
Autophagy Requirements for Eye Lens Differentiation and Transparency

 ,
,  ,
,  ,
, 
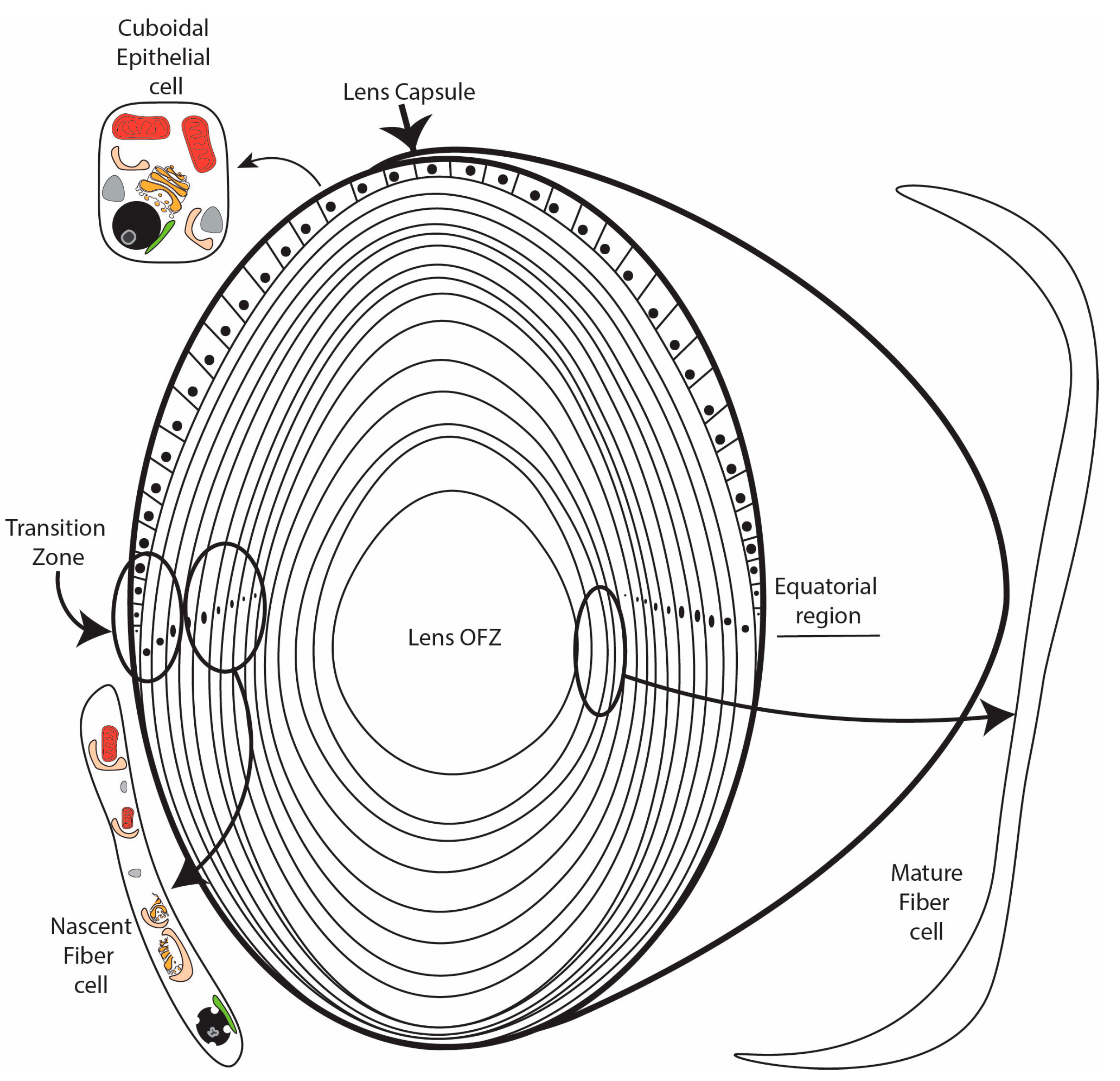{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Autophagy Overview
3. Signaling Regulators Control the Induction of Autophagy to Form the OFZ
4. Autophagy Mechanisms in Non-Nuclear Organelle Degradation
5. Lens Autophagy Structures
6. Evidence against Autophagy in Organelle Elimination
7. Role of Autophagy in Cataractogenesis
8. Summary
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Bassnett, S. On the mechanism of organelle degradation in the vertebrate lens. Exp. Eye Res. 2009, 88, 133–139. [Google Scholar] [CrossRef] [PubMed]
- Menko, A.S. Lens epithelial cell differentiation. Exp. Eye Res. 2002, 75, 485–490. [Google Scholar] [CrossRef] [PubMed]
- Wride, M.A. Lens fibre cell differentiation and organelle loss: Many paths lead to clarity. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2011, 366, 1219–1233. [Google Scholar] [CrossRef] [PubMed]
- Kupfer, C. The National Eye Institute’s low vision education program: Improving quality of life. Ophthalmology 2000, 107, 229–230. [Google Scholar] [CrossRef] [PubMed]
- Khairallah, M.; Kahloun, R.; Bourne, R.; Limburg, H.; Flaxman, S.R.; Jonas, J.B.; Keeffe, J.; Leasher, J.; Naidoo, K.; Pesudovs, K.; et al. Number of People Blind or Visually Impaired by Cataract Worldwide and in World Regions, 1990 to 2010. Investig. Ophthalmol. Vis. Sci. 2015, 56, 6762–6769. [Google Scholar] [CrossRef]
- Li, H.; Mao, Y.; Bouaziz, M.; Yu, H.; Qu, X.; Wang, F.; Feng, G.-S.; Shawber, C.; Zhang, X. Lens differentiation is controlled by the balance between PDGF and FGF signaling. PLoS Biol. 2019, 17, e3000133. [Google Scholar] [CrossRef]
- Lovicu, F.J.; McAvoy, J.W. FGF-induced lens cell proliferation and differentiation is dependent on MAPK (ERK1/2) signalling. Development 2001, 128, 5075–5084. [Google Scholar] [CrossRef]
- Chamberlain, C.G.; McAvoy, J.W. Induction of lens fibre differentiation by acidic and basic fibroblast growth factor (FGF). Growth Factors 1989, 1, 125–134. [Google Scholar] [CrossRef]
- Klok, E.j.; Lubsen, N.H.; Chamberlain, C.G.; McAvoy, J.W. Induction and Maintenance of Differentiation of Rat Lens Epithelium by FGF-2, Insulin and IGF-1. Exp. Eye Res. 1998, 67, 425–431. [Google Scholar] [CrossRef]
- Basu, S.; Rajakaruna, S.; De Arcangelis, A.; Zhang, L.; Georges-Labouesse, E.; Menko, A.S. α6 integrin transactivates insulin-like growth factor receptor-1 (IGF-1R) to regulate caspase-3-mediated lens epithelial cell differentiation initiation. J. Biol. Chem. 2014, 289, 3842–3855. [Google Scholar] [CrossRef] [Green Version]
- Mochizuki, T.; Masai, I. The lens equator: A platform for molecular machinery that regulates the switch from cell proliferation to differentiation in the vertebrate lens. Dev. Growth Differ. 2014, 56, 387–401. [Google Scholar] [CrossRef]
- Brennan, L.; Disatham, J.; Kantorow, M. Mechanisms of organelle elimination for lens development and differentiation. Exp. Eye Res. 2021, 209, 108682. [Google Scholar] [CrossRef]
- Hejtmancik, J.F.; Riazuddin, S.A.; McGreal, R.; Liu, W.; Cvekl, A.; Shiels, A. Lens Biology and Biochemistry. Prog. Mol. Biol. Transl. Sci. 2015, 134, 169–201. [Google Scholar]
- Yu, G.; Klionsky, D.J. Life and Death Decisions—The Many Faces of Autophagy in Cell Survival and Cell Death. Biomolecules 2022, 12, 866. [Google Scholar] [CrossRef]
- Gómez-Virgilio, L.; Silva-Lucero, M.-D.; Flores-Morelos, D.-S.; Gallardo-Nieto, J.; Lopez-Toledo, G.; Abarca-Fernandez, A.-M.; Zacapala-Gómez, A.-E.; Luna-Muñoz, J.; Montiel-Sosa, F.; Soto-Rojas, L.O.; et al. Autophagy: A Key Regulator of Homeostasis and Disease: An Overview of Molecular Mechanisms and Modulators. Cells 2022, 11, 2262. [Google Scholar] [CrossRef]
- Xie, Z.; Klionsky, D.J. Autophagosome formation: Core machinery and adaptations. Nat. Cell Biol. 2007, 9, 1102–1109. [Google Scholar] [CrossRef]
