
Targeted Sequencing Approach and Its Clinical Applications for the Molecular Diagnosis of Human Diseases
 , , ,
, , ,
Abstract
:1. Introduction
2. Targeted Sequencing
2.1. The History of Sequencing and Discovery of TS
2.2. Assay Design Consideration for TS
2.2.1. Genetic Heterogeneity
2.2.2. Pre-Analytical Considerations
2.2.3. Sequencing Cost-Effectiveness
2.3. Method of TS
3. Clinical Applications of TS
3.1. SARS-CoV-2 Surveillance and COVID-19 Research
3.2. Bacteria
3.2.1. Usefulness and Clinical Benefits of Targeted 16S rRNA Gene Sequencing
3.2.2. Limitations and Challenges of Targeted 16S rRNA Gene Sequencing
3.3. Human
3.3.1. FFPE
3.3.2. cfDNA and Circulating Tumour DNA (ctDNA)
3.3.3. TS Approaches for Gene Fusion
3.3.4. TS Applications in Rare Disease
4. Challenging in Mutation Identification Genes/Diseases for Target Panels
4.1. Inborn Error of Metabolism NGS
4.2. Mitochondrial DNA NGS
4.3. Polycystic Kidney Disease NGS(PKD1/PKD2)
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Bewicke-Copley, F.; Kumar, E.A.; Palladino, G.; Korfi, K.; Wang, J. Applications and analysis of targeted genomic sequencing in cancer studies. Comput. Struct. Biotechnol. J. 2019, 17, 1348–1359. [Google Scholar] [CrossRef] [PubMed]
- Kulkarni, P.; Frommolt, P. Challenges in the setup of large-scale next-generation sequencing analysis workflows. Comput. Struct. Biotechnol. J. 2017, 15, 471–477. [Google Scholar] [CrossRef] [PubMed]
- Nakagawa, H.; Wardell, C.; Furuta, M.; Taniguchi, H.; Fujimoto, A. Cancer whole-genome sequencing: Present and future. Oncogene 2015, 34, 5943–5950. [Google Scholar] [CrossRef] [PubMed]
- Petersen, B.-S.; Fredrich, B.; Hoeppner, M.P.; Ellinghaus, D.; Franke, A. Opportunities and challenges of whole-genome and-exome sequencing. BMC Genet. 2017, 18, 1–13. [Google Scholar] [CrossRef]
- Paskey, A.C.; Frey, K.G.; Schroth, G.; Gross, S.; Hamilton, T.; Bishop-Lilly, K.A. Enrichment post-library preparation enhances the sensitivity of high-throughput sequencing-based detection and characterization of viruses from complex samples. BMC Genom. 2019, 20, 155. [Google Scholar] [CrossRef]
- Mertes, F.; Elsharawy, A.; Sauer, S.; van Helvoort, J.M.; van der Zaag, P.J.; Franke, A.; Nilsson, M.; Lehrach, H.; Brookes, A.J. Targeted enrichment of genomic DNA regions for next-generation sequencing. Brief Funct Genom. 2011, 10, 374–386. [Google Scholar] [CrossRef]
- Berger, M.F.; Mardis, E.R. The emerging clinical relevance of genomics in cancer medicine. Nat. Rev. Clin. Oncol. 2018, 15, 353–365. [Google Scholar] [CrossRef]
- John, G.; Sahajpal, N.S.; Mondal, A.K.; Ananth, S.; Williams, C.; Chaubey, A.; Rojiani, A.M.; Kolhe, R. Next-generation sequencing (NGS) in COVID-19: A tool for SARS-CoV-2 diagnosis, monitoring new strains and phylodynamic modeling in molecular epidemiology. Curr. Issues Mol. Biol. 2021, 43, 61. [Google Scholar] [CrossRef]
- Sanger, F.; Nicklen, S.; Coulson, A.R. DNA sequencing with chain-terminating inhibitors. Proc. Natl. Acad. Sci. USA 1977, 74, 5463–5467. [Google Scholar] [CrossRef]
- Heather, J.M.; Chain, B. The sequence of sequencers: The history of sequencing DNA. Genomics 2016, 107, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Ronaghi, M.; Uhlén, M.; Nyrén, P. A sequencing method based on real-time pyrophosphate. Science 1998, 281, 363–365. [Google Scholar] [CrossRef]
- Liu, L.; Li, Y.; Li, S.; Hu, N.; He, Y.; Pong, R.; Lin, D. Comparison of next-generation sequencing systems. In The Role of Bioinformatics in Agriculture; Apple Academic Press: Palm Bay, FL, USA, 2014; pp. 31–56. [Google Scholar]
- Wheeler, D.A.; Srinivasan, M.; Egholm, M.; Shen, Y.; Chen, L.; McGuire, A.; He, W.; Chen, Y.-J.; Makhijani, V.; Roth, G.T. The complete genome of an individual by massively parallel DNA sequencing. Nature 2008, 452, 872–876. [Google Scholar] [CrossRef]
- Lupski, J.R.; Reid, J.G.; Gonzaga-Jauregui, C.; Rio Deiros, D.; Chen, D.C.; Nazareth, L.; Bainbridge, M.; Dinh, H.; Jing, C.; Wheeler, D.A. Whole-genome sequencing in a patient with Charcot–Marie–Tooth neuropathy. N. Engl. J. Med. 2010, 362, 1181–1191. [Google Scholar] [CrossRef]
- Wong, A.N.N.; He, Z.; Leung, K.L.; To, C.C.K.; Wong, C.Y.; Wong, S.C.C.; Yoo, J.S.; Chan, C.K.R.; Chan, A.Z.; Lacambra, M.D.; et al. Current Developments of Artificial Intelligence in Digital Pathology and Its Future Clinical Applications in Gastrointestinal Cancers. Cancers 2022, 14, 3780. [Google Scholar] [CrossRef]
- McCabe, M.J.; Gauthier, M.-E.A.; Chan, C.-L.; Thompson, T.J.; De Sousa, S.; Puttick, C.; Grady, J.P.; Gayevskiy, V.; Tao, J.; Ying, K. Development and validation of a targeted gene sequencing panel for application to disparate cancers. Sci. Rep. 2019, 9, 17052. [Google Scholar] [CrossRef]
- Leung, H.Y.; Yeung, M.H.Y.; Leung, W.T.; Wong, K.H.; Tang, W.Y.; Cho, W.C.S.; Wong, H.T.; Tsang, H.F.; Wong, Y.K.E.; Pei, X.M.; et al. The current and future applications of in situ hybridization technologies in anatomical pathology. Expert Rev. Mol. Diagn. 2022, 22, 5–18. [Google Scholar] [CrossRef]
- Sagaert, X.; Vanstapel, A.; Verbeek, S. Tumor heterogeneity in colorectal cancer: What do we know so far? Pathobiology 2018, 85, 72–84. [Google Scholar] [CrossRef]
