Physiol. 2023, 14, 1110926. [Google Scholar] [CrossRef]
- Eckel, J. Myokines in Metabolic Homeostasis and Diabetes. Diabetologia 2019, 62, 1523–1528. [Google Scholar] [CrossRef]
- Maimon, R.; Ankol, L.; Weissova, R.; Tank, E.; Pery, T.G.; Opatowsky, Y.; Barmada, S.; Balastik, M.; Perlson, E. Sema3A Facil-itates a Retrograde Death Signal via CRMP4-Dynein Complex Formation in ALS Motor Axons. bioRxiv 2019. [Google Scholar] [CrossRef]
- Anakor, E.; Duddy, W.J.; Duguez, S. The Cellular and Molecular Signature of ALS in Muscle. J. Pers. Med. 2022, 12, 1868. [Google Scholar] [CrossRef]
- Chal, J.; Pourquié, O. Making Muscle: Skeletal Myogenesis in Vivo and in Vitro. Development 2017, 144, 2104–2122. [Google Scholar] [CrossRef] [PubMed]
- Badu-Mensah, A.; Guo, X.; McAleer, C.W.; Rumsey, J.W.; Hickman, J.J. Functional Skeletal Muscle Model Derived from SOD1-Mutant ALS Patient IPSCS Recapitulates Hallmarks of Disease Progression. Sci. Rep. 2020, 10, 14302. [Google Scholar] [CrossRef] [PubMed]
- Norante, R.P.; Massimino, M.L.; Lorenzon, P.; De Mario, A.; Peggion, C.; Vicario, M.; Albiero, M.; Sorgato, M.C.; Lopreiato, R.; Bertoli, A. Generation and Validation of Novel Adeno-Associated Viral Vectors for the Analysis of Ca2+ Homeostasis in Motor Neurons. Sci. Rep. 2017, 7, 6521. [Google Scholar] [CrossRef] [PubMed]
- Massimino, M.L.; Ferrari, J.; Sorgato, M.C.; Bertoli, A. Heterogeneous PrPC Metabolism in Skeletal Muscle Cells. FEBS Lett. 2006, 580, 878–884. [Google Scholar] [CrossRef]
- Schneider, C.A.; Rasband, W.S.; Eliceiri, K.W. NIH Image to ImageJ: 25 Years of Image Analysis. Nat. Methods 2012, 9, 671–675. [Google Scholar] [CrossRef] [PubMed]
- Colella, A.D.; Chegenii, N.; Tea, M.N.; Gibbins, I.L.; Williams, K.A.; Chataway, T.K. Comparison of Stain-Free Gels with Traditional Immunoblot Loading Control Methodology. Anal. Biochem. 2012, 430, 108–110. [Google Scholar] [CrossRef]
- Stella, R.; Bonadio, R.S.; Cagnin, S.; Massimino, M.L.; Bertoli, A.; Peggion, C. Perturbations of the Proteome and of Secreted Metabolites in Primary Astrocytes from the HSOD1(G93A) ALS Mouse Model. Int. J. Mol. Sci. 2021, 22, 7028. [Google Scholar] [CrossRef]
- Xia, J.; Sinelnikov, I.V.; Han, B.; Wishart, D.S. MetaboAnalyst 3.0—Making Metabolomics More Meaningful. Nucleic Acids Res. 2015, 43, W251–W257. [Google Scholar] [CrossRef]
- Weintraub, H.; Davis, R.; Tapscott, S.; Thayer, M.; Krause, M.; Benezra, R.; Blackwell, T.K.; Turner, D.; Rupp, R.; Hollenberg, S.; et al. The MyoD Gene Family: Nodal Point during Specification of the Muscle Cell Lineage. Science 1991, 251, 761–766. [Google Scholar] [CrossRef]
- Berkes, C.A.; Tapscott, S.J. MyoD and the Transcriptional Control of Myogenesis. Semin. Cell Dev. Biol. 2005, 16, 585–595. [Google Scholar] [CrossRef]
- Brennan, C.M.; Emerson, C.P.; Owens, J.; Christoforou, N. P38 MAPKs—Roles in Skeletal Muscle Physiology, Disease Mech-anisms, and as Potential Therapeutic Targets. JCI Insight 2021, 6, e149915. [Google Scholar] [CrossRef] [PubMed]
- Gil-de-la-Fuente, A.; Godzien, J.; Saugar, S.; Garcia-Carmona, R.; Badran, H.; Wishart, D.S.; Barbas, C.; Otero, A. CEU Mass Mediator 3.0: A Metabolite Annotation Tool. J. Proteome Res. 2019, 18, 797–802. [Google Scholar] [CrossRef] [PubMed]