- Hubbard, V.M.; Valdor, R.; Macian, F.; Cuervo, A.M. Selective autophagy in the maintenance of cellular homeostasis in aging organisms. Biogerontology 2012, 13, 21–35. [Google Scholar] [CrossRef]
- Kaushik, S.; Cuervo, A.M. The coming of age of chaperone-mediated autophagy. Nat. Rev. Mol. Cell Biol. 2018, 19, 365–381. [Google Scholar] [CrossRef]
- Vargas, J.N.S.; Hamasaki, M.; Kawabata, T.; Youle, R.J.; Yoshimori, T. The mechanisms and roles of selective autophagy in mammals. Nat. Rev. Mol. Cell Biol. 2022, 1–19. [Google Scholar] [CrossRef]
- Wang, L.; Daniel, D.J.; Klionsky, J.; Han-Ming Shen, H.M. The emerging mechanisms and functions of microautophagy. Nat. Rev. Mol. Cell Biol. 2022, 1–18. [Google Scholar] [CrossRef]
- Kroemer, G.; Marino, G.; Levine, B. Autophagy and the integrated stress response. Mol. Cell 2010, 40, 280–293. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Levine, B.; Yuan, J. Autophagy Cell Death: Innocent Convict? J. Clin. Investig. 2005, 115, 2679–2688. [Google Scholar] [CrossRef] [PubMed]
- Mizushima, N.; Komatsu, M. Autophagy: Renovation of cells and tissues. Cell 2011, 147, 728–741. [Google Scholar] [CrossRef] [PubMed]
- Lu, G.; Wang, Y.; Shi, Y.; Zhang, Z.; Huang, C.; He, W.; Wang, C.; Shen, H. Autophagy in health and disease: From molecular mechanisms to therapeutic target. MedComm 2022, 3, e150. [Google Scholar] [CrossRef] [PubMed]
- Shibutani, S.T.; Yoshimori, T. A current perspective of autophagosome biogenesis. Cell Res. 2014, 24, 58–68. [Google Scholar] [CrossRef]
- Majeed, S.T.; Majeed, R.; Andrabi, K.I. Expanding the view of the molecular mechanisms of autophagy pathway. J. Cell. Physiol. 2022, 237, 3257–3277. [Google Scholar] [CrossRef]
- Parzych, K.R.; Klionsky, D.J. An overview of autophagy: Morphology, mechanism, and regulation. Antioxid. Redox Signal. 2014, 20, 460–473. [Google Scholar] [CrossRef]
- Yang, Z.; Klionsky, D.J. An overview of the molecular mechanism of autophagy. In Autophagy in Infection and Immunity. Current Topics in Microbiology and Immunology; Springer: Berlin/Heidelberg, Germany, 2009; Volume 335, pp. 1–32. [Google Scholar]
- Hayashi-Nishino, M.; Fujita, N.; Noda, T.; Yamaguchi, A.; Yoshimori, T.; Yamamoto, A. A subdomain of the endoplasmic reticulum forms a cradle for autophagosome formation. Nat. Cell Biol. 2009, 11, 1433–1437. [Google Scholar] [CrossRef]
- Ylä-Anttila, P.; Vihinen, H.; Jokitalo, E.; Eskelinen, E.-L. 3D tomography reveals connections between the phagophore and endoplasmic reticulum. Autophagy 2009, 5, 1180–1185. [Google Scholar] [CrossRef]
- Kabeya, Y.; Mizushima, N.; Yamamoto, A.; Oshitani-Okamoto, S.; Ohsumi, Y.; Yoshimori, T. LC3, GABARAP and GATE16 localize to autophagosomal membrane depending on form-II formation. J. Cell. Sci. 2004, 117, 2805–2812. [Google Scholar] [CrossRef]
- Shpilka, T.; Weidberg, H.; Pietrokovski, S.; Elazar, Z. Atg8: An autophagy-related ubiquitin-like protein family. Genome Biol. 2011, 12, 226. [Google Scholar] [CrossRef]
- Klionsky, D.J.; Abdel-Aziz, A.K.; Abdelfatah, S.; Abdellatif, M.; Abdoli, A.; Abel, S.; Abeliovich, H.; Abildgaard, M.H.; Abudu, Y.P.; Acevedo-Arozena, A.; et al. Guidelines for the use and interpretation of assays for monitoring autophagy. In Autophag, 4th ed.; Taylor & Francis: Abingdon, UK, 2021; Volume 17, pp. 1–382. [Google Scholar]
- Noda, N.N. Atg2 and Atg9: Intermembrane and interleaflet lipid transporters driving autophagy. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2021, 1866, 158956. [Google Scholar] [CrossRef]
- Kakuta, S.; Yamamoto, H.; Negishi, L.; Kondo-Kakuta, C.; Hayashi, N.; Ohsumi, Y. Atg9 vesicles recruit vesicle-tethering proteins Trs85 and Ypt1 to the autophagosome formation site. J. Biol. Chem. 2012, 287, 44261–44269. [Google Scholar] [CrossRef]
- Matoba, K.; Kotani, T.; Tsutsumi, A.; Tsuji, T.; Mori, T.; Noshiro, D.; Sugita, Y.; Nomura, N.; Iwata, S.; Ohsumi, Y.; et al. Atg9 is a lipid scramblase that mediates autophagosomal membrane expansion. Nat. Struct. Mol. Biol. 2020, 27, 1185–1193. [Google Scholar] [CrossRef]