- Kalman, L.V.; Datta, V.; Williams, M.; Zook, J.M.; Salit, M.L.; Han, J.-Y. Development and characterization of reference materials for genetic testing: Focus on public partnerships. Ann. Lab. Med. 2016, 36, 513. [Google Scholar] [CrossRef]
- Petersen, J.L.; Coleman, S.J. Next-Generation Sequencing in Equine Genomics. Vet. Clin. Equine Pract. 2020, 36, 195–209. [Google Scholar] [CrossRef]
- Wadman, M. James Watson’s genome sequenced at high speed. Nature 2008, 452, 788. [Google Scholar] [CrossRef] [Green Version]
- Muir, P.; Li, S.; Lou, S.; Wang, D.; Spakowicz, D.J.; Salichos, L.; Zhang, J.; Weinstock, G.M.; Isaacs, F.; Rozowsky, J.; et al. The real cost of sequencing: Scaling computation to keep pace with data generation. Genome Biol. 2016, 17, 53. [Google Scholar] [CrossRef] [PubMed]
- Schwarze, K.; Buchanan, J.; Taylor, J.C.; Wordsworth, S. Are whole-exome and whole-genome sequencing approaches cost-effective? A systematic review of the literature. Genet. Med. 2018, 20, 1122–1130. [Google Scholar] [CrossRef] [PubMed]
- Alfares, A.; Aloraini, T.; Subaie, L.A.; Alissa, A.; Qudsi, A.A.; Alahmad, A.; Mutairi, F.A.; Alswaid, A.; Alothaim, A.; Eyaid, W.; et al. Whole-genome sequencing offers additional but limited clinical utility compared with reanalysis of whole-exome sequencing. Genet. Med. 2018, 20, 1328–1333. [Google Scholar] [CrossRef] [PubMed]
- Klau, J.; Abou Jamra, R.; Radtke, M.; Oppermann, H.; Lemke, J.R.; Beblo, S.; Popp, B. Exome first approach to reduce diagnostic costs and time-retrospective analysis of 111 individuals with rare neurodevelopmental disorders. Eur. J. Hum. Genet. 2022, 30, 117–125. [Google Scholar] [CrossRef]
- Masri, A.; Hamamy, H. Cost Effectiveness of Whole Exome Sequencing for Children with Developmental Delay in a Developing Country: A Study from Jordan. J. Pediatr. Neurol. 2021, 20, 20–23. [Google Scholar] [CrossRef]
- Gaudin, M.; Desnues, C. Hybrid capture-based next generation sequencing and its application to human infectious diseases. Front. Microbiol. 2018, 9, 2924. [Google Scholar] [CrossRef]
- Nagy-Szakal, D.; Couto-Rodriguez, M.; Wells, H.L.; Barrows, J.E.; Debieu, M.; Butcher, K.; Chen, S.; Berki, A.; Hager, C.; Boorstein, R.J. Targeted Hybridization Capture of SARS-CoV-2 and Metagenomics Enables Genetic Variant Discovery and Nasal Microbiome Insights. Microbiol. Spectr. 2021, 9, e00197-21. [Google Scholar] [CrossRef]
- Klempt, P.; Brož, P.; Kašný, M.; Novotný, A.; Kvapilová, K.; Kvapil, P. Performance of targeted library preparation solutions for SARS-CoV-2 whole genome analysis. Diagnostics 2020, 10, 769. [Google Scholar] [CrossRef]
- Schenk, D.; Song, G.; Ke, Y.; Wang, Z. Amplification of overlapping DNA amplicons in a single-tube multiplex PCR for targeted next-generation sequencing of BRCA1 and BRCA2. PLoS ONE 2017, 12, e0181062. [Google Scholar] [CrossRef]
- Samorodnitsky, E.; Jewell, B.M.; Hagopian, R.; Miya, J.; Wing, M.R.; Lyon, E.; Damodaran, S.; Bhatt, D.; Reeser, J.W.; Datta, J. Evaluation of hybridization capture versus amplicon-based methods for whole-exome sequencing. Hum. Mutat. 2015, 36, 903–914. [Google Scholar] [CrossRef] [Green Version]
- Zhu, N.; Zhang, D.; Wang, W.; Li, X.; Yang, B.; Song, J.; Zhao, X.; Huang, B.; Shi, W.; Lu, R. A novel coronavirus from patients with pneumonia in China, 2019. N. Engl. J. Med. 2020, 382, 727–733. [Google Scholar] [CrossRef]
- Tsang, H.F.; Chan, L.W.C.; Cho, W.C.S.; Yu, A.C.S.; Yim, A.K.Y.; Chan, A.K.C.; Ng, L.P.W.; Wong, Y.K.E.; Pei, X.M.; Li, M.J.W. An update on COVID-19 pandemic: The epidemiology, pathogenesis, prevention and treatment strategies. Expert Rev. Anti-Infect. Ther. 2021, 19, 877–888. [Google Scholar] [CrossRef]
- Wu, S.Y.; Yau, H.S.; Yu, M.Y.; Tsang, H.F.; Chan, L.W.C.; Cho, W.C.S.; Shing Yu, A.C.; Yuen Yim, A.K.; Li, M.J.; Wong, Y.K.E. The diagnostic methods in the COVID-19 pandemic, today and in the future. Expert Rev. Mol. Diagn. 2020, 20, 985–993. [Google Scholar] [CrossRef]
- Tsang, H.F.; Leung, W.M.S.; Chan, L.W.C.; Cho, W.C.S.; Wong, S.C.C. Performance comparison of the Cobas® Liat® and Cepheid® GeneXpert® systems on SARS-CoV-2 detection in nasopharyngeal swab and posterior oropharyngeal saliva. Expert Rev. Mol. Diagn. 2021, 21, 515–518. [Google Scholar] [CrossRef]
- Tsang, H.F.; Yu, A.C.S.; Wong, H.T.; Leung, W.M.S.; Chiou, J.; Wong, Y.K.E.; Yim, A.K.Y.; Tsang, D.N.C.; Tsang, A.K.; Wong, W.T. Whole genome amplicon sequencing and phylogenetic analysis of severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) from lineage B. 1.36. 27 isolated in Hong Kong. Expert Rev. Mol. Diagn. 2022, 22, 119–124. [Google Scholar] [CrossRef]
- Meredith, L.W.; Hamilton, W.L.; Warne, B.; Houldcroft, C.J.; Hosmillo, M.; Jahun, A.S.; Curran, M.D.; Parmar, S.; Caller, L.G.; Caddy, S.L. Rapid implementation of SARS-CoV-2 sequencing to investigate cases of health-care associated COVID-19: A prospective genomic surveillance study. Lancet Infect. Dis. 2020, 20, 1263–1271. [Google Scholar] [CrossRef]
- Illumina. COVIDSeq Test | SARS-CoV-2 NGS test (for the COVID-19 Coronavirus). Available online: https://www.illumina.com/products/by-type/ivd-products/covidseq.html (accessed on 27 May 2021).