- Horai, H.; Arita, M.; Kanaya, S.; Nihei, Y.; Ikeda, T.; Suwa, K.; Ojima, Y.; Tanaka, K.; Tanaka, S.; Aoshima, K.; et al. MassBank: A Public Repository for Sharing Mass Spectral Data for Life Sciences. J. Mass Spectrom. 2010, 45, 703–714. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.; Pang, Z.; Xia, J. Comprehensive Investigation of Pathway Enrichment Methods for Functional Interpretation of LC–MS Global Metabolomics Data. Brief. Bioinform. 2023, 24, bbac553. [Google Scholar] [CrossRef] [PubMed]
- Manzano, R.; Toivonen, J.M.; Moreno-Martínez, L.; de la Torre, M.; Moreno-García, L.; López-Royo, T.; Molina, N.; Zaragoza, P.; Calvo, A.C.; Osta, R. What skeletal muscle has to say in amyotrophic lateral sclerosis: Implications for therapy. Br. J. Pharmacol. 2021, 17, 1279–1297. [Google Scholar] [CrossRef] [PubMed]
- Chargé, S.B.P.; Rudnicki, M.A. Cellular and Molecular Regulation of Muscle Regeneration. Physiol. Rev. 2004, 84, 209–238. [Google Scholar] [CrossRef] [PubMed]
- Quessada, C.; Bouscary, A.; René, F.; Valle, C.; Ferri, A.; Ngo, S.T.; Loeffler, J.-P. Skeletal Muscle Metabolism: Origin or Prognostic Factor for Amyotrophic Lateral Sclerosis (ALS) Development? Cells 2021, 10, 1449. [Google Scholar] [CrossRef]
- Meroni, M.; Crippa, V.; Cristofani, R.; Rusmini, P.; Cicardi, M.E.; Messi, E.; Piccolella, M.; Tedesco, B.; Ferrari, V.; Sorarù, G.; et al. Transforming Growth Factor Beta 1 Signaling Is Altered in the Spinal Cord and Muscle of Amyotrophic Lateral Sclerosis Mice and Patients. Neurobiol. Aging 2019, 82, 48–59. [Google Scholar] [CrossRef]
- Dobrowolny, G.; Aucello, M.; Molinaro, M.; Musarò, A. Local Expression of MIgf-1 Modulates Ubiquitin, Caspase and CDK5 Expression in Skeletal Muscle of an ALS Mouse Model. Neurol. Res. 2008, 30, 131–136. [Google Scholar] [CrossRef]
- Van Dyke, J.M.; Smit-Oistad, I.M.; Macrander, C.; Krakora, D.; Meyer, M.G.; Suzuki, M. Macrophage-Mediated Inflammation and Glial Response in the Skeletal Muscle of a Rat Model of Familial Amyotrophic Lateral Sclerosis (ALS). Exp. Neurol. 2016, 277, 275–282. [Google Scholar] [CrossRef]
- Le Gall, L.; Duddy, W.J.; Martinat, C.; Mariot, V.; Connolly, O.; Milla, V.; Anakor, E.; Ouandaogo, Z.G.; Millecamps, S.; Lainé, J.; et al. Muscle Cells of Sporadic Amyotrophic Lateral Sclerosis Patients Secrete Neurotoxic Vesicles. J. Cachex Sarcopenia Muscle 2022, 13, 1385–1402. [Google Scholar] [CrossRef]
- Cruzat, V.F. Glutamine and Skeletal Muscle. In Nutrition and Skeletal Muscle; Elsevier: Amsterdam, The Netherlands, 2019; pp. 299–313. [Google Scholar]
- Tedesco, B.; Ferrari, V.; Cozzi, M.; Chierichetti, M.; Casarotto, E.; Pramaggiore, P.; Mina, F.; Galbiati, M.; Rusmini, P.; Crippa, V.; et al. The Role of Small Heat Shock Proteins in Protein Misfolding Associated Motoneuron Diseases. Int. J. Mol. Sci. 2022, 23, 11759. [Google Scholar] [CrossRef] [PubMed]
