Therapeutic Monoclonal Antibodies against Cancer: Present and Future



Abstract
:
1. Introduction
2. An Example: HER2 Targeting
3. Future
3.1. Choice of the Tumor Type
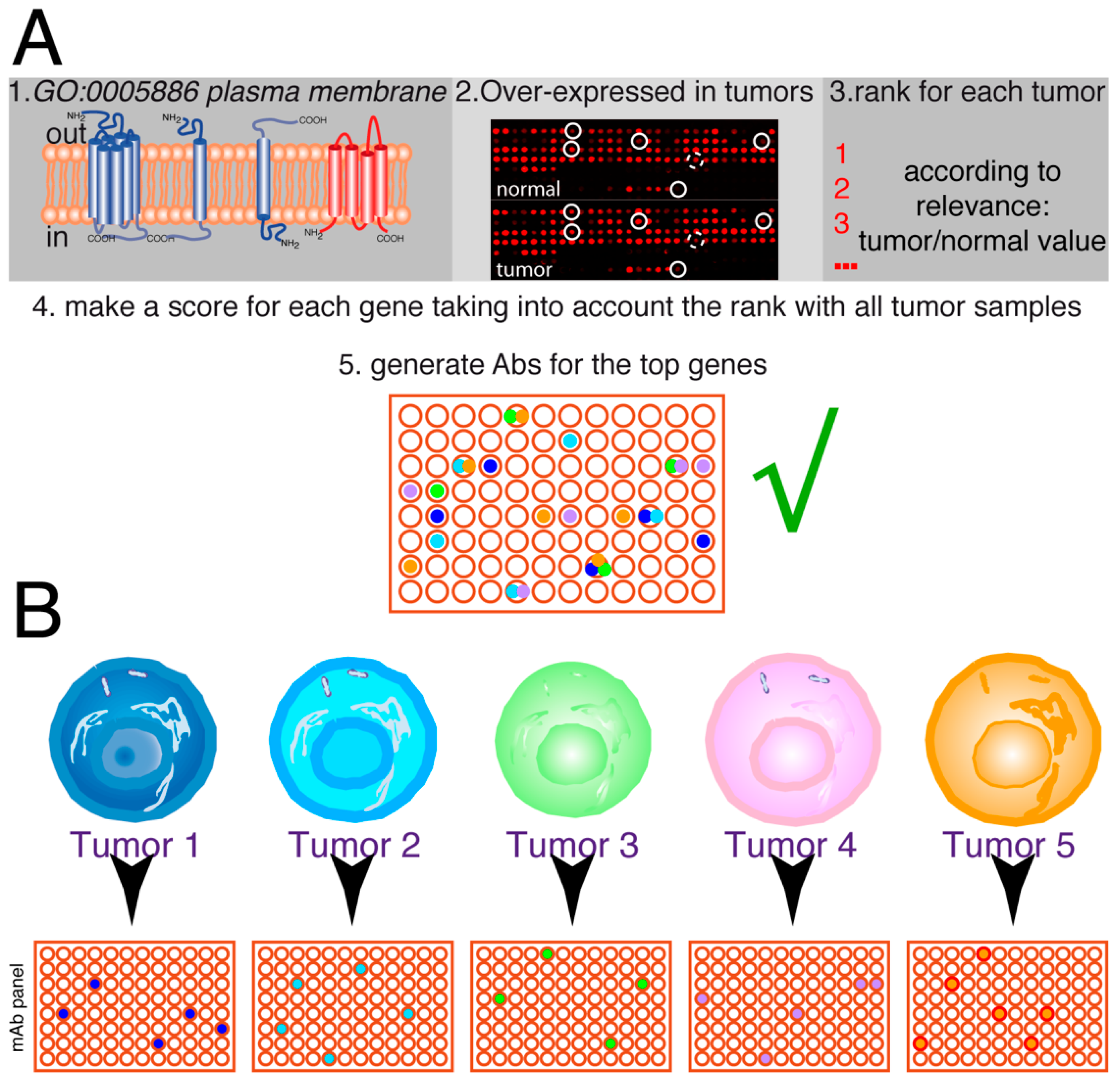
3.2. Analysis of Cell Surface Proteins
3.3. Choice of Target Genes
3.4. Antibody Generation Strategies
3.5. Antibody Screening
3.6. Challenges
4. The Goal
5. Concluding Remarks
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Köhler, G.; Milstein, C. Continuous cultures of fused cells secreting antibody of predefined specificity. Nature 1975, 256, 495–497. [Google Scholar] [CrossRef] [PubMed]
- Mach, J.P.; Carrel, S.; Forni, M.; Ritschard, J.; Donath, A.; Alberto, P. Tumor localization of radiolabeled antibodies against carcinoembryonic antigen in patients with carcinoma: A critical evaluation. N. Engl. J. Med. 1980, 303, 5–10. [Google Scholar] [CrossRef]
- Buchegger, F.; Vacca, A.; Carrel, S.; Schreyer, M.; Mach, J.P. Radioimmunotherapy of human colon carcinoma by 131I-labelled monoclonal anti-CEA antibodies in a nude mouse model. Int. J. Cancer 1988, 41, 127–134. [Google Scholar] [CrossRef]
- Hanack, K.; Messerschmidt, K.; Listek, M. Antibodies and Selection of Monoclonal Antibodies. Adv. Exp. Med. Biol. 2016, 917, 11–22. [Google Scholar] [CrossRef]
- Corraliza-Gorjón, I.; Somovilla-Crespo, B.; Santamaria, S.; Garcia-Sanz, J.A.; Kremer, L. New Strategies Using Antibody Combinations to Increase Cancer Treatment Effectiveness. Front. Immunol. 2017, 8, 1804. [Google Scholar] [CrossRef]
- Argollo, M.; Kotze, P.G.; Kakkadasam, P.; D’Haens, G. Optimizing biologic therapy in IBD: How essential is therapeutic drug monitoring? Nat. Rev. Gastroenterol. Hepatol. 2020, 17, 702–710. [Google Scholar] [CrossRef]
- Maleki, A.; Manhapra, A.; Asgari, S.; Chang, P.Y.; Foster, C.S.; Anesi, S.D. Tocilizumab Employment in the Treatment of Resistant Juvenile Idiopathic Arthritis Associated Uveitis. Ocul. Immunol. Inflamm. 2021, 29, 14–20. [Google Scholar] [CrossRef]
- Lemiere, C.; Taille, C.; Lee, J.K.; Smith, S.G.; Mallett, S.; Albers, F.C.; Bradford, E.S.; Yancey, S.W.; Liu, M.C. Impact of baseline clinical asthma characteristics on the response to mepolizumab: A post hoc meta-analysis of two Phase III trials. Respir. Res. 2021, 22, 184. [Google Scholar] [CrossRef]
- Edvinsson, L. CGRP Antibodies as Prophylaxis in Migraine. Cell 2018, 175, 1719. [Google Scholar] [CrossRef]
- Reichert, J.M. Antibodies to watch in 2010. MAbs 2010, 2, 84–100. [Google Scholar] [CrossRef] [PubMed]
- Reichert, J.M. Which are the antibodies to watch in 2012? MAbs 2012, 4, 1–3. [Google Scholar] [CrossRef]
- Reichert, J.M. Which are the antibodies to watch in 2013? MAbs 2013, 5, 1–4. [Google Scholar] [CrossRef]
- Reichert, J.M. Antibodies to watch in 2013: Mid-year update. MAbs 2013, 5, 513–517. [Google Scholar] [CrossRef]
- Reichert, J.M. Antibodies to watch in 2014. MAbs 2014, 6, 5–14. [Google Scholar] [CrossRef]
- Reichert, J.M. Antibodies to watch in 2014: Mid-year update. MAbs 2014, 6, 799–802. [Google Scholar] [CrossRef]
- Reichert, J.M. Antibodies to watch in 2015. MAbs 2015, 7, 1–8. [Google Scholar] [CrossRef]
- Reichert, J.M. Antibodies to watch in 2016. MAbs 2016, 8, 197–204. [Google Scholar] [CrossRef]
- Reichert, J.M. Antibodies to watch in 2017. MAbs 2017, 9, 167–181. [Google Scholar] [CrossRef]
- Kaplon, H.; Reichert, J.M. Antibodies to watch in 2018. MAbs 2018, 10, 183–203. [Google Scholar] [CrossRef] [PubMed]
- Kaplon, H.; Reichert, J.M. Antibodies to watch in 2019. MAbs 2019, 11, 219–238. [Google Scholar] [CrossRef] [PubMed]
- Kaplon, H.; Muralidharan, M.; Schneider, Z.; Reichert, J.M. Antibodies to watch in 2020. MAbs 2020, 12, 1703531. [Google Scholar] [CrossRef] [PubMed]
- Kaplon, H.; Reichert, J.M. Antibodies to watch in 2021. MAbs 2021, 13, 1860476. [Google Scholar] [CrossRef] [PubMed]
- Kaplon, H.; Chenoweth, A.; Crescioli, S.; Reichert, J.M. Antibodies to watch in 2022. MAbs 2022, 14, 2014296. [Google Scholar] [CrossRef]
- Kaplon, H.; Crescioli, S.; Chenoweth, A.; Visweswaraiah, J.; Reichert, J.M. Antibodies to watch in 2023. MAbs 2023, 15, 2153410. [Google Scholar] [CrossRef] [PubMed]
- The Antibody Society. Therapeutic Monoclonal Antibodies Approved or in Regulatory Review. Available online: www.antibodysociety.org/antibody-therapeutics-product-data (accessed on 30 October 2023).