- Bhoyar, R.C.; Jain, A.; Sehgal, P.; Divakar, M.K.; Sharma, D.; Imran, M.; Jolly, B.; Ranjan, G.; Rophina, M.; Sharma, S. High throughput detection and genetic epidemiology of SARS-CoV-2 using COVIDSeq next-generation sequencing. PLoS ONE 2021, 16, e0247115. [Google Scholar] [CrossRef]
- Yang, Y.; Walls, S.D.; Gross, S.M.; Schroth, G.P.; Jarman, R.G.; Hang, J. Targeted sequencing of respiratory viruses in clinical specimens for pathogen identification and genome-wide analysis. In The Human Virome; Springer: Berlin/Heidelberg, Germany, 2018; pp. 125–140. [Google Scholar]
- Tsang, H.F.; Yu, A.C.S.; Jin, N.; Yim, A.K.Y.; Leung, W.M.S.; Lam, K.W.; Cho, W.C.S.; Chiou, J.; Wong, S.C.C. The clinical application of metagenomic next-generation sequencing for detecting pathogens in bronchoalveolar lavage fluid: Case reports and literature review. Expert Rev. Mol. Diagn. 2022, 22, 575–582. [Google Scholar] [CrossRef]
- Charre, C.; Ginevra, C.; Sabatier, M.; Regue, H.; Destras, G.; Brun, S.; Burfin, G.; Scholtes, C.; Morfin, F.; Valette, M. Evaluation of NGS-based approaches for SARS-CoV-2 whole genome characterisation. Virus Evol. 2020, 6, veaa075. [Google Scholar] [CrossRef]
- Thermo Fisher Scientific. Advances in Epidemiological Research Using Next-Generation Sequencing. Available online: https://assets.thermofisher.com/TFS-Assets/CSD/brochures/Advances-epidemiological-research-next-generation-sequencing-ebook.pdf (accessed on 27 May 2021).
- Biosciences, A. Targeted Sequencing of SARS-CoV-2: Swift RNA Library Kit and Arbor Biosciences Mybaits Expert Virus Panel (1st ed.). Available online: https://arborbiosci.com/genomics/targeted-sequencing/mybaits/mybaits-expert/mybaits-expert-virus-sars-cov-2/ (accessed on 27 May 2021).
- Illumina. Respiratory Pathogen ID/AMR Panel (with COVID-19) | NGS Enrichment Kit. Available online: https://www.illumina.com/products/by-type/sequencing-kits/library-prep-kits/respiratory-pathogen-id-panel.html (accessed on 27 May 2021).
- Illumina. Respiratory Virus Oligo Panel. Available online: https://www.illumina.com/products/by-type/sequencing-kits/library-prep-kits/respiratory-virus-oligo-panel.html (accessed on 27 May 2021).
- Papoutsis, A.; Borody, T.; Dolai, S.; Daniels, J.; Steinberg, S.; Barrows, B.; Hazan, S. Detection of SARS-CoV-2 from patient fecal samples by whole genome sequencing. Gut Pathog. 2021, 13, 1–8. [Google Scholar] [CrossRef]
- Roche. KAPA Target Enrichment Panel for COVID-19 Research. Available online: https://sequencing.roche.com/en-us/products-solutions/by-category/target-enrichment/hybridization/kapa-target-enrichment/kapa-te-custom-panel-covid-19.html (accessed on 27 May 2021).
- Alessandrini, F.; Caucci, S.; Onofri, V.; Melchionda, F.; Tagliabracci, A.; Bagnarelli, P.; Di Sante, L.; Turchi, C.; Menzo, S. Evaluation of the ion AmpliSeq SARS-CoV-2 research panel by massive parallel sequencing. Genes 2020, 11, 929. [Google Scholar] [CrossRef] [PubMed]
- Marine, R.L.; Magaña, L.C.; Castro, C.J.; Zhao, K.; Montmayeur, A.M.; Schmidt, A.; Diez-Valcarce, M.; Ng, T.F.F.; Vinjé, J.; Burns, C.C. Comparison of Illumina MiSeq and the Ion Torrent PGM and S5 platforms for whole-genome sequencing of picornaviruses and caliciviruses. J. Virol. Methods 2020, 280, 113865. [Google Scholar] [CrossRef] [PubMed]
- Tringe, S.G.; Hugenholtz, P. A renaissance for the pioneering 16S rRNA gene. Curr. Opin. Microbiol. 2008, 11, 442–446. [Google Scholar] [CrossRef] [PubMed]
- Chakravorty, S.; Helb, D.; Burday, M.; Connell, N.; Alland, D. A detailed analysis of 16S ribosomal RNA gene segments for the diagnosis of pathogenic bacteria. J. Microbiol. Methods 2007, 69, 330–339. [Google Scholar] [CrossRef]
- D’Amore, R.; Ijaz, U.Z.; Schirmer, M.; Kenny, J.G.; Gregory, R.; Darby, A.C.; Shakya, M.; Podar, M.; Quince, C.; Hall, N. A comprehensive benchmarking study of protocols and sequencing platforms for 16S rRNA community profiling. BMC Genom. 2016, 17, 1–20. [Google Scholar] [CrossRef]
- Patel, J.B. 16S rRNA gene sequencing for bacterial pathogen identification in the clinical laboratory. Mol. Diagn. 2001, 6, 313–321. [Google Scholar] [CrossRef]
- Biosystems, A. MicroSEQ 500 16S rDNA Identification [User Guide] (G ed.). Available online: https://assets.thermofisher.com/TFS-Assets/LSG/manuals/4346298-MicroSEQ500-16S-rDNA-ID-UG.pdf (accessed on 27 May 2021).
- Illumina. 16S and ITS rRNA Sequencing | Identify Bacteria & Fungi with NGS. Available online: https://www.illumina.com/areas-of-interest/microbiology/microbial-sequencing-methods/16s-rrna-sequencing.html (accessed on 27 May 2021).
- Hao, D.; Gu, X.; Xiao, P.; Peng, Y. Chemical and biological research of Clematis medicinal resources. Chin. Sci. Bull. 2013, 58, 1120–1129. [Google Scholar] [CrossRef]
- Schoch, C.L.; Seifert, K.A.; Huhndorf, S.; Robert, V.; Spouge, J.L.; Levesque, C.A.; Chen, W.; Fungal Barcoding Consortium. Nuclear ribosomal internal transcribed spacer (ITS) region as a universal DNA barcode marker for Fungi. Proc. Natl. Acad. Sci. USA 2012, 109, 6241–6246. [Google Scholar] [CrossRef]
- Qiagen. QIAseq 16S/ITS Screening Panels and Index Kits. Available online: https://www.qiagen.com/ve/products/next-generation-sequencing/qiaseq-16s-its-index-kits/?clear=true#orderinginformation (accessed on 27 May 2021).
- Scientific., T. Ion 16S Metagenomics Solution | Thermo Fisher Scientific-NL. Available online: https://www.thermofisher.com/nl/en/home/life-science/sequencing/dna-sequencing/microbial-sequencing/microbial-identification-ion-torrent-next-generation-sequencing/ion-16s-metagenomics-solution.html (accessed on 27 May 2021).