- Orii, M.; Tsuji, T.; Ogasawara, Y.; Fujimoto, T. Transmembrane phospholipid translocation mediated by Atg9 is involved in autophagosome formation. J. Cell Biol. 2021, 220, e202009194. [Google Scholar] [CrossRef]
- Nakamura, S.; Yoshimori, T. New insights into autophagosome-lysosome fusion. J. Cell Sci. 2017, 130, 1209–1216. [Google Scholar] [CrossRef]
- Nishida, Y.; Arakawa, S.; Fujitani, K.; Yamaguchi, H.; Mizuta, T.; Kanaseki, T.; Komatsu, M.; Otsu, K.; Tsujimoto, Y.; Shimizu, S. Discovery of Atg5/Atg7-independent alternative macroautophagy. Nature 2009, 461, 654–658. [Google Scholar] [CrossRef]
- Bassnett, S.; Mataic, D. Chromatin Degradation in Differentiating Fiber Cells of the Eye Lens. J. Cell Biol. 1997, 137, 37–49. [Google Scholar] [CrossRef]
- Basu, S.; Rajakaruna, S.; Reyes, B.; Van Bockstaele, E.; Menko, A.S. Suppression of MAPK/JNK-MTORC1 signaling leads to premature loss of organelles and nuclei by autophagy during terminal differentiation of lens fiber cells. Autophagy 2014, 10, 1193–1211. [Google Scholar] [CrossRef]
- Costello, M.J.; Brennan, L.A.; Basu, S.; Chauss, D.; Mohamed, A.; Gilliland, K.O.; Johnsen, S.; Menko, A.S.; Kantorow, M. Autophagy and mitophagy participate in ocular lens organelle degradation. Exp. Eye Res. 2013, 116, 141–150. [Google Scholar] [CrossRef]
- Gheyas, R.; Ortega-Alvarez, R.; Chauss, D.; Kantorow, M.; Menko, A.S. Suppression of PI3K signaling is linked to autophagy activation and the spatiotemporal induction of the lens organelle free zone. Exp. Cell Res. 2022, 412, 113043. [Google Scholar] [CrossRef] [PubMed]
- Menko, A.S. The link between inhibition of PI3K signaling, induction of autophagy, and elimination of organelles to form the lens organelle-free zone. Autophagy Rep. 2022, 1, 238–241. [Google Scholar] [CrossRef]
- Quan, Y.; Du, Y.; Tong, Y.; Gu, S.; Jiang, J.X. Connexin Gap Junctions and Hemichannels in Modulating Lens Redox Homeostasis and Oxidative Stress in Cataractogenesis. Antioxidants 2021, 10, 1374. [Google Scholar] [CrossRef] [PubMed]
- Cheng, C.; Nowak, R.B.; Gao, J.; Sun, X.; Biswas, S.K.; Lo, W.-K.; Mathias, R.T.; Fowler, V.M. Lens ion homeostasis relies on the assembly and/or stability of large connexin 46 gap junction plaques on the broad sides of differentiating fiber cells. Am. J. Physiol. Cell Physiol. 2015, 308, C835–C847. [Google Scholar] [CrossRef]
- Mathias, R.T.; Kistler, J.; Donaldson, P. The lens circulation. J. Membr. Biol. 2007, 216, 1–16. [Google Scholar] [CrossRef]
- Donaldson, P.; Kistler, J.; Mathias, R. Molecular solutions to mammalian lens transparency. Physiology 2001, 16, 118–123. [Google Scholar] [CrossRef]
- Morishita, H.; Eguchi, S.; Kimura, H.; Sasaki, J.; Sakamaki, Y.; Robinson, M.L.; Sasaki, T.; Mizushima, N. Deletion of autophagy-related 5 (Atg5) and Pik3c3 genes in the lens causes cataract independent of programmed organelle degradation. J. Biol. Chem. 2013, 288, 11436–11447. [Google Scholar] [CrossRef]
- Levine, B.; Klionsky, D.J. Development by Self-Digestion: Molecular Mechanisms and Biological Functions of Autophagy. Dev. Cell 2004, 6, 463–477. [Google Scholar] [CrossRef]
- Yu, X.; Long, Y.C.; Shen, H.M. Differential regulatory functions of three classes of phosphatidylinositol and phosphoinositide 3-kinases in autophagy. Autophagy 2015, 11, 1711–1728. [Google Scholar] [CrossRef]
- Vanhaesebroeck, B.; Stephens, L.; Hawkins, P. PI3K signalling: The path to discovery and understanding. Nat. Rev. Mol. Cell Biol. 2012, 13, 195–203. [Google Scholar] [CrossRef]
- Menko, A.S.; Klukas, K.A.; Johnson, R.G. Chicken embryo lens cultures mimic differentiation in the lens. Dev. Biol. 1984, 103, 129–141. [Google Scholar] [CrossRef]
- Weber, G.F.; Menko, A.S. Phosphatidylinositol 3-kinase is necessary for lens fiber cell differentiation and survival. Investig. Ophthalmol. Vis. Sci. 2006, 47, 4490–4499. [Google Scholar] [CrossRef]
- Kim, Y.C.; Guan, K.L. mTOR: A pharmacologic target for autophagy regulation. J. Clin. Investig. 2015, 125, 25–32. [Google Scholar] [CrossRef]