- Zahavi, D.; Weiner, L. Monoclonal Antibodies in Cancer Therapy. Antibodies 2020, 9, 34. [Google Scholar] [CrossRef] [PubMed]
- Jin, S.; Sun, Y.; Liang, X.; Gu, X.; Ning, J.; Xu, Y.; Chen, S.; Pan, L. Emerging new therapeutic antibody derivatives for cancer treatment. Signal Transduct. Target. Ther. 2022, 7, 39. [Google Scholar] [CrossRef] [PubMed]
- Li, D.-K.; Wang, W. Characteristics and clinical trial results of agonistic anti-CD40 antibodies in the treatment of malignancies (Review). Oncol. Lett. 2020, 20, 176. [Google Scholar] [CrossRef]
- Larbouret, C.; Gros, L.; Pelegrin, A.; Chardes, T. Improving Biologics’ Effectiveness in Clinical Oncology: From the Combination of Two Monoclonal Antibodies to Oligoclonal Antibody Mixtures. Cancers 2021, 13, 4620. [Google Scholar] [CrossRef]
- Vela, M.; Aris, M.; Llorente, M.; Garcia-Sanz, J.A.; Kremer, L. Chemokine receptor-specific antibodies in cancer immunotherapy: Achievements and challenges. Front. Immunol. 2015, 6, 12. [Google Scholar] [CrossRef]
- Redman, J.M.; Hill, E.M.; AlDeghaither, D.; Weiner, L.M. Mechanisms of action of therapeutic antibodies for cancer. Mol. Immunol. 2015, 67, 28–45. [Google Scholar] [CrossRef]
- Xu-Monette, Z.Y.; Zhang, M.; Li, J.; Young, K.H. PD-1/PD-L1 Blockade: Have We Found the Key to Unleash the Antitumor Immune Response? Front. Immunol. 2017, 8, 1597. [Google Scholar] [CrossRef] [PubMed]
- Chiorean, E.G.; Sweeney, C.; Youssoufian, H.; Qin, A.; Dontabhaktuni, A.; Loizos, N.; Nippgen, J.; Amato, R. A phase I study of olaratumab, an anti-platelet-derived growth factor receptor alpha (PDGFRalpha) monoclonal antibody, in patients with advanced solid tumors. Cancer Chemother. Pharmacol. 2014, 73, 595–604. [Google Scholar] [CrossRef] [PubMed]
- Deshpande, H.A.; Cecchini, M.; Ni Choileain, S.; Jones, R. Olaratumab for the treatment of soft tissue sarcoma. Drugs Today 2017, 53, 247–255. [Google Scholar] [CrossRef] [PubMed]
- Joglekar-Javadekar, M.; Van Laere, S.; Bourne, M.; Moalwi, M.; Finetti, P.; Vermeulen, P.B.; Birnbaum, D.; Dirix, L.Y.; Ueno, N.; Carter, M.; et al. Characterization and Targeting of Platelet-Derived Growth Factor Receptor alpha (PDGFRA) in Inflammatory Breast Cancer (IBC). Neoplasia 2017, 19, 564–573. [Google Scholar] [CrossRef] [PubMed]
- Shirley, M. Olaratumab: First Global Approval. Drugs 2017, 77, 107–112. [Google Scholar] [CrossRef] [PubMed]
- Baselga, J. The EGFR as a target for anticancer therapy—Focus on cetuximab. Eur. J. Cancer 2001, 37 (Suppl. 4), S16–S22. [Google Scholar] [CrossRef]
- Hudis, C.A. Trastuzumab—Mechanism of action and use in clinical practice. N. Engl. J. Med. 2007, 357, 39–51. [Google Scholar] [CrossRef] [PubMed]
- Agus, D.B.; Gordon, M.S.; Taylor, C.; Natale, R.B.; Karlan, B.; Mendelson, D.S.; Press, M.F.; Allison, D.E.; Sliwkowski, M.X.; Lieberman, G.; et al. Phase I clinical study of pertuzumab, a novel HER dimerization inhibitor, in patients with advanced cancer. J. Clin. Oncol. 2005, 23, 2534–2543. [Google Scholar] [CrossRef]
- O’Sullivan Coyne, G.; Burotto, M. MABp1 for the treatment of colorectal cancer. Expert. Opin. Biol. Ther. 2017, 17, 1155–1161. [Google Scholar] [CrossRef]
- Savvidou, O.D.; Bolia, I.K.; Chloros, G.D.; Papanastasiou, J.; Koutsouradis, P.; Papagelopoulos, P.J. Denosumab: Current Use in the Treatment of Primary Bone Tumors. Orthopedics 2017, 40, 204–210. [Google Scholar] [CrossRef]
- Jhajj, H.S.; Lwo, T.S.; Yao, E.L.; Tessier, P.M. Unlocking the potential of agonist antibodies for treating cancer using antibody engineering. Trends Mol. Med. 2023, 29, 48–60. [Google Scholar] [CrossRef] [PubMed]
- Djureinovic, D.; Wang, M.; Kluger, H.M. Agonistic CD40 Antibodies in Cancer Treatment. Cancers 2021, 13, 1302. [Google Scholar] [CrossRef] [PubMed]
- Siu, L.L.; Steeghs, N.; Meniawy, T.; Joerger, M.; Spratlin, J.L.; Rottey, S.; Nagrial, A.; Cooper, A.; Meier, R.; Guan, X.; et al. Preliminary results of a phase I/IIa study of BMS-986156 (glucocorticoid-induced tumor necrosis factor receptor–related gene [GITR] agonist), alone and in combination with nivolumab in pts with advanced solid tumors. J. Clin. Oncol. 2017, 35, 104. [Google Scholar] [CrossRef]
- Krupitskaya, Y.; Wakelee, H.A. Ramucirumab, a fully human mAb to the transmembrane signaling tyrosine kinase VEGFR-2 for the potential treatment of cancer. Curr. Opin. Investig. Drugs 2009, 10, 597–605. [Google Scholar]
- Ferrara, N.; Hillan, K.J.; Gerber, H.P.; Novotny, W. Discovery and development of bevacizumab, an anti-VEGF antibody for treating cancer. Nat. Rev. Drug Discov. 2004, 3, 391–400. [Google Scholar] [CrossRef] [PubMed]
- Perez Horta, Z.; Goldberg, J.L.; Sondel, P.M. Anti-GD2 mAbs and next-generation mAb-based agents for cancer therapy. Immunotherapy 2016, 8, 1097–1117. [Google Scholar] [CrossRef]
- Patriarca, C.; Macchi, R.M.; Marschner, A.K.; Mellstedt, H. Epithelial cell adhesion molecule expression (CD326) in cancer: A short review. Cancer Treat. Rev. 2012, 38, 68–75. [Google Scholar] [CrossRef]
- Godwin, C.D.; Gale, R.P.; Walter, R.B. Gemtuzumab ozogamicin in acute myeloid leukemia. Leukemia 2017, 31, 1855–1868. [Google Scholar] [CrossRef]
- de Weers, M.; Tai, Y.T.; van der Veer, M.S.; Bakker, J.M.; Vink, T.; Jacobs, D.C.; Oomen, L.A.; Peipp, M.; Valerius, T.; Slootstra, J.W.; et al. Daratumumab, a novel therapeutic human CD38 monoclonal antibody, induces killing of multiple myeloma and other hematological tumors. J. Immunol. 2011, 186, 1840–1848. [Google Scholar] [CrossRef]