- Schröttner, P.; Gunzer, F.; Schüppel, J.; Rudolph, W.W. Identification of rare bacterial pathogens by 16S rRNA gene sequencing and MALDI-TOF MS. JoVE (J. Vis. Exp.) 2016, 113, e53176. [Google Scholar]
- Woo, P.C.; Lau, S.K.; Teng, J.L.; Tse, H.; Yuen, K.-Y. Then and now: Use of 16S rDNA gene sequencing for bacterial identification and discovery of novel bacteria in clinical microbiology laboratories. Clin. Microbiol. Infect. 2008, 14, 908–934. [Google Scholar] [CrossRef]
- Boers, S.A.; Jansen, R.; Hays, J.P. Understanding and overcoming the pitfalls and biases of next-generation sequencing (NGS) methods for use in the routine clinical microbiological diagnostic laboratory. Eur. J. Clin. Microbiol. Infect. Dis. 2019, 38, 1059–1070. [Google Scholar] [CrossRef] [Green Version]
- Deurenberg, R.H.; Bathoorn, E.; Chlebowicz, M.A.; Couto, N.; Ferdous, M.; García-Cobos, S.; Kooistra-Smid, A.M.; Raangs, E.C.; Rosema, S.; Veloo, A.C. Application of next generation sequencing in clinical microbiology and infection prevention. J. Biotechnol. 2017, 243, 16–24. [Google Scholar] [CrossRef]
- Muhamad Rizal, N.S.; Neoh, H.-M.; Ramli, R.; A/LK Periyasamy, P.R.; Hanafiah, A.; Abdul Samat, M.N.; Tan, T.L.; Wong, K.K.; Nathan, S.; Chieng, S. Advantages and limitations of 16S rRNA next-generation sequencing for pathogen identification in the diagnostic microbiology laboratory: Perspectives from a middle-income country. Diagnostics 2020, 10, 816. [Google Scholar] [CrossRef]
- Janda, J.M.; Abbott, S.L. 16S rRNA gene sequencing for bacterial identification in the diagnostic laboratory: Pluses, perils, and pitfalls. J. Clin. Microbiol. 2007, 45, 2761–2764. [Google Scholar] [CrossRef]
- Fox, G.E.; Wisotzkey, J.D.; Jurtshuk, P., Jr. How close is close: 16S rRNA sequence identity may not be sufficient to guarantee species identity. Int. J. Syst. Evol. Microbiol. 1992, 42, 166–170. [Google Scholar] [CrossRef]
- McDonough, S.J.; Bhagwate, A.; Sun, Z.; Wang, C.; Zschunke, M.; Gorman, J.A.; Kopp, K.J.; Cunningham, J.M. Use of FFPE-derived DNA in next generation sequencing: DNA extraction methods. PloS ONE 2019, 14, e0211400. [Google Scholar] [CrossRef]
- So, A.P.; Vilborg, A.; Bouhlal, Y.; Koehler, R.T.; Grimes, S.M.; Pouliot, Y.; Mendoza, D.; Ziegle, J.; Stein, J.; Goodsaid, F. A robust targeted sequencing approach for low input and variable quality DNA from clinical samples. NPJ Genom. Med. 2018, 3, 1–10. [Google Scholar] [CrossRef]
- Roychowdhury, S.; Iyer, M.K.; Robinson, D.R.; Lonigro, R.J.; Wu, Y.-M.; Cao, X.; Kalyana-Sundaram, S.; Sam, L.; Balbin, O.A.; Quist, M.J. Personalized oncology through integrative high-throughput sequencing: A pilot study. Sci. Transl. Med. 2011, 3, 111ra121. [Google Scholar] [CrossRef]
- Wong, S.Q.; Li, J.; Tan, A.Y.; Vedururu, R.; Pang, J.-M.B.; Do, H.; Ellul, J.; Doig, K.; Bell, A.; McArthur, G.A. Sequence artefacts in a prospective series of formalin-fixed tumours tested for mutations in hotspot regions by massively parallel sequencing. BMC Med. Genom. 2014, 7, 1–10. [Google Scholar] [CrossRef]
- Kerick, M.; Isau, M.; Timmermann, B.; Sültmann, H.; Herwig, R.; Krobitsch, S.; Schaefer, G.; Verdorfer, I.; Bartsch, G.; Klocker, H. Targeted high throughput sequencing in clinical cancer settings: Formaldehyde fixed-paraffin embedded (FFPE) tumor tissues, input amount and tumor heterogeneity. BMC Med. Genom. 2011, 4, 68. [Google Scholar] [CrossRef]
- Miller, E.M.; Patterson, N.E.; Zechmeister, J.M.; Bejerano-Sagie, M.; Delio, M.; Patel, K.; Ravi, N.; Quispe-Tintaya, W.; Maslov, A.; Simmons, N. Development and validation of a targeted next generation DNA sequencing panel outperforming whole exome sequencing for the identification of clinically relevant genetic variants. Oncotarget 2017, 8, 102033. [Google Scholar] [CrossRef] [PubMed]
- Schwarzenbach, H.; Hoon, D.S.; Pantel, K. Cell-free nucleic acids as biomarkers in cancer patients. Nat. Rev. Cancer 2011, 11, 426–437. [Google Scholar] [CrossRef] [PubMed]
- Verma, S.; Moore, M.W.; Ringler, R.; Ghosal, A.; Horvath, K.; Naef, T.; Anvari, S.; Cotter, P.D.; Gunn, S. Analytical performance evaluation of a commercial next generation sequencing liquid biopsy platform using plasma ctDNA, reference standards, and synthetic serial dilution samples derived from normal plasma. BMC Cancer 2020, 20, 945. [Google Scholar] [CrossRef] [PubMed]
- Breveglieri, G.; D’Aversa, E.; Finotti, A.; Borgatti, M. Non-invasive prenatal testing using fetal DNA. Mol. Diagn. Ther. 2019, 23, 291–299. [Google Scholar] [CrossRef] [PubMed]
- Gil, M.; Quezada, M.; Revello, R.; Akolekar, R.; Nicolaides, K. Analysis of cell-free DNA in maternal blood in screening for fetal aneuploidies: Updated meta-analysis. Ultrasound Obstet. Gynecol. 2015, 45, 249–266. [Google Scholar] [CrossRef]
- Das, S. Hemolytic Disease of the Fetus and Newborn. In Blood Groups; Tombak, A., Ed.; Intechopen: London, UK, 2019. [Google Scholar]
- Rieneck, K.; Clausen, F.B.; Dziegiel, M.H. Noninvasive antenatal determination of fetal blood group using next-generation sequencing. Cold Spring Harb. Perspect. Med. 2016, 6, a023093. [Google Scholar] [CrossRef]
- Pisapia, P.; Pepe, F.; Smeraglio, R.; Russo, M.; Rocco, D.; Sgariglia, R.; Nacchio, M.; De Luca, C.; Vigliar, E.; Bellevicine, C. Cell free DNA analysis by SiRe® next generation sequencing panel in non small cell lung cancer patients: Focus on basal setting. J. Thorac. Dis. 2017, 9, S1383. [Google Scholar] [CrossRef]
- Alborelli, I.; Generali, D.; Jermann, P.; Cappelletti, M.R.; Ferrero, G.; Scaggiante, B.; Bortul, M.; Zanconati, F.; Nicolet, S.; Haegele, J. Cell-free DNA analysis in healthy individuals by next-generation sequencing: A proof of concept and technical validation study. Cell Death Dis. 2019, 10, 1–11. [Google Scholar] [CrossRef]
- Shen, S.Y.; Singhania, R.; Fehringer, G.; Chakravarthy, A.; Roehrl, M.H.; Chadwick, D.; Zuzarte, P.C.; Borgida, A.; Wang, T.T.; Li, T. Sensitive tumour detection and classification using plasma cell-free DNA methylomes. Nature 2018, 563, 579–583. [Google Scholar] [CrossRef]