- Girven, M.; Dugdale, H.F.; Owens, D.J.; Hughes, D.C.; Stewart, C.E.; Sharples, A.P. l-glutamine Improves Skeletal Muscle Cell Differentiation and Prevents Myotube Atrophy After Cytokine (TNF-α) Stress Via Reduced P38 MAPK Signal Transduction. J. Cell. Physiol. 2016, 231, 2720–2732. [Google Scholar] [CrossRef] [PubMed]
- Dobrowolny, G.; Lepore, E.; Martini, M.; Barberi, L.; Nunn, A.; Scicchitano, B.M.; Musarò, A. Metabolic Changes Associated with Muscle Expression of SOD1G93A. Front. Physiol. 2018, 9, 831. [Google Scholar] [CrossRef]
- Corbett, A.J.; Griggs, R.C.; Moxley, R.T. Skeletal Muscle Catabolism in Amyotrophic Lateral Sclerosis and Chronic Spinal Muscular Atrophy. Neurology 1982, 32, 550–552. [Google Scholar] [CrossRef] [PubMed]
- Niebroj-Dobosz, I.; Janik, P.; Sokołowska, B.; Kwiecinski, H. Matrix Metalloproteinases and Their Tissue Inhibitors in Serum and Cerebrospinal Fluid of Patients with Amyotrophic Lateral Sclerosis. Eur. J. Neurol. 2010, 17, 226–231. [Google Scholar] [CrossRef] [PubMed]
- Schoser, B.G.; Blottner, D. Matrix Metalloproteinases MMP-2, MMP-7 and MMP-9 in Denervated Human Muscle. Neuroreport 1999, 10, 2795–2797. [Google Scholar] [CrossRef] [PubMed]
- Geraldo, L.H.M.; de Sampaio Spohr, T.C.L.; do Amaral, R.F.; da Fonseca, A.C.C.; Garcia, C.; de Almeida Mendes, F.; Freitas, C.; Fabio dosSantos, M.; Lima, F.R.S. Role of Lysophosphatidic Acid and Its Receptors in Health and Disease: Novel Therapeutic Strategies. Signal Transduct. Target. Ther. 2021, 6, 45. [Google Scholar] [CrossRef]
- Ray, R.; Sinha, S.; Aidinis, V.; Rai, V. Atx Regulates Skeletal Muscle Regeneration via LPAR1 and Promotes Hypertrophy. Cell Rep. 2021, 34, 108809. [Google Scholar] [CrossRef]
- Crofford, L.J. Prostaglandin biology. Gastroenterol. Clin. N. Am. 2001, 30, 863–876. [Google Scholar] [CrossRef]
- McLennan, I. E and Fα Series Prostaglandins in Developing Muscles. Prostaglandins Leukot. Essent. Fat. Acids 1991, 43, 77–82. [Google Scholar] [CrossRef] [PubMed]
- Murray, J.; Whitson, R.H.; Itakura, K. Reduced Prostaglandin I 2 Signaling in Arid5b−/− Primary Skeletal Muscle Cells Attenuates Myogenesis. FASEB J. 2018, 32, 1868–1879. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.Z.; Jemiolo, B.; Lavin, K.M.; Lester, B.E.; Trappe, S.W.; Trappe, T.A.; Ratchford, S.M.; Perkins, R.K.; Caligiuri, S.P.B.; Parikh, M.; et al. Prostaglandin E2/Cyclooxygenase Pathway in Human Skeletal Muscle: Influence of Muscle Fiber Type and Age. J. Appl. Physiol. 2016, 120, 546–551. [Google Scholar] [CrossRef] [PubMed]
- Ho, A.T.V.; Palla, A.R.; Blake, M.R.; Yucel, N.D.; Wang, Y.X.; Magnusson, K.E.G.; Holbrook, C.A.; Kraft, P.E.; Delp, S.L.; Blau, H.M. Prostaglandin E2 Is Essential for Efficacious Skeletal Muscle Stem-Cell Function, Augmenting Regeneration and Strength. Proc. Natl. Acad. Sci. USA 2017, 114, 6675–6684. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| m/z | Retention Time (min) | Adduct | Formula | Polarity | Annotation | Ratio [hSOD1(G93A)/hSOD1(WT)] | p-Value |
|---|---|---|---|---|---|---|---|
| 81.0452 | 9.31 | [M+H]+ | C4H4N2 | positive | Pyrimidine | 0.17 | 1.70 × 10−6 |
| 95.0607 | 7.62 | - | - | positive | - | 0.14 | 6.20 × 10−9 |
| 121.0648 | 7.09 | [M+H]+ | C8H8O | positive | 4-Hydroxystyrene | 0.13 | 7.44 × 10−9 |