- Yang, C.; Wang, X. Lysosome biogenesis: Regulation and functions. J. Cell Biol. 2021, 220, e202102001. [Google Scholar] [CrossRef]
- Uyama, T.; Tsuboi, K.; Ueda, N. An involvement of phospholipase A/acyltransferase family proteins in peroxisome regulation and plasmalogen metabolism. FEBS Lett. 2017, 591, 2745–2760. [Google Scholar] [CrossRef]
- Morishita, H.; Eguchi, T.; Tsukamoto, S.; Sakamaki, Y.; Takahashi, S.; Saito, C.; Koyama-Honda, I.; Mizushima, N. Organelle degradation in the lens by PLAAT phospholipases. Nature 2021, 592, 634–638. [Google Scholar] [CrossRef]
- Watanabe, S.; Nihongaki, Y.; Itoh, K.; Uyama, T.; Toda, S.; Watanabe, S.; Inoue, T. Defunctionalizing intracellular organelles such as mitochondria and peroxisomes with engineered phospholipase A/acyltransferases. Nat. Commun. 2022, 13, 4413. [Google Scholar] [CrossRef]
- Weber, G.F.; Menko, A.S. The canonical intrinsic mitochondrial death pathway has a non-apoptotic role in signaling lens cell differentiation. J. Biol. Chem. 2005, 280, 22135–22145. [Google Scholar] [CrossRef]
- Zhu, Z.; He, X.; Johnson, C.; Stoops, J.; Eaker, A.E.; Stoffer, D.S.; Bell, A.; Zarnegar, R.; DeFrances, M.C. PI3K is negatively regulated by PIK3IP1, a novel p110 interacting protein. Biochem. Biophys. Res. Commun. 2007, 358, 66–72. [Google Scholar] [CrossRef]
- Novak, I.; Kirkin, V.; McEwan, D.G.; Zhang, J.; Wild, P.; Rozenknop, A.; Rogov, V.; Löhr, F.; Popovic, D.; Occhipinti, A.; et al. Nix is a selective autophagy receptor for mitochondrial clearance. EMBO Rep. 2010, 11, 45–51. [Google Scholar] [CrossRef]
- Sandoval, H.; Thiagarajan, P.; Dasgupta, S.K.; Schumacher, A.; Prchal, J.T.; Chen, M.; Wang, J. Essential role for Nix in autophagic maturation of erythroid cells. Nature 2008, 454, 232–235. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schweers, R.L.; Zhang, J.; Randall, M.S.; Loyd, M.R.; Li, W.; Dorsey, F.C.; Kundu, M.; Opferman, J.T.; Cleveland, J.L.; Miller, J.L.; et al. NIX is required for programmed mitochondrial clearance during reticulocyte maturation. Proc. Natl. Acad. Sci. USA 2007, 104, 19500–19505. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Ney, P.A. Mechanisms and biology of B-cell leukemia/lymphoma 2/adenovirus E1B interacting protein 3 and Nip-like protein X. Antioxid. Redox Signal. 2011, 14, 1959–1969. [Google Scholar] [CrossRef] [PubMed]
- Onishi, M.; Yamano, K.; Sato, M.; Matsuda, N.; Okamoto, K. Molecular mechanisms and physiological functions of mitophagy. EMBO J. 2021, 40, e104705. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Ney, P.A. Role of BNIP3 and NIX in cell death, autophagy, and mitophagy. Cell Death Differ. 2009, 16, 939–946. [Google Scholar] [CrossRef]
- Diwan, A.; Koesters, A.G.; Odley, A.M.; Pushkaran, S.; Baines, C.P.; Spike, B.T.; Daria, D.; Jegga, A.G.; Geiger, H.; Aronow, B.J.; et al. Unrestrained erythroblast development in Nix−/− mice reveals a mechanism for apoptotic modulation of erythropoiesis. Proc. Natl. Acad. Sci. USA 2007, 104, 6794–6799. [Google Scholar] [CrossRef]
- Marinkovic, M.; Sprung, M.; Novak, I. Dimerization of mitophagy receptor BNIP3L/NIX is essential for recruitment of autophagic machinery. Autophagy 2021, 17, 1232–1243. [Google Scholar] [CrossRef]
- Chauss, D.; Basu, S.; Rajakaruna, S.; Ma, Z.; Gau, V.; Anastas, S.; Brennan, L.A.; Hejtmancik, J.F.; Menko, A.S.; Kantorow, M. Differentiation state-specific mitochondrial dynamic regulatory networks are revealed by global transcriptional analysis of the developing chicken lens. G3 Genes Genomes Genet. 2014, 4, 1515–1527. [Google Scholar] [CrossRef]
- Brennan, L.A.; McGreal-Estrada, R.; Logan, C.M.; Cvekl, A.; Menko, A.S.; Kantorow, M. BNIP3L/NIX is required for elimination of mitochondria, endoplasmic reticulum and Golgi apparatus during eye lens organelle-free zone formation. Exp. Eye Res. 2018, 174, 173–184. [Google Scholar] [CrossRef]
- Diwan, A.; Matkovich, S.J.; Yuan, Q.; Zhao, W.; Yatani, A.; Brown, J.H.; Molkentin, J.D.; Kranias, E.G.; Dorn, G.W. Endoplasmic reticulum-mitochondria crosstalk in NIX-mediated murine cell death. J. Clin. Investig. 2009, 119, 203–212. [Google Scholar] [CrossRef]