- Lanman, R.B.; Mortimer, S.A.; Zill, O.A.; Sebisanovic, D.; Lopez, R.; Blau, S.; Collisson, E.A.; Divers, S.G.; Hoon, D.S.; Kopetz, E.S. Analytical and clinical validation of a digital sequencing panel for quantitative, highly accurate evaluation of cell-free circulating tumor DNA. PloS ONE 2015, 10, e0140712. [Google Scholar] [CrossRef]
- Guo, Q.; Wang, J.; Xiao, J.; Wang, L.; Hu, X.; Yu, W.; Song, G.; Lou, J.; Chen, J. Heterogeneous mutation pattern in tumor tissue and circulating tumor DNA warrants parallel NGS panel testing. Mol. Cancer 2018, 17, 1–5. [Google Scholar] [CrossRef]
- Verhein, K.C.; Hariani, G.; Hastings, S.B.; Hurban, P. Analytical validation of Illumina’s TruSight Oncology 500 ctDNA assay. Cancer Res. 2020, 80, 3114. [Google Scholar] [CrossRef]
- Birkenkamp-Demtröder, K.; Nordentoft, I.; Christensen, E.; Høyer, S.; Reinert, T.; Vang, S.; Borre, M.; Agerbæk, M.; Jensen, J.B.; Ørntoft, T.F. Genomic alterations in liquid biopsies from patients with bladder cancer. Eur. Urol. 2016, 70, 75–82. [Google Scholar] [CrossRef]
- Christensen, E.; Nordentoft, I.; Vang, S.; Birkenkamp-Demtröder, K.; Jensen, J.B.; Agerbæk, M.; Pedersen, J.S.; Dyrskjøt, L. Optimized targeted sequencing of cell-free plasma DNA from bladder cancer patients. Sci. Rep. 2018, 8, 1–11. [Google Scholar] [CrossRef]
- Bruno, R.; Fontanini, G. Next generation sequencing for gene fusion analysis in lung cancer: A literature review. Diagnostics 2020, 10, 521. [Google Scholar] [CrossRef]
- Sakai, K.; Ohira, T.; Matsubayashi, J.; Yoneshige, A.; Ito, A.; Mitsudomi, T.; Nagao, T.; Iwamatsu, E.; Katayama, J.; Ikeda, N. Performance of Oncomine Fusion Transcript kit for formalin-fixed, paraffin-embedded lung cancer specimens. Cancer Sci. 2019, 110, 2044–2049. [Google Scholar] [CrossRef]
- Hindi, I.; Shen, G.; Tan, Q.; Cotzia, P.; Snuderl, M.; Feng, X.; Jour, G. Feasibility and clinical utility of a pan-solid tumor targeted RNA fusion panel: A single center experience. Exp. Mol. Pathol. 2020, 114, 104403. [Google Scholar] [CrossRef]
- Solomon, J.; Benayed, R.; Hechtman, J.; Ladanyi, M. Identifying patients with NTRK fusion cancer. Ann. Oncol. 2019, 30, viii16–viii22. [Google Scholar] [CrossRef]
- Davies, K.D.; Le, A.T.; Sheren, J.; Nijmeh, H.; Gowan, K.; Jones, K.L.; Varella-Garcia, M.; Aisner, D.L.; Doebele, R.C. Comparison of molecular testing modalities for detection of ROS1 rearrangements in a cohort of positive patient samples. J. Thorac. Oncol. 2018, 13, 1474–1482. [Google Scholar] [CrossRef]
- Robinson, P.N.; Köhler, S.; Oellrich, A.; Wang, K.; Mungall, C.J.; Lewis, S.E.; Washington, N.; Bauer, S.; Seelow, D.; Krawitz, P.J.; et al. Improved exome prioritization of disease genes through cross-species phenotype comparison. Genome Res. 2014, 24, 340–348. [Google Scholar] [CrossRef]
- Washington, N.L.; Haendel, M.A.; Mungall, C.J.; Ashburner, M.; Westerfield, M.; Lewis, S.E. Linking human diseases to animal models using ontology-based phenotype annotation. PLoS Biol. 2009, 7, e1000247. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Swaminathan, G.J.; Bragin, E.; Chatzimichali, E.A.; Corpas, M.; Bevan, A.P.; Wright, C.F.; Carter, N.P.; Hurles, M.E.; Firth, H.V. DECIPHER: Web-based, community resource for clinical interpretation of rare variants in developmental disorders. Hum. Mol. Genet. 2012, 21, R37–R44. [Google Scholar] [CrossRef] [PubMed]
- Buske, O.J.; Girdea, M.; Dumitriu, S.; Gallinger, B.; Hartley, T.; Trang, H.; Misyura, A.; Friedman, T.; Beaulieu, C.; Bone, W.P.; et al. PhenomeCentral: A portal for phenotypic and genotypic matchmaking of patients with rare genetic diseases. Hum. Mutat. 2015, 36, 931–940. [Google Scholar] [CrossRef] [PubMed]
- Philippakis, A.A.; Azzariti, D.R.; Beltran, S.; Brookes, A.J.; Brownstein, C.A.; Brudno, M.; Brunner, H.G.; Buske, O.J.; Carey, K.; Doll, C.; et al. The Matchmaker Exchange: A platform for rare disease gene discovery. Hum. Mutat. 2015, 36, 915–921. [Google Scholar] [CrossRef] [PubMed]
- Firth, H.V.; Richards, S.M.; Bevan, A.P.; Clayton, S.; Corpas, M.; Rajan, D.; Van Vooren, S.; Moreau, Y.; Pettett, R.M.; Carter, N.P. DECIPHER: Database of chromosomal imbalance and phenotype in humans using ensembl resources. Am. J. Hum. Genet. 2009, 84, 524–533. [Google Scholar] [CrossRef]
- Sobreira, N.; Schiettecatte, F.; Valle, D.; Hamosh, A. GeneMatcher: A matching tool for connecting investigators with an interest in the same gene. Hum. Mutat. 2015, 36, 928–930. [Google Scholar] [CrossRef]
- Chong, J.X.; Yu, J.-H.; Lorentzen, P.; Park, K.M.; Jamal, S.M.; Tabor, H.K.; Rauch, A.; Saenz, M.S.; Boltshauser, E.; Patterson, K.E.; et al. Gene discovery for Mendelian conditions via social networking: De novo variants in KDM1A cause developmental delay and distinctive facial features. Anesthesia Analg. 2016, 18, 788–795. [Google Scholar] [CrossRef]
- Pais, L.S.; Snow, H.; Weisburd, B.; Zhang, S.; Baxter, S.M.; DiTroia, S.; O’Heir, E.; England, E.; Chao, K.R.; Lemire, G.; et al. seqr: A web-based analysis and collaboration tool for rare disease genomics. Hum. Mutat. 2022, 43, 698–707. [Google Scholar] [CrossRef]
- Adachi, T.; Kawamura, K.; Furusawa, Y.; Nishizaki, Y.; Imanishi, N.; Umehara, S.; Izumi, K.; Suematsu, M. Japan’s initiative on rare and undiagnosed diseases (IRUD): Towards an end to the diagnostic odyssey. Eur. J. Hum. Genet. 2017, 25, 1025–1028. [Google Scholar] [CrossRef]
- Rasi, C.; Nilsson, D.; Magnusson, M.; Lesko, N.; Lagerstedt-Robinson, K.; Wedell, A.; Lindstrand, A.; Wirta, V.; Stranneheim, H. PatientMatcher: A customizable Python-based open-source tool for matching undiagnosed rare disease patients via the Matchmaker Exchange network. Hum. Mutat. 2022, 43, 708–716. [Google Scholar] [CrossRef]
- Laurie, S.; Piscia, D.; Matalonga, L.; Corvó, A.; Fernández-Callejo, M.; Garcia-Linares, C.; Hernandez-Ferrer, C.; Luengo, C.; Martínez, I.; Papakonstantinou, A.; et al. The RD-Connect Genome-Phenome Analysis Platform: Accelerating diagnosis, research, and gene discovery for rare diseases. Hum. Mutat. 2022, 43, 717–733. [Google Scholar] [CrossRef]
- Matchmaker Exchange. Exchange Statistics and Publications - Matchmaker Exchange. Available online: https://www.matchmakerexchange.org/statistics.html (accessed on 20 January 2023).