- Warner, J.L.; Arnason, J.E. Alemtuzumab use in relapsed and refractory chronic lymphocytic leukemia: A history and discussion of future rational use. Ther. Adv. Hematol. 2012, 3, 375–389. [Google Scholar] [CrossRef]
- Guo, L.; Zhang, H.; Chen, B. Nivolumab as Programmed Death-1 (PD-1) Inhibitor for Targeted Immunotherapy in Tumor. J. Cancer 2017, 8, 410–416. [Google Scholar] [CrossRef]
- Lee, H.T.; Lee, J.Y.; Lim, H.; Lee, S.H.; Moon, Y.J.; Pyo, H.J.; Ryu, S.E.; Shin, W.; Heo, Y.S. Molecular mechanism of PD-1/PD-L1 blockade via anti-PD-L1 antibodies atezolizumab and durvalumab. Sci. Rep. 2017, 7, 5532. [Google Scholar] [CrossRef] [PubMed]
- Movva, S.; Verschraegen, C. The monoclonal antibody to cytotoxic T lymphocyte antigen 4, ipilimumab (MDX-010), a novel treatment strategy in cancer management. Expert. Opin. Biol. Ther. 2009, 9, 231–241. [Google Scholar] [CrossRef] [PubMed]
- Schmitt, C.; Marie-Cardine, A.; Bensussan, A. Therapeutic Antibodies to KIR3DL2 and Other Target Antigens on Cutaneous T-Cell Lymphomas. Front. Immunol. 2017, 8, 1010. [Google Scholar] [CrossRef] [PubMed]
- Muntasell, A.; Cabo, M.; Servitja, S.; Tusquets, I.; Martinez-Garcia, M.; Rovira, A.; Rojo, F.; Albanell, J.; Lopez-Botet, M. Interplay between Natural Killer Cells and Anti-HER2 Antibodies: Perspectives for Breast Cancer Immunotherapy. Front. Immunol. 2017, 8, 1544. [Google Scholar] [CrossRef] [PubMed]
- Lee, E.C.; Liang, Q.; Ali, H.; Bayliss, L.; Beasley, A.; Bloomfield-Gerdes, T.; Bonoli, L.; Brown, R.; Campbell, J.; Carpenter, A.; et al. Complete humanization of the mouse immunoglobulin loci enables efficient therapeutic antibody discovery. Nat. Biotechnol. 2014, 32, 356–363. [Google Scholar] [CrossRef] [PubMed]
- Macdonald, L.E.; Karow, M.; Stevens, S.; Auerbach, W.; Poueymirou, W.T.; Yasenchak, J.; Frendewey, D.; Valenzuela, D.M.; Giallourakis, C.C.; Alt, F.W.; et al. Precise and in situ genetic humanization of 6 Mb of mouse immunoglobulin genes. Proc. Natl. Acad. Sci. USA 2014, 111, 5147–5152. [Google Scholar] [CrossRef] [PubMed]
- Baumann, N.; Arndt, C.; Petersen, J.; Lustig, M.; Rosner, T.; Klausz, K.; Kellner, C.; Bultmann, M.; Bastian, L.; Vogiatzi, F.; et al. Myeloid checkpoint blockade improves killing of T-acute lymphoblastic leukemia cells by an IgA2 variant of daratumumab. Front. Immunol. 2022, 13, 949140. [Google Scholar] [CrossRef] [PubMed]
- Gruijs, M.; Sewnath, C.A.N.; van Egmond, M. Therapeutic exploitation of neutrophils to fight cancer. Semin. Immunol. 2021, 57, 101581. [Google Scholar] [CrossRef]
- Heemskerk, N.; Gruijs, M.; Temming, A.R.; Heineke, M.H.; Gout, D.Y.; Hellingman, T.; Tuk, C.W.; Winter, P.J.; Lissenberg-Thunnissen, S.; Bentlage, A.E.; et al. Augmented antibody-based anticancer therapeutics boost neutrophil cytotoxicity. J. Clin. Investig. 2021, 131, e134680. [Google Scholar] [CrossRef]
- Brandsma, A.M.; Bondza, S.; Evers, M.; Koutstaal, R.; Nederend, M.; Jansen, J.H.M.; Rosner, T.; Valerius, T.; Leusen, J.H.W.; Ten Broeke, T. Potent Fc Receptor Signaling by IgA Leads to Superior Killing of Cancer Cells by Neutrophils Compared to IgG. Front. Immunol. 2019, 10, 704. [Google Scholar] [CrossRef] [PubMed]
- Hart, F.; Danielczyk, A.; Goletz, S. Human Cell Line-Derived Monoclonal IgA Antibodies for Cancer Immunotherapy. Bioengineering 2017, 4, 42. [Google Scholar] [CrossRef] [PubMed]
- van Egmond, M.; Bakema, J.E. Neutrophils as effector cells for antibody-based immunotherapy of cancer. Semin. Cancer Biol. 2013, 23, 190–199. [Google Scholar] [CrossRef] [PubMed]
- Chan, C.; Lustig, M.; Baumann, N.; Valerius, T.; van Tetering, G.; Leusen, J.H.W. Targeting Myeloid Checkpoint Molecules in Combination with Antibody Therapy: A Novel Anti-Cancer Strategy with IgA Antibodies? Front. Immunol. 2022, 13, 932155. [Google Scholar] [CrossRef] [PubMed]
- Xing, P.X.; Hu, X.F.; Pietersz, G.A.; Hosick, H.L.; McKenzie, I.F. Cripto: A novel target for antibody-based cancer immunotherapy. Cancer Res. 2004, 64, 4018–4023. [Google Scholar] [CrossRef] [PubMed]
- Luiten, R.M.; Fleuren, G.J.; Warnaar, S.O.; Litvinov, S.V. Target-specific activation of mast cells by immunoglobulin E reactive with a renal cell carcinoma-associated antigen. Lab. Investig. 1996, 74, 467–475. [Google Scholar] [PubMed]
- Karagiannis, P.; Singer, J.; Hunt, J.; Gan, S.K.; Rudman, S.M.; Mechtcheriakova, D.; Knittelfelder, R.; Daniels, T.R.; Hobson, P.S.; Beavil, A.J.; et al. Characterisation of an engineered trastuzumab IgE antibody and effector cell mechanisms targeting HER2/neu-positive tumour cells. Cancer Immunol. Immunother. 2009, 58, 915–930. [Google Scholar] [CrossRef]
- Singer, J.; Jensen-Jarolim, E. IgE-based Immunotherapy of Cancer -A Comparative Oncology Approach. J. Carcinog. Mutagen. 2014, 5, 1000176. [Google Scholar] [CrossRef]
- Nicodemus, C.F. Antibody-based immunotherapy of solid cancers: Progress and possibilities. Immunotherapy 2015, 7, 923–939. [Google Scholar] [CrossRef]
- Williams, I.P.; Crescioli, S.; Sow, H.S.; Bax, H.J.; Hobbs, C.; Ilieva, K.M.; French, E.; Pellizzari, G.; Cox, V.; Josephs, D.H.; et al. In vivo safety profile of a CSPG4-directed IgE antibody in an immunocompetent rat model. MAbs 2020, 12, 1685349. [Google Scholar] [CrossRef]
- Mimoto, F.; Igawa, T.; Kuramochi, T.; Katada, H.; Kadono, S.; Kamikawa, T.; Shida-Kawazoe, M.; Hattori, K. Novel asymmetrically engineered antibody Fc variant with superior FcgammaR binding affinity and specificity compared with afucosylated Fc variant. MAbs 2013, 5, 229–236. [Google Scholar] [CrossRef] [PubMed]