- Bellot, G.; Garcia-Medina, R.; Gounon, P.; Chiche, J.; Roux, D.; Pouysségur, J.; Mazure, N.M. Hypoxia-induced autophagy is mediated through hypoxia-inducible factor induction of BNIP3 and BNIP3L via their BH3 domains. Mol. Cell. Biol. 2009, 29, 2570–2581. [Google Scholar] [CrossRef] [Green Version]
- Cummins, E.P.; Taylor, C.T. Hypoxia-responsive transcription factors. Pflügers Arch. 2005, 450, 363–371. [Google Scholar] [CrossRef]
- Kenneth, N.S.; Rocha, S. Regulation of gene expression by hypoxia. Biochem. J. 2008, 414, 19–29. [Google Scholar] [CrossRef]
- Brahimi-Horn, M.C.; Pouysségur, J. HIF1 at a glance. J. Cell Sci. 2009, 122, 1055–1057. [Google Scholar] [CrossRef]
- Lutty, G.A.; McLeod, D.S. Development of the hyaloid, choroidal and retinal vasculatures in the fetal human eye. Prog. Retin. Eye Res. 2018, 62, 58–76. [Google Scholar] [CrossRef]
- Goldberg, M.F. Persistent Fetal Vasculature (PFV): An Integrated Interpretation of Signs and Symptoms Associated with Persistent Hyperplastic Primary Vitreous (PHPV) LIV Edward Jackson Memorial Lecture. Am. J. Ophthalmol. 1997, 124, 587–626. [Google Scholar] [CrossRef]
- Bassnett, S.; McNulty, R. The effect of elevated intraocular oxygen on organelle degradation in the embryonic chicken lens. J. Exp. Biol. 2003, 206, 4353–4361. [Google Scholar] [CrossRef]
- Beebe, D.C.; Shui, Y.-B.; Siegfried, C.J.; Holekamp, N.M.; Bai, F. Preserve the (intraocular) environment: The importance of maintaining normal oxygen gradients in the eye. Jpn. J. Ophthalmol. 2014, 58, 225–231. [Google Scholar] [CrossRef]
- McNulty, R.; Wang, H.; Mathias, R.T.; Ortwerth, B.J.; Truscott, R.J.W.; Bassnett, S. Regulation of tissue oxygen levels in the mammalian lens. J. Physiol. 2004, 559, 883–898. [Google Scholar] [CrossRef]
- Shestopalov, V.I.; Bassnett, S. Expression of autofluorescent proteins reveals a novel protein permeable pathway between cells in the lens core. J. Cell Sci. 2000, 113, 1913–1921. [Google Scholar] [CrossRef]
- Brennan, L.; Disatham, J.; Kantorow, M. Hypoxia regulates the degradation of non-nuclear organelles during lens differentiation through activation of HIF1a. Exp. Eye Res. 2020, 198, 108129. [Google Scholar] [CrossRef] [PubMed]
- Baudhuin, P.; Beaufay, H.; de Duve, C. Combined biochemical and morphological study of particulate fractions from rat liver. J. Cell Biol. 1965, 26, 219–243. [Google Scholar] [CrossRef] [PubMed]
- De Duve, C.; Wattiaux, R. Functions of Lysosomes. Ann. Rev. Physiol. 1966, 28, 435–492. [Google Scholar] [CrossRef] [PubMed]
- Costello, M.J.; Brennan, L.A.; Mohamed, A.; Gilliland, K.O.; Johnsen, S.; Kantorow, M. Identification and Ultrastructural Characterization of a Novel Nuclear Degradation Complex in Differentiating Lens Fiber Cells. PLoS ONE 2016, 11, e0160785. [Google Scholar] [CrossRef]
- Costello, M.J.; Gilliland, K.O.; Mohamed, A.; Schey, K.L.; Johnsen, S.; Brennan, L.A.; Kantorow, M. Novel mitochondrial derived Nuclear Excisosome degrades nuclei during differentiation of prosimian Galago (bush baby) monkey lenses. PLoS ONE 2020, 15, e0241631. [Google Scholar] [CrossRef]
- Wignes, J.A.; Goldman, J.W.; Weihl, C.C.; Bartley, M.G.; Andley, U.P. p62 expression and autophagy in alphaB-crystallin R120G mutant knock-in mouse model of hereditary cataract. Exp. Eye Res. 2013, 115, 263–273. [Google Scholar] [CrossRef]
- Costello, M.J.; Johnsen, S.; Metlapally, S.; Gilliland, K.O.; Frame, L.; Balasubramanian, D. Multilamellar spherical particles as potential sources of excessive light scattering in human age-related nuclear cataracts. Exp. Eye Res. 2010, 91, 881–889. [Google Scholar] [CrossRef]
- Pavel, M.; Rubinsztein, D.C. Mammalian autophagy and the plasma membrane. FEBS J. 2017, 284, 672–679. [Google Scholar] [CrossRef]
- Matsui, M.; Yamamoto, A.; Kuma, A.; Ohsumi, Y.; Mizushima, N. Organelle degradation during the lens and erythroid differentiation is independent of autophagy. Biochem. Biophys. Res. Commun. 2006, 339, 485–489. [Google Scholar] [CrossRef]