- Azzariti, D.R.; Hamosh, A. Genomic data sharing for novel Mendelian disease gene discovery: The matchmaker exchange. Annu. Rev. Genom. Hum. Genet. 2020, 21, 305–326. [Google Scholar] [CrossRef]
- Palmer, E.E.; Kumar, R.; Gordon, C.T.; Shaw, M.; Hubert, L.; Carroll, R.; Rio, M.; Murray, L.; Leffler, M.; Dudding-Byth, T.; et al. A recurrent de novo nonsense variant in ZSWIM6 results in severe intellectual disability without frontonasal or limb malformations. Am. J. Hum. Genet. 2017, 101, 995–1005. [Google Scholar] [CrossRef]
- Ito, Y.; Carss, K.J.; Duarte, S.T.; Hartley, T.; Keren, B.; Kurian, M.A.; Marey, I.; Charles, P.; Mendonça, C.; Nava, C.; et al. De novo truncating mutations in WASF1 cause intellectual disability with seizures. Am. J. Hum. Genet. 2018, 103, 144–153. [Google Scholar] [CrossRef]
- Carapito, R.; Ivanova, E.L.; Morlon, A.; Meng, L.; Molitor, A.; Erdmann, E.; Kieffer, B.; Pichot, A.; Naegely, L.; Kolmer, A.; et al. ZMIZ1 variants cause a syndromic neurodevelopmental disorder. Am. J. Hum. Genet. 2019, 104, 319–330. [Google Scholar] [CrossRef]
- Friedman, J.; Smith, D.E.; Issa, M.Y.; Stanley, V.; Wang, R.; Mendes, M.I.; Wright, M.S.; Wigby, K.; Hildreth, A.; Crawford, J.R. Biallelic mutations in valyl-tRNA synthetase gene VARS are associated with a progressive neurodevelopmental epileptic encephalopathy. Nat. Commun. 2019, 10, 1–10. [Google Scholar] [CrossRef]
- Fischer-Zirnsak, B.; Segebrecht, L.; Schubach, M.; Charles, P.; Alderman, E.; Brown, K.; Cadieux-Dion, M.; Cartwright, T.; Chen, Y.; Costin, C.; et al. Haploinsufficiency of the notch ligand DLL1 causes variable neurodevelopmental disorders. Am. J. Hum. Genet. 2019, 105, 631–639. [Google Scholar] [CrossRef]
- Yıldırım, M.; Bektaş, Ö.; Tunçez, E.; Süt, N.Y.; Sayar, Y.; Öncül, Ü.; Teber, S. A Case of Combined Oxidative Phosphorylation Deficiency 35 Associated with a Novel Missense Variant of the TRIT1 Gene. Mol. Syndr. 2022, 13, 164–170. [Google Scholar] [CrossRef]
- Skraban, C.M.; Wells, C.F.; Markose, P.; Cho, M.T.; Nesbitt, A.I.; Au, P.B.; Begtrup, A.; Bernat, J.A.; Bird, L.M.; Cao, K.; et al. WDR26 haploinsufficiency causes a recognizable syndrome of intellectual disability, seizures, abnormal gait, and distinctive facial features. Am. J. Hum. Genet. 2017, 101, 139–148. [Google Scholar] [CrossRef]
- Gurovich, Y.; Hanani, Y.; Bar, O.; Nadav, G.; Fleischer, N.; Gelbman, D.; Basel-Salmon, L.; Krawitz, P.M.; Kamphausen, S.B.; Zenker, M.; et al. Identifying facial phenotypes of genetic disorders using deep learning. Nat. Med. 2019, 25, 60–64. [Google Scholar] [CrossRef]
- Hsieh, T.-C.; Bar-Haim, A.; Moosa, S.; Ehmke, N.; Gripp, K.W.; Pantel, J.T.; Danyel, M.; Mensah, M.A.; Horn, D.; Rosnev, S.; et al. GestaltMatcher facilitates rare disease matching using facial phenotype descriptors. Nat. Genet. 2022, 54, 349–357. [Google Scholar] [CrossRef]
- Auron, A.; Brophy, P.D. Hyperammonemia in review: Pathophysiology, diagnosis, and treatment. Pediatr. Nephrol. 2012, 27, 207–222. [Google Scholar] [CrossRef] [PubMed]
- Quinonez, S.C.; Thoene, J.G. Citrullinemia Type I. Available online: https://www.ncbi.nlm.nih.gov/books/NBK1458/ (accessed on 27 May 2021).
- Saheki, T.; Song, Y.Z. Citrin Deficiency. Available online: https://www.ncbi.nlm.nih.gov/books/NBK1181/ (accessed on 27 May 2021).
- Hudak, M.L.; Jones, M.D., Jr.; Brusilow, S.W. Differentiation of transient hyperammonemia of the newborn and urea cycle enzyme defects by clinical presentation. J. Pediatr. 1985, 107, 712–719. [Google Scholar] [CrossRef] [PubMed]
- Genetics ACoM. Newborn Screening ACT Sheet [Increased Citrulline] Amino Aciduria/Urea Cycle Disorder 2012. Available online: https://www.acmg.net//PDFLibrary/Citrullinemia.pdf (accessed on 27 May 2021).
- McCormick, E.M.; Lott, M.T.; Dulik, M.C.; Shen, L.; Attimonelli, M.; Vitale, O.; Karaa, A.; Bai, R.; Pineda-Alvarez, D.E.; Singh, L.N. Specifications of the ACMG/AMP standards and guidelines for mitochondrial DNA variant interpretation. Hum. Mutat. 2020, 41, 2028–2057. [Google Scholar] [CrossRef] [PubMed]
- Parr, R.L.; Maki, J.; Reguly, B.; Dakubo, G.D.; Aguirre, A.; Wittock, R.; Robinson, K.; Jakupciak, J.P.; Thayer, R.E. The pseudo-mitochondrial genome influences mistakes in heteroplasmy interpretation. BMC Genom. 2006, 7, 1–13. [Google Scholar] [CrossRef]
- Illumina. Mitochondrial DNA Sequencing on the iSeqTM 100 Sequencing System [Analyze Data]. Available online: https://www.illumina.com/content/dam/illumina-marketing/documents/products/appnotes/iseq100-mitochondrial-app-note-770-2017-033.pdf (accessed on 27 May 2021).
- Santibanez-Koref, M.; Griffin, H.; Turnbull, D.M.; Chinnery, P.F.; Herbert, M.; Hudson, G. Assessing mitochondrial heteroplasmy using next generation sequencing: A note of caution. Mitochondrion 2019, 46, 302–306. [Google Scholar] [CrossRef]
- El-Hattab, A.W.; Almannai, M.; Scaglia, F. Melas. Available online: https://www.ncbi.nlm.nih.gov/books/NBK1233/ (accessed on 27 May 2021).