- Josephs, D.H.; Spicer, J.F.; Karagiannis, P.; Gould, H.J.; Karagiannis, S.N. IgE immunotherapy: A novel concept with promise for the treatment of cancer. MAbs 2014, 6, 54–72. [Google Scholar] [CrossRef] [PubMed]
- Karagiannis, S.N.; Karagiannis, P.; Josephs, D.H.; Saul, L.; Gilbert, A.E.; Upton, N.; Gould, H.J. Immunoglobulin E and Allergy: Antibodies in Immune Inflammation and Treatment. Microbiol. Spectr. 2013, 1, AID-0006-2012. [Google Scholar] [CrossRef] [PubMed]
- Karagiannis, S.N.; Josephs, D.H.; Karagiannis, P.; Gilbert, A.E.; Saul, L.; Rudman, S.M.; Dodev, T.; Koers, A.; Blower, P.J.; Corrigan, C.; et al. Recombinant IgE antibodies for passive immunotherapy of solid tumours: From concept towards clinical application. Cancer Immunol. Immunother. 2012, 61, 1547–1564. [Google Scholar] [CrossRef]
- Rudman, S.M.; Josephs, D.H.; Cambrook, H.; Karagiannis, P.; Gilbert, A.E.; Dodev, T.; Hunt, J.; Koers, A.; Montes, A.; Taams, L.; et al. Harnessing engineered antibodies of the IgE class to combat malignancy: Initial assessment of FcεRI-mediated basophil activation by a tumour-specific IgE antibody to evaluate the risk of type I hypersensitivity. Clin. Exp. Allergy 2011, 41, 1400–1413. [Google Scholar] [CrossRef] [PubMed]
- Josephs, D.H.; Nakamura, M.; Bax, H.J.; Dodev, T.S.; Muirhead, G.; Saul, L.; Karagiannis, P.; Ilieva, K.M.; Crescioli, S.; Gazinska, P.; et al. An immunologically relevant rodent model demonstrates safety of therapy using a tumour-specific IgE. Allergy 2018, 73, 2328–2341. [Google Scholar] [CrossRef] [PubMed]
- Josephs, D.H.; Bax, H.J.; Dodev, T.; Georgouli, M.; Nakamura, M.; Pellizzari, G.; Saul, L.; Karagiannis, P.; Cheung, A.; Herraiz, C.; et al. Anti-Folate Receptor-alpha IgE but not IgG Recruits Macrophages to Attack Tumors via TNFalpha/MCP-1 Signaling. Cancer Res. 2017, 77, 1127–1141. [Google Scholar] [CrossRef]
- Chauhan, J.; Grandits, M.; Palhares, L.; Mele, S.; Nakamura, M.; Lopez-Abente, J.; Crescioli, S.; Laddach, R.; Romero-Clavijo, P.; Cheung, A.; et al. Anti-cancer pro-inflammatory effects of an IgE antibody targeting the melanoma-associated antigen chondroitin sulfate proteoglycan 4. Nat. Commun. 2023, 14, 2192. [Google Scholar] [CrossRef]
- Trauth, B.C.; Klas, C.; Peters, A.M.; Matzku, S.; Moller, P.; Falk, W.; Debatin, K.M.; Krammer, P.H. Monoclonal antibody-mediated tumor regression by induction of apoptosis. Science 1989, 245, 301–305. [Google Scholar] [CrossRef]
- Horner, S.; Moustafa-Oglou, M.; Teppert, K.; Hagelstein, I.; Kauer, J.; Pflugler, M.; Neumann, K.; Rammensee, H.G.; Metz, T.; Herrmann, A.; et al. IgG-Based Bispecific Anti-CD95 Antibodies for the Treatment of B Cell-Derived Malignancies and Autoimmune Diseases. Cancers 2022, 14, 3941. [Google Scholar] [CrossRef]
- Petricevic, B.; Laengle, J.; Singer, J.; Sachet, M.; Fazekas, J.; Steger, G.; Bartsch, R.; Jensen-Jarolim, E.; Bergmann, M. Trastuzumab mediates antibody-dependent cell-mediated cytotoxicity and phagocytosis to the same extent in both adjuvant and metastatic HER2/neu breast cancer patients. J. Transl. Med. 2013, 11, 307. [Google Scholar] [CrossRef]
- Santamaria, S.; Delgado, M.; Botas, M.; Castellano, E.; Corraliza-Gorjon, I.; Lafuente, P.; Muñoz-Calleja, C.; Toribio, M.L.; Kremer, L.; Garcia-Sanz, J.A. Therapeutic potential of an anti-CCR9 mAb evidenced in xenografts of human CCR9+ tumors. Front. Immunol. 2022, 13, 825635. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Q.; Tran, H.; Dimitrov, D.S.; Cheung, N.K. A dual-specific anti-IGF-1/IGF-2 human monoclonal antibody alone and in combination with temsirolimus for therapy of neuroblastoma. Int. J. Cancer 2015, 137, 2243–2252. [Google Scholar] [CrossRef] [PubMed]
- Straughn, J.M., Jr.; Oliver, P.G.; Zhou, T.; Wang, W.; Alvarez, R.D.; Grizzle, W.E.; Buchsbaum, D.J. Anti-tumor activity of TRA-8 anti-death receptor 5 (DR5) monoclonal antibody in combination with chemotherapy and radiation therapy in a cervical cancer model. Gynecol. Oncol. 2006, 101, 46–54. [Google Scholar] [CrossRef]
- Cheung, N.K.; Modak, S. Oral (1-->3),(1-->4)-beta-D-glucan synergizes with antiganglioside GD2 monoclonal antibody 3F8 in the therapy of neuroblastoma. Clin. Cancer Res. 2002, 8, 1217–1223. [Google Scholar] [PubMed]
- Warren, T.L.; Dahle, C.E.; Weiner, G.J. CpG oligodeoxynucleotides enhance monoclonal antibody therapy of a murine lymphoma. Clin. Lymphoma 2000, 1, 57–61. [Google Scholar] [CrossRef]
- Takahashi, N.; Haba, A.; Matsuno, F.; Seon, B.K. Antiangiogenic therapy of established tumors in human skin/severe combined immunodeficiency mouse chimeras by anti-endoglin (CD105) monoclonal antibodies, and synergy between anti-endoglin antibody and cyclophosphamide. Cancer Res. 2001, 61, 7846–7854. [Google Scholar]
- Militerno, G.; Gugenheim, J.; Cuomo, O.; Hofman, P.; Mouiel, J.; Tovey, M. Synergistic interaction between anti-IFN alpha/beta antibody and low doses of cyclosporine therapy prolongs heart transplants in rats. Transpl. Transplant. Proc. 1994, 26, 3050–3051. [Google Scholar]
- Tellides, G.; Dallman, M.J.; Morris, P.J. Synergistic interaction of cyclosporine A with interleukin 2 receptor monoclonal antibody therapy. Transpl. Transplant. Proc. 1988, 20, 202–206. [Google Scholar]
- Ahmad, G.; Mackenzie, G.G.; Egan, J.; Amiji, M.M. DHA-SBT-1214 Taxoid Nanoemulsion and Anti-PD-L1 Antibody Combination Therapy Enhances Antitumor Efficacy in a Syngeneic Pancreatic Adenocarcinoma Model. Mol. Cancer Ther. 2019, 18, 1961–1972. [Google Scholar] [CrossRef]
- Huang, C.; Li, H.; Feng, Y.; Li, X.; Zhang, Z.; Jiang, C.; Wang, J.; Yang, C.; Fu, Y.; Mu, M.; et al. Combination therapy with B7H3-redirected bispecific antibody and Sorafenib elicits enhanced synergistic antitumor efficacy. Theranostics 2020, 10, 10498–10512. [Google Scholar] [CrossRef] [PubMed]