- Chen, J.; Ma, Z.; Jiao, X.; Fariss, R.; Kantorow, W.L.; Kantorow, M.; Pras, E.; Frydman, M.; Pras, E.; Riazuddin, S.; et al. Mutations in FYCO1 cause autosomal-recessive congenital cataracts. Am. J. Hum. Genet. 2011, 88, 827–838. [Google Scholar] [CrossRef]
- Khan, S.Y.; Ali, M.; Kabir, F.; Na, C.H.; Delannoy, M.; Ma, Y.; Qiu, C.; Costello, M.J.; Hejtmancik, J.F.; Riazuddin, S.A. The role of FYCO1-dependent autophagy in lens fiber cell differentiation. Autophagy 2022, 18, 2198–2215. [Google Scholar] [CrossRef]
- Satoh, K.; Takemura, Y.; Satoh, M.; Ozaki, K.; Kubota, S. Loss of FYCO1 leads to cataract formation. Sci. Rep. 2021, 11, 13771. [Google Scholar] [CrossRef]
- McWilliams, T.G.; Prescott, A.R.; Villarejo-Zori, B.; Ball, G.; Boya, P.; Ganley, I.G. A comparative map of macroautophagy and mitophagy in the vertebrate eye. Autophagy 2019, 15, 1296–1308. [Google Scholar] [CrossRef]
- Shiels, A.; Bennett, T.M.; Knopf, H.L.; Yamada, K.; Yoshiura, K.-I.; Niikawa, N.; Shim, S.; Hanson, P.I. CHMP4B, a novel gene for autosomal dominant cataracts linked to chromosome 20q. Am. J. Hum. Genet. 2007, 81, 596–606. [Google Scholar] [CrossRef]
- Lefebvre, C.; Legouis, R.; Culetto, E. ESCRT and autophagies: Endosomal functions and beyond. Semin. Cell Dev. Biol. 2018, 74, 21–28. [Google Scholar] [CrossRef]
- Olmos, Y. The ESCRT Machinery: Remodeling, Repairing, and Sealing Membranes. Membranes 2022, 12, 633. [Google Scholar] [CrossRef]
- Lee, J.-A.; Beigneux, A.; Ahmad, S.T.; Young, S.G.; Gao, F.-B. ESCRT-III dysfunction causes autophagosome accumulation and neurodegeneration. Curr. Biol. 2007, 17, 1561–1567. [Google Scholar] [CrossRef]
- Lee, J.A.; Gao, F.B. Roles of ESCRT in autophagy-associated neurodegeneration. Autophagy 2008, 4, 230–232. [Google Scholar] [CrossRef]
- Sagona, A.P.; Nezis, I.P.; Stenmark, H. Association of CHMP4B and autophagy with micronuclei: Implications for cataract formation. BioMed Res. Int. 2014, 2014, 974393. [Google Scholar] [CrossRef]
- Zhang, X.H.; Da Wang, J.; Jia, H.Y.; Zhang, J.S.; Li, Y.; Xiong, Y.; Li, J.; Li, X.X.; Huang, Y.; Zhu, G.Y.; et al. Mutation profiles of congenital cataract genes in 21 northern Chinese families. Mol. Vis. 2018, 24, 471–477. [Google Scholar]
- Jackson, D.; Malka, S.; Harding, P.; Palma, J.; Dunbar, H.; Moosajee, M. Molecular diagnostic challenges for non-retinal developmental eye disorders in the United Kingdom. Am. J. Med. Genet. C Semin. Med. Genet. 2020, 184, 578–589. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Wang, D.; Wang, Q.; Huang, W.; Dongye, M.; Zhang, X.; Lin, D.; Lin, Z.; Li, J.; Hu, W.; et al. Broadening the Mutation Spectrum in GJA8 and CHMP4B: Novel Missense Variants and the Associated Phenotypes in Six Chinese Han Congenital Cataracts Families. Front. Med. 2021, 8, 713284. [Google Scholar] [CrossRef] [PubMed]
- Yonova-Doing, E.; Zhao, W.; Igo, R.P.; Wang, C.; Sundaresan, P.; Lee, K.E.; Jun, G.R.; Alves, A.C.; Chai, X.; Chan, A.S.Y.; et al. Common variants in SOX-2 and congenital cataract genes contribute to age-related nuclear cataract. Commun. Biol. 2020, 3, 755. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Bennett, T.M.; Shiels, A. A charged multivesicular body protein (CHMP4B) is required for lens growth and differentiation. Differentiation 2019, 109, 16–27. [Google Scholar] [CrossRef]
- Skowyra, M.L.; Schlesinger, P.H.; Naismith, T.V.; Hanson, P.I. Triggered recruitment of ESCRT machinery promotes endolysosomal repair. Science 2018, 360, eaar5078. [Google Scholar] [CrossRef]
- Radulovic, M.; O Schink, K.; Wenzel, E.M.; Nähse, V.; Bongiovanni, A.; Lafont, F.; Stenmark, H. ESCRT-mediated lysosome repair precedes lysophagy and promotes cell survival. EMBO J. 2018, 37, e99753. [Google Scholar] [CrossRef]
- Loi, M.; Raimondi, A.; Morone, D.; Molinari, M. ESCRT-III-driven piecemeal micro-ER-phagy remodels the ER during recovery from ER stress. Nat. Commun. 2019, 10, 5058. [Google Scholar] [CrossRef]