- Jones, B.E.; Mkhaimer, Y.G.; Rangel, L.J.; Chedid, M.; Schulte, P.J.; Mohamed, A.K.; Neal, R.M.; Zubidat, D.; Randhawa, A.K.; Hanna, C. Asymptomatic Pyuria as a Prognostic Biomarker in Autosomal Dominant Polycystic Kidney Disease. Kidney360 2022, 3, 465. [Google Scholar] [CrossRef]
- Bogdanova, N.; Markoff, A.; Gerke, V.; McCluskey, M.; Horst, J.; Dworniczak, B. Homologues to the first gene for autosomal dominant polycystic kidney disease are pseudogenes. Genomics 2001, 74, 333–341. [Google Scholar] [CrossRef]
- Harris, P.C.; Rossetti, S. Molecular diagnostics for autosomal dominant polycystic kidney disease. Nat. Rev. Nephrol. 2010, 6, 197–206. [Google Scholar] [CrossRef]
- Tan, A.Y.; Michaeel, A.; Liu, G.; Elemento, O.; Blumenfeld, J.; Donahue, S.; Parker, T.; Levine, D.; Rennert, H. Molecular diagnosis of autosomal dominant polycystic kidney disease using next-generation sequencing. J. Mol. Diagn. 2014, 16, 216–228. [Google Scholar] [CrossRef]
- Ali, H.; Al-Mulla, F.; Hussain, N.; Naim, M.; Asbeutah, A.M.; AlSahow, A.; Abu-Farha, M.; Abubaker, J.; Al Madhoun, A.; Ahmad, S. PKD1 duplicated regions limit clinical utility of whole exome sequencing for genetic diagnosis of autosomal dominant polycystic kidney disease. Sci. Rep. 2019, 9, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Ros-Freixedes, R.; Battagin, M.; Johnsson, M.; Gorjanc, G.; Mileham, A.J.; Rounsley, S.D.; Hickey, J.M. Impact of index hopping and bias towards the reference allele on accuracy of genotype calls from low-coverage sequencing. Genet. Sel. Evol. 2018, 50, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Illumina. Effects of Index Misassignment on Multiplexing and Downstream Analysis [Analyze Data]. 2018. Available online: https://www.illumina.com/content/dam/illumina-marketing/documents/products/whitepapers/index-hopping-white-paper-770-2017-004.pdf?linkId=36607862 (accessed on 27 May 2021).




{kind=link}
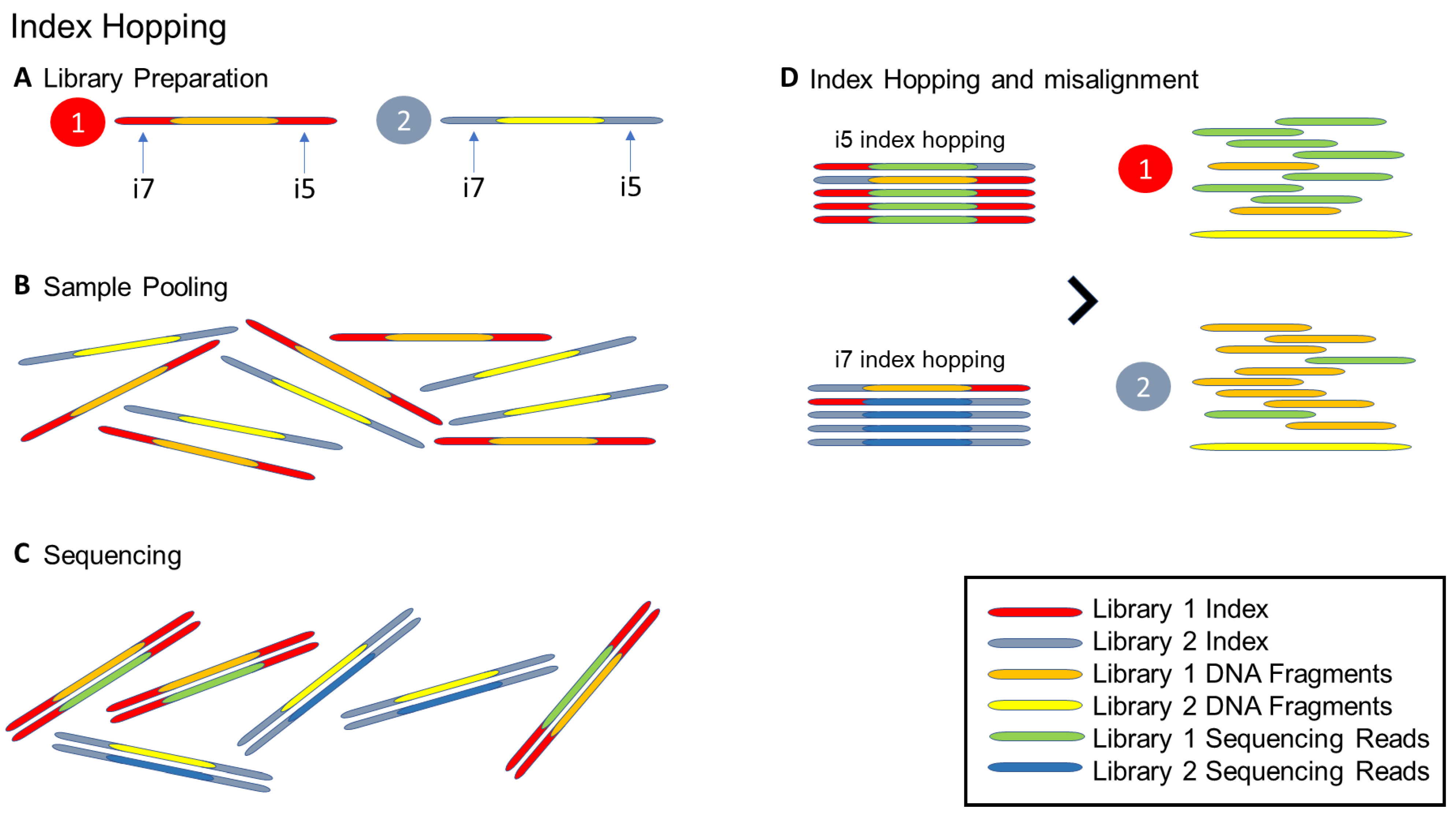{kind=link}
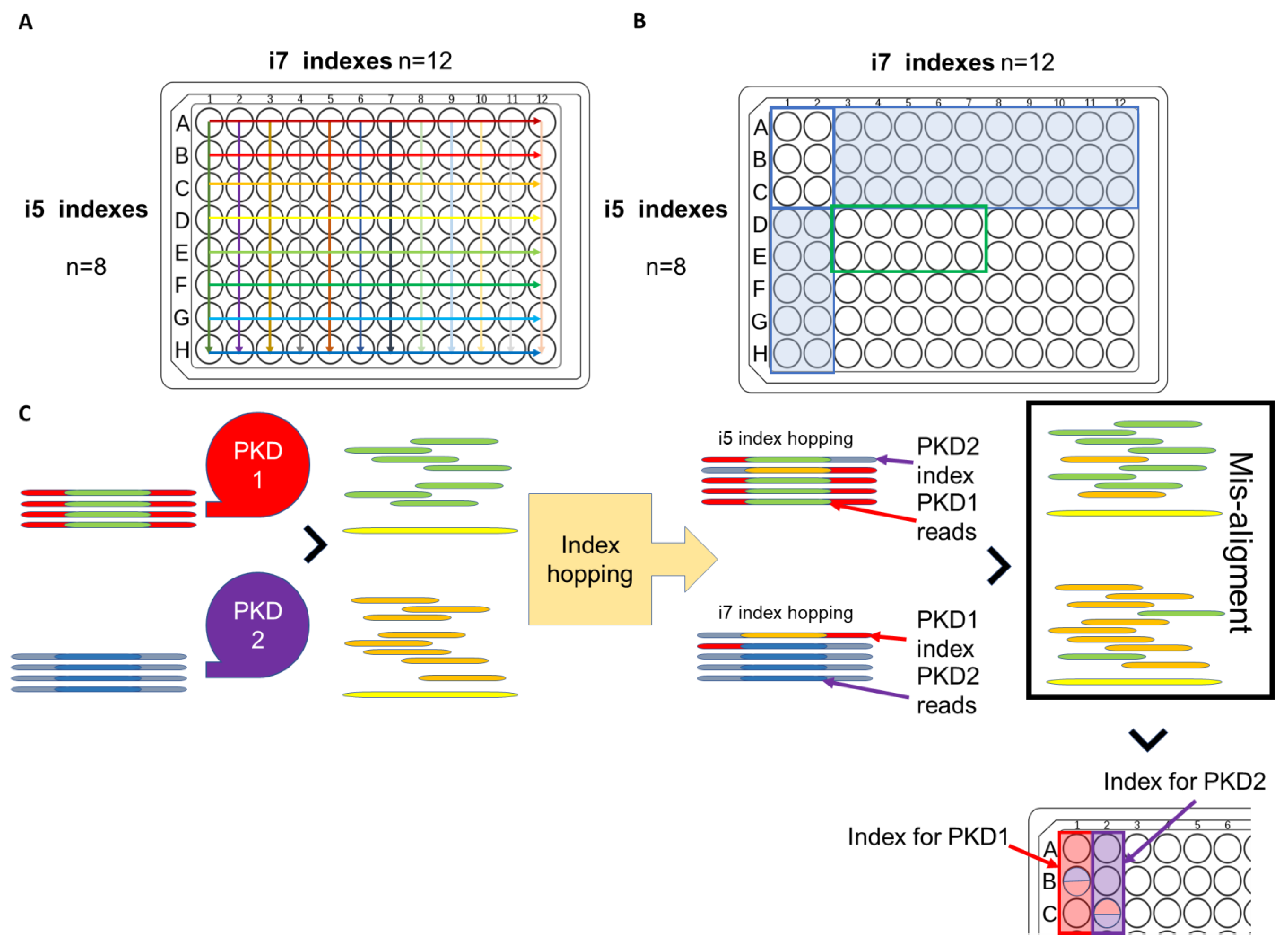{kind=link}
| Shotgun Metagenomics | Capture-Based Enrichment Targeted Sequencing | Amplicon-Based Enrichment Targeted Sequencing | |
|---|---|---|---|
| Examples of commercial kits | Illumina Stranded Total RNA Prep with Ribo-Zero Plus | Illumina Respiratory Virus Oligo Panel Illumina Respiratory Pathogen ID/AMR Enrichment Panel Kit Roche KAPA SARS-CoV-2 Target Enrichment Panel Biosystem TWIST. SARS-CoV-2 Research Panel | Illumina COVIDSeq Test ThermoFisher Ion AmpliSeq™ SARS-CoV-2 Research Panel Paragon Genomics CleanPlex® SARS-CoV-2 Panel Qiagen QIAseq SARS-CoV-2 Primer Panel |
| Characteristics | |||
| Turnaround time | Long | Moderate | Short |
| Cost | High | Moderate to Low | Low |
| The complexity of the workflow | Moderate | Moderate to Low | Low |
| Coverage of the genome | High | Moderate with high uniformity | Low. with variable uniformity |
| Sequence depth | Low | High | High |
| The amount of starting material | High | Moderate to Low | Low |
| Sensitivity to the target | Low | High | High |
| Sensitivity to the background | High | Low | Low |