- Gadri, Z.; Kukulansky, T.; Bar-Or, E.; Haimovich, J.; Hollander, N. Synergistic effect of dendritic cell vaccination and anti-CD20 antibody treatment in the therapy of murine lymphoma. J. Immunother. 2009, 32, 333–340. [Google Scholar] [CrossRef] [PubMed]
- Guedj, M.; Marisa, L.; de Reynies, A.; Orsetti, B.; Schiappa, R.; Bibeau, F.; Macgrogan, G.; Lerebours, F.; Finetti, P.; Longy, M.; et al. A refined molecular taxonomy of breast cancer. Oncogene 2012, 31, 1196–1206. [Google Scholar] [CrossRef] [PubMed]
- Slamon, D.J.; Godolphin, W.; Jones, L.A.; Holt, J.A.; Wong, S.G.; Keith, D.E.; Levin, W.J.; Stuart, S.G.; Udove, J.; Ullrich, A.; et al. Studies of the HER-2/neu proto-oncogene in human breast and ovarian cancer. Science 1989, 244, 707–712. [Google Scholar] [CrossRef] [PubMed]
- Slamon, D.J.; Clark, G.M.; Wong, S.G.; Levin, W.J.; Ullrich, A.; McGuire, W.L. Human breast cancer: Correlation of relapse and survival with amplification of the HER-2/neu oncogene. Science 1987, 235, 177–182. [Google Scholar] [CrossRef]
- Goldenberg, M.M. Trastuzumab, a recombinant DNA-derived humanized monoclonal antibody, a novel agent for the treatment of metastatic breast cancer. Clin. Ther. 1999, 21, 309–318. [Google Scholar] [CrossRef]
- Molina, M.A.; Codony-Servat, J.; Albanell, J.; Rojo, F.; Arribas, J.; Baselga, J. Trastuzumab (herceptin), a humanized anti-HER2 receptor monoclonal antibody, inhibits basal and activated HER2 ectodomain cleavage in breast cancer cells. Cancer Res. 2001, 61, 4744–4749. [Google Scholar]
- Gall, V.A.; Philips, A.V.; Qiao, N.; Clise-Dwyer, K.; Perakis, A.A.; Zhang, M.; Clifton, G.T.; Sukhumalchandra, P.; Ma, Q.; Reddy, S.M.; et al. Trastuzumab Increases HER2 Uptake and Cross-Presentation by Dendritic Cells. Cancer Res. 2017, 77, 5374–5383. [Google Scholar] [CrossRef]
- Rexer, B.N.; Arteaga, C.L. Intrinsic and acquired resistance to HER2-targeted therapies in HER2 gene-amplified breast cancer: Mechanisms and clinical implications. Crit. Rev. Oncog. 2012, 17, 1–16. [Google Scholar] [CrossRef]
- Valabrega, G.; Montemurro, F.; Aglietta, M. Trastuzumab: Mechanism of action, resistance and future perspectives in HER2-overexpressing breast cancer. Ann. Oncol. 2007, 18, 977–984. [Google Scholar] [CrossRef]
- Li, G.; Guo, J.; Shen, B.Q.; Yadav, D.B.; Sliwkowski, M.X.; Crocker, L.M.; Lacap, J.A.; Phillips, G.D.L. Mechanisms of Acquired Resistance to Trastuzumab Emtansine in Breast Cancer Cells. Mol. Cancer Ther. 2018, 17, 1441–1453. [Google Scholar] [CrossRef] [PubMed]
- Bitar, L.; Zouein, J.; Haddad, F.G.; Eid, R.; Kourie, H.R. HER2 in metastatic colorectal cancer: A new to target to remember. Biomark. Med. 2021, 15, 133–136. [Google Scholar] [CrossRef] [PubMed]
- Batman, S.; Bohn, J.; Weisenberger, M.W.; Hersh, A.; Bruegl, A.; Caughey, A.; Winter, W., 3rd. Trastuzumab with carboplatin/paclitaxel for treatment of advanced stage and recurrent uterine papillary serous carcinoma: A cost-effectiveness analysis. Gynecol. Oncol. 2021, 160, 214–218. [Google Scholar] [CrossRef]
- Assenat, E.; Mineur, L.; Mollevi, C.; Lopez-Crapez, E.; Lombard-Bohas, C.; Samalin, E.; Portales, F.; Walter, T.; de Forges, H.; Dupuy, M.; et al. Phase II study evaluating the association of gemcitabine, trastuzumab and erlotinib as first-line treatment in patients with metastatic pancreatic adenocarcinoma (GATE 1). Int. J. Cancer 2021, 148, 682–691. [Google Scholar] [CrossRef]
- Javle, M.; Borad, M.J.; Azad, N.S.; Kurzrock, R.; Abou-Alfa, G.K.; George, B.; Hainsworth, J.; Meric-Bernstam, F.; Swanton, C.; Sweeney, C.J.; et al. Pertuzumab and trastuzumab for HER2-positive, metastatic biliary tract cancer (MyPathway): A multicentre, open-label, phase 2a, multiple basket study. Lancet Oncol. 2021, 22, 1290–1300. [Google Scholar] [CrossRef]
- Isnard, C.; Plantier, F.; Theriaut, M.; Avril, M.F.; Moyal-Barracco, M. Complete but transient clinical remission of vulvar Paget’s disease with paclitaxel and trastuzumab. Ann. Dermatol. Venereol. 2021, 148, 47–48. [Google Scholar] [CrossRef]
- Shen, G.; Gao, Q.; Liu, F.; Zhang, Y.; Dai, M.; Zhao, T.; Cheng, M.; Xu, T.; Jin, P.; Yin, W.; et al. The Wnt3a/beta-catenin/TCF7L2 signaling axis reduces the sensitivity of HER2-positive epithelial ovarian cancer to trastuzumab. Biochem. Biophys. Res. Commun. 2020, 526, 685–691. [Google Scholar] [CrossRef] [PubMed]
- Park, S.J.; Yoon, B.H.; Kim, S.K.; Kim, S.Y. GENT2: An updated gene expression database for normal and tumor tissues. BMC Med. Genom. 2019, 12, 101. [Google Scholar] [CrossRef]
- Nikolaou, M.; Pavlopoulou, A.; Georgakilas, A.G.; Kyrodimos, E. The challenge of drug resistance in cancer treatment: A current overview. Clin. Exp. Metastasis 2018, 35, 309–318. [Google Scholar] [CrossRef]
- Santamaria, S.; Delgado, M.; Kremer, L.; Garcia-Sanz, J.A. Will a mAb-Based Immunotherapy Directed against Cancer Stem Cells Be Feasible? Front. Immunol. 2017, 8, 1509. [Google Scholar] [CrossRef]
- Baselga, J.; Cortés, J.; Kim, S.-B.; Im, S.-A.; Hegg, R.; Im, Y.-H.; Roman, L.; Pedrini, J.L.; Pienkowski, T.; Knott, A.; et al. Pertuzumab plus Trastuzumab plus Docetaxel for Metastatic Breast Cancer. N. Engl. J. Med. 2011, 366, 109–119. [Google Scholar] [CrossRef] [PubMed]
- National Cancer Institute. Center for Cancer Genomics. The Cancer Genome Atlas Program (TCGA). Available online: https://www.cancer.gov/ccg/research/genome-sequencing/tcga (accessed on 10 October 2023).