- Zhen, Y.; Spangenberg, H.; Munson, M.J.; Brech, A.; Schink, K.O.; Tan, K.-W.; Sørensen, V.; Wenzel, E.M.; Radulovic, M.; Engedal, N.; et al. ESCRT-mediated phagophore sealing during mitophagy. Autophagy 2020, 16, 826–841. [Google Scholar] [CrossRef]
- Gulluni, F.; Prever, L.; Li, H.; Krafcikova, P.; Corrado, I.; Lo, W.-T.; Margaria, J.P.; Chen, A.; De Santis, M.C.; Cnudde, S.J.; et al. PI(3,4)P2-mediated cytokinetic abscission prevents early senescence and cataract formation. Science 2021, 374, eabk0410. [Google Scholar] [CrossRef]
- Chen, J.; Wang, Q.; Cabrera, P.E.; Zhong, Z.; Sun, W.; Jiao, X.; Chen, Y.; Govindarajan, G.; Naeem, M.A.; Khan, S.N.; et al. Molecular Genetic Analysis of Pakistani Families with Autosomal Recessive Congenital Cataracts by Homozygosity Screening. Invest. Ophthalmol. Vis. Sci. 2017, 58, 2207–2217. [Google Scholar] [CrossRef]
- Barashkov, N.A.; Konovalov, F.A.; Borisova, T.V.; Teryutin, F.M.; Solovyev, A.V.; Pshennikova, V.G.; Sapojnikova, N.V.; Vychuzhina, L.S.; Romanov, G.P.; Gotovtsev, N.N.; et al. Autosomal recessive cataract (CTRCT18) in the Yakut population isolate of Eastern Siberia: A novel founder variant in the FYCO1 gene. Eur. J. Hum. Genet. 2021, 29, 965–976. [Google Scholar] [CrossRef]
- Pankiv, S.; Alemu, E.A.; Brech, A.; Bruun, J.-A.; Lamark, T.; Øvervatn, A.; Bjørkøy, G.; Johansen, T. FYCO1 is a Rab7 effector that binds to LC3 and PI3P to mediate microtubule plus end-directed vesicle transport. J. Cell Biol. 2010, 188, 253–269. [Google Scholar] [CrossRef] [Green Version]
- Choquet, H.; Melles, R.B.; Anand, D.; Yin, J.; Cuellar-Partida, G.; Wang, W.; Hoffmann, T.J.; Nair, K.S.; Hysi, P.G.; Lachke, S.A.; et al. A large multiethnic GWAS meta-analysis of cataract identifies new risk loci and sex-specific effects. Nat. Commun. 2021, 12, 3595. [Google Scholar] [CrossRef]
- Xiao, W.; Yeerken, D.; Li, J.; Li, Z.; Jiang, L.; Li, D.; Fu, M.; Ma, L.; Song, Y.; Zhang, W.; et al. Nlp promotes autophagy through facilitating the interaction of Rab7 and FYCO1. Signal Transduct. Target. Ther. 2021, 6, 152. [Google Scholar] [CrossRef]
- Nieto-Torres, J.L.; Shanahan, S.-L.; Chassefeyre, R.; Chaiamarit, T.; Zaretski, S.; Landeras-Bueno, S.; Verhelle, A.; Encalada, S.E.; Hansen, M. LC3B phosphorylation regulates FYCO1 binding and directional transport of autophagosomes. Curr. Biol. 2021, 31, 3440–3449.e7. [Google Scholar] [CrossRef]
- Olsvik, H.L.; Lamark, T.; Takagi, K.; Larsen, K.B.; Evjen, G.; Øvervatn, A.; Mizushima, T.; Johansen, T. FYCO1 Contains a C-terminally Extended, LC3A/B-preferring LC3-interacting Region (LIR) Motif Required for Efficient Maturation of Autophagosomes during Basal Autophagy. J. Biol. Chem. 2015, 290, 29361–29374. [Google Scholar] [CrossRef]
- Cheng, X.; Wang, Y.; Gong, Y.; Li, F.; Guo, Y.; Hu, S.; Liu, J.; Pan, L. Structural basis of FYCO1 and MAP1LC3A interaction reveals a novel binding mode for Atg8-family proteins. Autophagy 2016, 12, 1330–1339. [Google Scholar] [CrossRef]
- Hong, Z.; Pedersen, N.M.; Wang, L.; Torgersen, M.L.; Stenmark, H.; Raiborg, C. PtdIns3P controls mTORC1 signaling through lysosomal positioning. J. Cell Biol. 2017, 216, 4217–4233. [Google Scholar] [CrossRef]
- Chen, J.H.; Huang, C.; Zhang, B.; Yin, S.; Liang, J.; Xu, C.; Huang, Y.; Cen, L.P.; Ng, T.K.; Zheng, C.; et al. Mutations of RagA GTPase in mTORC1 Pathway Are Associated with Autosomal Dominant Cataracts. PLoS Genet. 2016, 12, e1006090. [Google Scholar] [CrossRef]
- Lachke, S.A.; Alkuraya, F.S.; Kneeland, S.C.; Ohn, T.; Aboukhalil, A.; Howell, G.R.; Saadi, I.; Cavallesco, R.; Yue, Y.; Tsai, A.C.; et al. Mutations in the RNAGranule Component TDRD7 Cause Cataract and Glaucoma. Science 2011, 331, 1571–1576. [Google Scholar] [CrossRef]
- Tan, Y.-Q.; Tu, C.; Meng, L.; Yuan, S.; Sjaarda, C.; Luo, A.; Du, J.; Li, W.; Gong, F.; Zhong, C.; et al. Loss-of-function mutations in TDRD7 lead to a rare novel syndrome combining congenital cataract and nonobstructive azoospermia in humans. Genet. Med. 2019, 21, 1209–1217. [Google Scholar] [CrossRef] [PubMed]