| Susceptibility to mutational effect | Low | High | High |
| Applications | |||
| Track transmission | Yes | Yes | Yes |
| Identification of novel pathogen | Yes | No | No |
| Identification of co-infections and complex disease | Yes | Only Illumina respiratory panels | No |
| Identification of new mutations | Yes | Yes | No |
| Genus | Species |
|---|---|
| Aeromonas | A. veronii |
| Bacillus | B. anthracis, B. cereus, B.globisporus, B. psychrophilus |
| Bordetella | B. bronchiseptica, B. parapertussis, B. pertussis |
| Burkholderia | B. cocovenenans, B. gladioli, B. pseudomallei, B. thailandensis |
| Campylobacter | Non-jejuni-coli group |
| Edwardsiella | E. tarda, E. hoshinae, E. ictaluri |
| Enterobacter | E. cloacae |
| Neisseria | N. cinerea, N. meningitidis |
| Pseudomonas | P. fluorescens, P. jessenii |
| Streptococcus | S. mitis, S. oralis, S. pneumoniae |
| Pros | Cons | |
|---|---|---|
| Hybrid capture | Characterization of both known and unknown fusion variants of target genes Easily scalable to large gene panels Adequate for DNA and RNA gene fusion analysis At the DNA level, it does not require RNA purification and allows the simultaneous analyses of different gene variants | Higher RNA input than amplicon-based methods Difficulty with fusion variants involving large DNA intronic regions with repetitive sequences |
| Amplicon-based: Classical multiplex PCR (mPCR) Anchored multiplex OCR | Low RNA input Particularly effective with small and mid-size panels Analysis of both known and unknown fusion variants of target genes (anchored mPCR) 5′ and 3′ imbalance evaluation can increase test diagnostic accuracy | Not adequate for gene fusion analysis at the DNA level Primer design can be complex Characterization of only known fusion variants included in the panel (classical mPCR) PCR biases such as allele dropout can impact analysis results |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pei, X.M.; Yeung, M.H.Y.; Wong, A.N.N.; Tsang, H.F.; Yu, A.C.S.; Yim, A.K.Y.; Wong, S.C.C. Targeted Sequencing Approach and Its Clinical Applications for the Molecular Diagnosis of Human Diseases. Cells 2023, 12, 493. https://doi.org/10.3390/cells12030493
Pei XM, Yeung MHY, Wong ANN, Tsang HF, Yu ACS, Yim AKY, Wong SCC. Targeted Sequencing Approach and Its Clinical Applications for the Molecular Diagnosis of Human Diseases. Cells. 2023; 12(3):493. https://doi.org/10.3390/cells12030493
Chicago/Turabian StylePei, Xiao Meng, Martin Ho Yin Yeung, Alex Ngai Nick Wong, Hin Fung Tsang, Allen Chi Shing Yu, Aldrin Kay Yuen Yim, and Sze Chuen Cesar Wong. 2023. "Targeted Sequencing Approach and Its Clinical Applications for the Molecular Diagnosis of Human Diseases" Cells 12, no. 3: 493. https://doi.org/10.3390/cells12030493
APA StylePei, X. M., Yeung, M. H. Y., Wong, A. N. N., Tsang, H. F., Yu, A. C. S., Yim, A. K. Y., & Wong, S. C. C. (2023). Targeted Sequencing Approach and Its Clinical Applications for the Molecular Diagnosis of Human Diseases. Cells, 12(3), 493. https://doi.org/10.3390/cells12030493