| 124.0394 | 4.88 | [M+H]+ | C6H5NO2 | positive | Nicotinic acid | 4.20 | 4.61 × 10−11 |
| 127.0503 | 9.31 | [M+H]+ | C5H6N2O2 | positive | Thymine | 0.18 | 2.87 × 10−7 |
| 129.0659 | 4.74 | [M+H]+ | C5H10N2O3 | positive | L-Glutamine | 0.16 | 1.08 × 10−5 |
| 157.0970 | 7.69 | [M-H2O+H]+ | C7H14N2O3 | positive | N2-Acetyl-L-ornithine | 0.16 | 8.57 × 10−6 |
| 160.0756 | 7.57 | [M+H]+ | C10H9NO | positive | Indoleacetaldehyde | 30.04 | 4.15 × 10−8 |
| 160.0756 | 11.64 | [M+H]+ | C10H9NO | positive | Indoleacetaldehyde | 23.15 | 2.06 × 10−6 |
| 308.1925 | 7.59 | [M+NH4]+ | C11H22N4O5 | positive | Ser Lys Gly | 6.29 | 8.23 × 10−6 |
| 392.1812 | 5.33 | [M-H2O+H]+ | C19H27N3O7 | positive | Glu Tyr Val/Tyr Asp Ile | 4.24 | 9.10 × 10−6 |
| 81.0457 | 9.29 | [M-H]− | C4H6N2 | negative | 4-Methylimidazole | 0.23 | 9.17 × 10−7 |
| 161.0123 | 9.31 | [M+Cl]− | C5H6N2O2 | negative | Thymine | 0.08 | 1.55 × 10−5 |
| 331.0784 | 4.99 | - | - | negative | - | 4.87 | 2.83 × 10−6 |
| 354.2032 | 4.75 | [M-H2O-H]− | C17H31N3O6 | negative | Glu Leu Ile | 6.49 | 1.04 × 10−4 |
| 374.1719 | 4.90 | [M-H2O-H]− | C19H27N3O6 | negative | Phe Asp Leu/Pro Pro Tyr | 8.89 | 1.70 × 10−6 |
| 374.1725 | 1.49 | [M-H]− | C19H25N3O5 | negative | Phe Asp Leu/Pro Pro Tyr | 4.43 | 2.04 × 10−9 |
| 390.1800 | 4.76 | [M-H2O-H]− | C23H27N3O4 | negative | Phe Pro Phe | 7.53 | 4.43 × 10−4 |
| 391.2260 | 1.45 | [M+Cl]− | C20H36O5 | negative | PGF1α | 0.13 | 1.22 × 10−6 |
| 396.1000 | 6.00 | [M+Cl]− | C14H23N3O6S | negative | Met Asp Pro | 5.76 | 5.08 × 10−5 |
| 410.1486 | 4.89 | [M+Cl]− | C19H25N3O5 | negative | Pro Pro Tyr | 14.70 | 6.57 × 10−5 |
| 419.2568 | 1.45 | [M−H2O−H]− | C21H43O7P | negative | LPA(0:0/18:0) | 0.15 | 2.19 × 10−9 |
| 426.1439 | 5.33 | [M+Cl]− | C19H25N3O6 | negative | Pro Phe Glu | 5.87 | 1.76 × 10−5 |
| 551.1812 | 4.89 | [M-H2O-H]− | C24H34N4O10S | negative | Met Glu Tyr Glu | 4.14 | 1.75 × 10−6 |
| 787.8521 | 7.69 | - | - | negative | - | 7.21 | 1.29 × 10−4 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Stella, R.; Bonadio, R.S.; Cagnin, S.; Andreotti, R.; Massimino, M.L.; Bertoli, A.; Peggion, C. Secreted Metabolome of ALS-Related hSOD1(G93A) Primary Cultures of Myocytes and Implications for Myogenesis. Cells 2023, 12, 2751. https://doi.org/10.3390/cells12232751
Stella R, Bonadio RS, Cagnin S, Andreotti R, Massimino ML, Bertoli A, Peggion C. Secreted Metabolome of ALS-Related hSOD1(G93A) Primary Cultures of Myocytes and Implications for Myogenesis. Cells. 2023; 12(23):2751. https://doi.org/10.3390/cells12232751
Chicago/Turabian StyleStella, Roberto, Raphael Severino Bonadio, Stefano Cagnin, Roberta Andreotti, Maria Lina Massimino, Alessandro Bertoli, and Caterina Peggion. 2023. "Secreted Metabolome of ALS-Related hSOD1(G93A) Primary Cultures of Myocytes and Implications for Myogenesis" Cells 12, no. 23: 2751. https://doi.org/10.3390/cells12232751
APA StyleStella, R., Bonadio, R. S., Cagnin, S., Andreotti, R., Massimino, M. L., Bertoli, A., & Peggion, C. (2023). Secreted Metabolome of ALS-Related hSOD1(G93A) Primary Cultures of Myocytes and Implications for Myogenesis. Cells, 12(23), 2751. https://doi.org/10.3390/cells12232751