- Cerami, E.; Gao, J.; Dogrusoz, U.; Gross, B.E.; Sumer, S.O.; Aksoy, B.A.; Jacobsen, A.; Byrne, C.J.; Heuer, M.L.; Larsson, E.; et al. The cBio cancer genomics portal: An open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012, 2, 401–404. [Google Scholar] [CrossRef] [PubMed]
- Gao, J.; Aksoy, B.A.; Dogrusoz, U.; Dresdner, G.; Gross, B.; Sumer, S.O.; Sun, Y.; Jacobsen, A.; Sinha, R.; Larsson, E.; et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci. Signal 2013, 6, pl1. [Google Scholar] [CrossRef] [PubMed]
- de Bruijn, I.; Kundra, R.; Mastrogiacomo, B.; Tran, T.N.; Sikina, L.; Mazor, T.; Li, X.; Ochoa, A.; Zhao, G.; Lai, B.; et al. Analysis and Visualization of Longitudinal Genomic and Clinical Data from the AACR Project GENIE Biopharma Collaborative in cBioPortal. Cancer Res. 2023, 83, 3861–3867. [Google Scholar] [CrossRef] [PubMed]
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Ferlay, J.; Ervik, M.; Lam, F.; Colombet, M.; Mery, L.; Piñeros, M.; Znaor, A.; Soerjomataram, I.; Bray, F. Global Cancer Observatory: Cancer Today; International Agency for Research on Cancer: Lyon, France. Available online: https://gco.iarc.fr/today (accessed on 22 September 2022).
- Siegel, R.L.; Miller, K.D.; Fuchs, H.E.; Jemal, A. Cancer statistics, 2022. CA Cancer J. Clin. 2022, 72, 7–33. [Google Scholar] [CrossRef] [PubMed]
- CancerResearch-UK. Cancer Statistics. Available online: https://www.cancerresearchuk.org/health-professional/cancer-statistics-for-the-uk (accessed on 1 September 2022).
- Ferlay, J.; Colombet, M.; Soerjomataram, I.; Parkin, D.M.; Pineros, M.; Znaor, A.; Bray, F. Cancer statistics for the year 2020: An overview. Int. J. Cancer 2021, 149, 778–789. [Google Scholar] [CrossRef]
- Trenevska, I.; Anderson, A.P.; Bentley, C.; Hassanali, T.; Wiblin, S.; Maguire, S.; Pezzella, F.; Banham, A.H.; Li, D. Comprehensive mutagenesis identifies the peptide repertoire of a p53 T-cell receptor mimic antibody that displays no toxicity in mice transgenic for human HLA-A*0201. PLoS ONE 2021, 16, e0249967. [Google Scholar] [CrossRef]
- Trenevska, I.; Li, D.; Banham, A.H. Therapeutic Antibodies against Intracellular Tumor Antigens. Front. Immunol. 2017, 8, 1001. [Google Scholar] [CrossRef]
- Boyd, D.; Schierle, C.; Beckwith, J. How many membrane proteins are there? Protein Sci. 1998, 7, 201–205. [Google Scholar] [CrossRef]
- Almen, M.S.; Nordstrom, K.J.; Fredriksson, R.; Schioth, H.B. Mapping the human membrane proteome: A majority of the human membrane proteins can be classified according to function and evolutionary origin. BMC Biol. 2009, 7, 50. [Google Scholar] [CrossRef]
- Dobson, L.; Remenyi, I.; Tusnady, G.E. The human transmembrane proteome. Biol. Direct 2015, 10, 31. [Google Scholar] [CrossRef]
- Fagerberg, L.; Jonasson, K.; von Heijne, G.; Uhlen, M.; Berglund, L. Prediction of the human membrane proteome. Proteomics 2010, 10, 1141–1149. [Google Scholar] [CrossRef] [PubMed]
- Gene Ontology, C. Gene Ontology Consortium: Going forward. Nucleic Acids Res. 2015, 43, D1049–D1056. [Google Scholar] [CrossRef] [PubMed]
- Ashburner, M.; Ball, C.A.; Blake, J.A.; Botstein, D.; Butler, H.; Cherry, J.M.; Davis, A.P.; Dolinski, K.; Dwight, S.S.; Eppig, J.T.; et al. Gene Ontology: Tool for the unification of biology. Nat. Genet. 2000, 25, 25–29. [Google Scholar] [CrossRef] [PubMed]
- Carbon, S.; Mungall, C. Gene Ontology Data Archive. Available online: https://zenodo.org/records/8436609 (accessed on 10 October 2023).
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Žídek, A.; Potapenko, A.; et al. Highly accurate protein structure prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef]
- Sjostedt, E.; Zhong, W.; Fagerberg, L.; Karlsson, M.; Mitsios, N.; Adori, C.; Oksvold, P.; Edfors, F.; Limiszewska, A.; Hikmet, F.; et al. An atlas of the protein-coding genes in the human, pig, and mouse brain. Science 2020, 367, eaay5947. [Google Scholar] [CrossRef]
- Uhlen, M.; Karlsson, M.J.; Hober, A.; Svensson, A.S.; Scheffel, J.; Kotol, D.; Zhong, W.; Tebani, A.; Strandberg, L.; Edfors, F.; et al. The human secretome. Sci. Signal 2019, 12, eaaz0274. [Google Scholar] [CrossRef]
- Uhlen, M.; Bjorling, E.; Agaton, C.; Szigyarto, C.A.; Amini, B.; Andersen, E.; Andersson, A.C.; Angelidou, P.; Asplund, A.; Asplund, C.; et al. A human protein atlas for normal and cancer tissues based on antibody proteomics. Mol. Cell Proteom. 2005, 4, 1920–1932. [Google Scholar] [CrossRef]
- Berglund, L.; Bjorling, E.; Oksvold, P.; Fagerberg, L.; Asplund, A.; Szigyarto, C.A.; Persson, A.; Ottosson, J.; Wernerus, H.; Nilsson, P.; et al. A genecentric Human Protein Atlas for expression profiles based on antibodies. Mol. Cell Proteom. 2008, 7, 2019–2027. [Google Scholar] [CrossRef]
- Ponten, F.; Jirstrom, K.; Uhlen, M. The Human Protein Atlas—A tool for pathology. J. Pathol. 2008, 216, 387–393. [Google Scholar] [CrossRef]
- Uhlen, M.; Karlsson, M.J.; Zhong, W.; Tebani, A.; Pou, C.; Mikes, J.; Lakshmikanth, T.; Forsstrom, B.; Edfors, F.; Odeberg, J.; et al. A genome-wide transcriptomic analysis of protein-coding genes in human blood cells. Science 2019, 366, eaax9198. [Google Scholar] [CrossRef]
- Maciocia, P.M.; Wawrzyniecka, P.A.; Maciocia, N.C.; Burley, A.; Karpanasamy, T.; Devereaux, S.; Hoekx, M.; O’Connor, D.; Leon, T.; Rapoz-D’Silva, T.; et al. Anti-CCR9 chimeric antigen receptor T cells for T-cell acute lymphoblastic leukemia. Blood 2022, 140, 25–37. [Google Scholar] [CrossRef]
- Wang, S.; Lu, S. DNA immunization. Curr. Protoc. Microbiol. 2013, 31, 18.13.11–18.13.24. [Google Scholar] [CrossRef]
- Hansen, D.T.; Craciunescu, F.M.; Fromme, P.; Johnston, S.A.; Sykes, K.F. Generation of High-Specificity Antibodies against Membrane Proteins Using DNA-Gold Micronanoplexes for Gene Gun Immunization. Curr. Protoc. Protein Sci. 2018, 91, 29.20.1–29.20.22. [Google Scholar] [CrossRef]
- Chamorro, S.; Vela, M.; Franco-Villanueva, A.; Carramolino, L.; Gutierrez, J.; Gomez, L.; Lozano, M.; Salvador, B.; Garcia-Gallo, M.; Martinez, A.C.; et al. Antitumor effects of a monoclonal antibody to human CCR9 in leukemia cell xenografts. MAbs 2014, 6, 1000–1012. [Google Scholar] [CrossRef]