- Zheng, C.; Wu, M.; He, C.-Y.; An, X.-J.; Sun, M.; Chen, C.-L.; Ye, J. RNA granule component TDRD7 gene polymorphisms in a Han Chinese population with age-related cataract. J. Int. Med. Res. 2014, 42, 153–163. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tu, C.; Li, H.; Liu, X.; Wang, Y.; Li, W.; Meng, L.; Wang, W.; Li, Y.; Li, D.; Du, J.; et al. TDRD7 participates in lens development and spermiogenesis by mediating autophagosome maturation. Autophagy 2021, 17, 3848–3864. [Google Scholar] [CrossRef] [PubMed]
- Liegel, R.P.; Handley, M.T.; Ronchetti, A.; Brown, S.; Langemeyer, L.; Linford, A.; Chang, B.; Morris-Rosendahl, D.J.; Carpanini, S.; Posmyk, R.; et al. Loss-of-function mutations in TBC1D20 cause cataracts and male infertility in blind sterile mice and Warburg micro syndrome in humans. Am. J. Hum. Genet. 2013, 93, 1001–1014. [Google Scholar] [CrossRef] [PubMed]
- Sidjanin, D.J.; Park, A.K.; Ronchetti, A.; Martins, J.; Jackson, W.T. TBC1D20 mediates autophagy as a key regulator of autophagosome maturation. Autophagy 2016, 12, 1759–1775. [Google Scholar] [CrossRef]
- Cullup, T.; Kho, A.L.; Dionisi-Vici, C.; Brandmeier, B.; Smith, F.; Urry, Z.; A Simpson, M.; Yau, S.; Bertini, E.; McClelland, V.; et al. Recessive mutations in EPG5 cause Vici syndrome, a multisystem disorder with defective autophagy. Nat. Genet. 2013, 45, 83–87. [Google Scholar] [CrossRef]
- Byrne, S.; Jansen, L.; U-King-Im, J.M.; Siddiqui, A.; Lidov, H.G.; Bodi, I.; Smith, L.; Mein, R.; Cullup, T.; Dionisi-Vici, C.; et al. EPG5-related Vici syndrome: A paradigm of neurodevelopmental disorders with defective autophagy. Brain 2016, 139, 765–781. [Google Scholar] [CrossRef]
- Ehmke, N.; Parvaneh, N.; Krawitz, P.; Ashrafi, M.-R.; Karimi, P.; Mehdizadeh, M.; Krüger, U.; Hecht, J.; Mundlos, S.; Robinson, P.N. First description of a patient with Vici syndrome due to a mutation affecting the penultimate exon of EPG5 and review of the literature. Am. J. Med. Genet. Part A 2014, 164, 3170–3175. [Google Scholar] [CrossRef]
- Rodger, C.; Flex, E.; Allison, R.J.; Sanchis-Juan, A.; Hasenahuer, M.A.; Cecchetti, S.; French, C.E.; Edgar, J.R.; Carpentieri, G.; Ciolfi, A.; et al. De Novo VPS4A Mutations Cause Multisystem Disease with Abnormal Neurodevelopment. Am. J. Hum. Genet. 2020, 107, 1129–1148. [Google Scholar] [CrossRef]
- Seu, K.G.; Trump, L.R.; Emberesh, S.; Lorsbach, R.B.; Johnson, C.; Meznarich, J.; Underhill, H.R.; Chou, S.T.; Sakthivel, H.; Nassar, N.N.; et al. VPS4A Mutations in Humans Cause Syndromic Congenital Dyserythropoietic Anemia due to Cytokinesis and Trafficking Defects. Am. J. Hum. Genet. 2020, 107, 1149–1156. [Google Scholar] [CrossRef]
- Ping, X.; Liang, J.; Shi, K.; Bao, J.; Wu, J.; Yu, X.; Tang, X.; Zou, J.; Shentu, X. Rapamycin relieves the cataract caused by ablation of Gja8b through stimulating autophagy in zebrafish. Autophagy 2021, 17, 3323–3337. [Google Scholar] [CrossRef]






Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Brennan, L.; Costello, M.J.; Hejtmancik, J.F.; Menko, A.S.; Riazuddin, S.A.; Shiels, A.; Kantorow, M. Autophagy Requirements for Eye Lens Differentiation and Transparency. Cells 2023, 12, 475. https://doi.org/10.3390/cells12030475
Brennan L, Costello MJ, Hejtmancik JF, Menko AS, Riazuddin SA, Shiels A, Kantorow M. Autophagy Requirements for Eye Lens Differentiation and Transparency. Cells. 2023; 12(3):475. https://doi.org/10.3390/cells12030475
Chicago/Turabian StyleBrennan, Lisa, M. Joseph Costello, J. Fielding Hejtmancik, A. Sue Menko, S. Amer Riazuddin, Alan Shiels, and Marc Kantorow. 2023. "Autophagy Requirements for Eye Lens Differentiation and Transparency" Cells 12, no. 3: 475. https://doi.org/10.3390/cells12030475
APA StyleBrennan, L., Costello, M. J., Hejtmancik, J. F., Menko, A. S., Riazuddin, S. A., Shiels, A., & Kantorow, M. (2023). Autophagy Requirements for Eye Lens Differentiation and Transparency. Cells, 12(3), 475. https://doi.org/10.3390/cells12030475