- Somovilla-Crespo, B.; Martín Monzón, M.T.; Vela, M.; Corraliza-Gorjón, I.; Santamaria, S.; Garcia-Sanz, J.A.; Kremer, L. 92R Monoclonal Antibody Inhibits Human CCR9+ Leukemia Cells Growth in NSG Mice Xenografts. Front. Immunol. 2018, 9, 77. [Google Scholar] [CrossRef]
- Zabel, B.A.; Agace, W.W.; Campbell, J.J.; Heath, H.M.; Parent, D.; Roberts, A.I.; Ebert, E.C.; Kassam, N.; Qin, S.; Zovko, M.; et al. Human G protein-coupled receptor GPR-9-6/CC chemokine receptor 9 is selectively expressed on intestinal homing T lymphocytes, mucosal lymphocytes, and thymocytes and is required for thymus-expressed chemokine-mediated chemotaxis. J. Exp. Med. 1999, 190, 1241–1256. [Google Scholar] [CrossRef]
- Almagro, J.C.; Daniels-Wells, T.R.; Perez-Tapia, S.M.; Penichet, M.L. Progress and Challenges in the Design and Clinical Development of Antibodies for Cancer Therapy. Front. Immunol. 2017, 8, 1751. [Google Scholar] [CrossRef]
- Green, L.L. Antibody engineering via genetic engineering of the mouse: XenoMouse strains are a vehicle for the facile generation of therapeutic human monoclonal antibodies. J. Immunol. Methods 1999, 231, 11–23. [Google Scholar] [CrossRef]
- Huse, W.D.; Sastry, L.; Iverson, S.A.; Kang, A.S.; Alting-Mees, M.; Burton, D.R.; Benkovic, S.J.; Lerner, R.A. Generation of a large combinatorial library of the immunoglobulin repertoire in phage lambda. Science 1989, 246, 1275–1281. [Google Scholar] [CrossRef] [PubMed]
- Sastry, L.; Alting-Mees, M.; Huse, W.D.; Short, J.M.; Sorge, J.A.; Hay, B.N.; Janda, K.D.; Benkovic, S.J.; Lerner, R.A. Cloning of the immunological repertoire in Escherichia coli for generation of monoclonal catalytic antibodies: Construction of a heavy chain variable region-specific cDNA library. Proc. Natl. Acad. Sci. USA 1989, 86, 5728–5732. [Google Scholar] [CrossRef] [PubMed]
- de Haard, H.J.; van Neer, N.; Reurs, A.; Hufton, S.E.; Roovers, R.C.; Henderikx, P.; de Bruine, A.P.; Arends, J.W.; Hoogenboom, H.R. A large non-immunized human Fab fragment phage library that permits rapid isolation and kinetic analysis of high affinity antibodies. J. Biol. Chem. 1999, 274, 18218–18230. [Google Scholar] [CrossRef] [PubMed]
- Nissim, A.; Hoogenboom, H.R.; Tomlinson, I.M.; Flynn, G.; Midgley, C.; Lane, D.; Winter, G. Antibody fragments from a ‘single pot’ phage display library as immunochemical reagents. EMBO J. 1994, 13, 692–698. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Katayama, A.; Wang, Y.; Yu, L.; Favoino, E.; Sakakura, K.; Favole, A.; Tsuchikawa, T.; Silver, S.; Watkins, S.C.; et al. Functional characterization of an scFv-Fc antibody that immunotherapeutically targets the common cancer cell surface proteoglycan CSPG4. Cancer Res. 2011, 71, 7410–7422. [Google Scholar] [CrossRef]
- Roguska, M.; Kaymakcalan, Z.; Salfeld, J. Overview on the use of therapeutic antibodies in drug discovery. Curr. Protoc. Pharmacol. 2005, 27, 9.7. [Google Scholar] [CrossRef]
- Jain, R.K. Transport of molecules, particles, and cells in solid tumors. Annu. Rev. Biomed. Eng. 1999, 1, 241–263. [Google Scholar] [CrossRef]
- Zhang, Y.; Pastan, I. High shed antigen levels within tumors: An additional barrier to immunoconjugate therapy. Clin. Cancer Res. 2008, 14, 7981–7986. [Google Scholar] [CrossRef]
- van der Velden, V.H.; Boeckx, N.; Jedema, I.; te Marvelde, J.G.; Hoogeveen, P.G.; Boogaerts, M.; van Dongen, J.J. High CD33-antigen loads in peripheral blood limit the efficacy of gemtuzumab ozogamicin (Mylotarg) treatment in acute myeloid leukemia patients. Leukemia 2004, 18, 983–988. [Google Scholar] [CrossRef]
- Kreitman, R.J.; Wilson, W.H.; Bergeron, K.; Raggio, M.; Stetler-Stevenson, M.; FitzGerald, D.J.; Pastan, I. Efficacy of the anti-CD22 recombinant immunotoxin BL22 in chemotherapy-resistant hairy-cell leukemia. N. Engl. J. Med. 2001, 345, 241–247. [Google Scholar] [CrossRef]
- Pak, Y.; Pastan, I.; Kreitman, R.J.; Lee, B. Effect of antigen shedding on targeted delivery of immunotoxins in solid tumors from a mathematical model. PLoS ONE 2014, 9, e110716. [Google Scholar] [CrossRef] [PubMed]
- Peggs, K.S.; Quezada, S.A.; Korman, A.J.; Allison, J.P. Principles and use of anti-CTLA4 antibody in human cancer immunotherapy. Curr. Opin. Immunol. 2006, 18, 206–213. [Google Scholar] [CrossRef] [PubMed]
- Quirk, S.K.; Shure, A.K.; Agrawal, D.K. Immune-mediated adverse events of anticytotoxic T lymphocyte-associated antigen 4 antibody therapy in metastatic melanoma. Transl. Res. 2015, 166, 412–424. [Google Scholar] [CrossRef] [PubMed]
- Dancey, J.E.; Chen, H.X. Strategies for optimizing combinations of molecularly targeted anticancer agents. Nat. Rev. Drug Discov. 2006, 5, 649–659. [Google Scholar] [CrossRef] [PubMed]
- Mahvi, D.M.; Meyers, W.C.; Bast, R.C.; Seigler, H.F.; Metzgar, R.S. Carcinoma of the pancreas. Therapeutic efficacy as defined by a serodiagnostic test utilizing a monoclonal antibody. Ann. Surg. 1985, 202, 440–445. [Google Scholar] [CrossRef] [PubMed]
- Renouf, D.J.; Loree, J.M.; Knox, J.J.; Topham, J.T.; Kavan, P.; Jonker, D.; Welch, S.; Couture, F.; Lemay, F.; Tehfe, M.; et al. The CCTG PA.7 phase II trial of gemcitabine and nab-paclitaxel with or without durvalumab and tremelimumab as initial therapy in metastatic pancreatic ductal adenocarcinoma. Nat. Commun. 2022, 13, 5020. [Google Scholar] [CrossRef] [PubMed]
- Zhu, M.; Costello, B.A.; Yin, J.; Pettinger, A.M.; Strosberg, J.R.; Erlichman, C.; Hobday, T.J. Phase II Trial of Bevacizumab Monotherapy in Pancreatic Neuroendocrine Tumors. Pancreas 2021, 50, 1435–1439. [Google Scholar] [CrossRef] [PubMed]
- Fraunhoffer, N.A.; Closa, D.; Folch-Puy, E.; Abuelafia, A.M.; Calvo, E.L.; Chuluyan, E.; Iovanna, J. Targeting REG3beta limits pancreatic ductal adenocarcinoma progression through CTGF downregulation. Cancer Lett. 2021, 521, 64–70. [Google Scholar] [CrossRef]
- Yao, H.P.; Hudson, R.; Wang, M.H. RON receptor tyrosine kinase in pancreatic ductal adenocarcinoma: Pathogenic mechanism in malignancy and pharmaceutical target for therapy. Biochim. Biophys. Acta Rev. Cancer 2020, 1873, 188360. [Google Scholar] [CrossRef]
- Ma, Y.; Li, J.; Wang, H.; Chiu, Y.; Kingsley, C.V.; Fry, D.; Delaney, S.N.; Wei, S.C.; Zhang, J.; Maitra, A.; et al. Combination of PD-1 Inhibitor and OX40 Agonist Induces Tumor Rejection and Immune Memory in Mouse Models of Pancreatic Cancer. Gastroenterology 2020, 159, 306–319.e12. [Google Scholar] [CrossRef]
- Nagaoka, K.; Bai, X.; Ogawa, K.; Dong, X.; Zhang, S.; Zhou, Y.; Carlson, R.I.; Jiang, Z.G.; Fuller, S.; Lebowitz, M.S.; et al. Anti-tumor activity of antibody drug conjugate targeting aspartate-beta-hydroxylase in pancreatic ductal adenocarcinoma. Cancer Lett. 2019, 449, 87–98. [Google Scholar] [CrossRef] [PubMed]
- Ye, J.; Yuen, S.M.; Murphy, G.; Xie, R.; Kwok, H.F. Anti-tumor effects of a ‘human & mouse cross-reactive’ anti-ADAM17 antibody in a pancreatic cancer model in vivo. Eur. J. Pharm. Sci. 2017, 110, 62–69. [Google Scholar] [CrossRef] [PubMed]
- Almhanna, K.; Wright, D.; Mercade, T.M.; Van Laethem, J.L.; Gracian, A.C.; Guillen-Ponce, C.; Faris, J.; Lopez, C.M.; Hubner, R.A.; Bendell, J.; et al. A phase II study of antibody-drug conjugate, TAK-264 (MLN0264) in previously treated patients with advanced or metastatic pancreatic adenocarcinoma expressing guanylyl cyclase C. Investig. New Drugs 2017, 35, 634–641. [Google Scholar] [CrossRef] [PubMed]
- Molejon, M.I.; Tellechea, J.I.; Loncle, C.; Gayet, O.; Gilabert, M.; Duconseil, P.; Lopez-Millan, M.B.; Moutardier, V.; Gasmi, M.; Garcia, S.; et al. Deciphering the cellular source of tumor relapse identifies CD44 as a major therapeutic target in pancreatic adenocarcinoma. Oncotarget 2015, 6, 7408–7423. [Google Scholar] [CrossRef] [PubMed]
- Fuchs, C.S.; Azevedo, S.; Okusaka, T.; Van Laethem, J.L.; Lipton, L.R.; Riess, H.; Szczylik, C.; Moore, M.J.; Peeters, M.; Bodoky, G.; et al. A phase 3 randomized, double-blind, placebo-controlled trial of ganitumab or placebo in combination with gemcitabine as first-line therapy for metastatic adenocarcinoma of the pancreas: The GAMMA trial. Ann. Oncol. 2015, 26, 921–927. [Google Scholar] [CrossRef]
- Cioffi, M.; Trabulo, S.; Hidalgo, M.; Costello, E.; Greenhalf, W.; Erkan, M.; Kleeff, J.; Sainz, B., Jr.; Heeschen, C. Inhibition of CD47 Effectively Targets Pancreatic Cancer Stem Cells via Dual Mechanisms. Clin. Cancer Res. 2015, 21, 2325–2337. [Google Scholar] [CrossRef] [PubMed]
- Arumugam, T.; Deng, D.; Bover, L.; Wang, H.; Logsdon, C.D.; Ramachandran, V. New Blocking Antibodies against Novel AGR2-C4.4A Pathway Reduce Growth and Metastasis of Pancreatic Tumors and Increase Survival in Mice. Mol. Cancer Ther. 2015, 14, 941–951. [Google Scholar] [CrossRef]
- Okusaka, T.; Ikeda, M.; Fukutomi, A.; Kobayashi, Y.; Shibayama, K.; Takubo, T.; Gansert, J. Safety, tolerability, pharmacokinetics and antitumor activity of ganitumab, an investigational fully human monoclonal antibody to insulin-like growth factor type 1 receptor, combined with gemcitabine as first-line therapy in patients with metastatic pancreatic cancer: A phase 1b study. Jpn. J. Clin. Oncol. 2014, 44, 442–447. [Google Scholar] [CrossRef]
- Royal, R.E.; Levy, C.; Turner, K.; Mathur, A.; Hughes, M.; Kammula, U.S.; Sherry, R.M.; Topalian, S.L.; Yang, J.C.; Lowy, I.; et al. Phase 2 trial of single agent Ipilimumab (anti-CTLA-4) for locally advanced or metastatic pancreatic adenocarcinoma. J. Immunother. 2010, 33, 828–833. [Google Scholar] [CrossRef]
- Philip, P.A.; Benedetti, J.; Corless, C.L.; Wong, R.; O’Reilly, E.M.; Flynn, P.J.; Rowland, K.M.; Atkins, J.N.; Mirtsching, B.C.; Rivkin, S.E.; et al. Phase III study comparing gemcitabine plus cetuximab versus gemcitabine in patients with advanced pancreatic adenocarcinoma: Southwest Oncology Group-directed intergroup trial S0205. J. Clin. Oncol. 2010, 28, 3605–3610. [Google Scholar] [CrossRef]
- Krempien, R.; Muenter, M.W.; Huber, P.E.; Nill, S.; Friess, H.; Timke, C.; Didinger, B.; Buechler, P.; Heeger, S.; Herfarth, K.K.; et al. Randomized phase II—Study evaluating EGFR targeting therapy with cetuximab in combination with radiotherapy and chemotherapy for patients with locally advanced pancreatic cancer—PARC: Study protocol [ISRCTN56652283]. BMC Cancer 2005, 5, 131. [Google Scholar] [CrossRef]
- Sinn, H.P.; Brown, S.A.; Thompson, J.S. Characterization of new monoclonal antibodies directed against normal human exocrine pancreas and pancreatic adenocarcinomas. Pancreas 1993, 8, 279–288. [Google Scholar] [CrossRef]
- Karanjawala, Z.E.; Illei, P.B.; Ashfaq, R.; Infante, J.R.; Murphy, K.; Pandey, A.; Schulick, R.; Winter, J.; Sharma, R.; Maitra, A.; et al. New markers of pancreatic cancer identified through differential gene expression analyses: Claudin 18 and annexin A8. Am. J. Surg. Pathol. 2008, 32, 188–196. [Google Scholar] [CrossRef]
- Sagiv, E.; Kazanov, D.; Arber, N. CD24 plays an important role in the carcinogenesis process of the pancreas. Biomed. Pharmacother. 2006, 60, 280–284. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Therapeutic Area | Number of Abs |
|---|---|
| Cancer | 91 |
| Immune-mediated disorders | 53 |
| Infectious diseases | 17 |
| Cardiovascular/hemostasis | 10 |
| Metabolic disorders | 8 |
| Neurological disorders | 7 |
| Ophthalmology | 4 |
| Genetic diseases | 3 |
| Musculoskeletal disorders | 3 |
| Hemostasis | 1 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Delgado, M.; Garcia-Sanz, J.A. Therapeutic Monoclonal Antibodies against Cancer: Present and Future. Cells 2023, 12, 2837. https://doi.org/10.3390/cells12242837
Delgado M, Garcia-Sanz JA. Therapeutic Monoclonal Antibodies against Cancer: Present and Future. Cells. 2023; 12(24):2837. https://doi.org/10.3390/cells12242837
Chicago/Turabian StyleDelgado, Marisa, and Jose A. Garcia-Sanz. 2023. "Therapeutic Monoclonal Antibodies against Cancer: Present and Future" Cells 12, no. 24: 2837. https://doi.org/10.3390/cells12242837
APA StyleDelgado, M., & Garcia-Sanz, J. A. (2023). Therapeutic Monoclonal Antibodies against Cancer: Present and Future. Cells, 12(24), 2837. https://doi.org/10.3390/cells12242837