
Innovative Therapeutic Approaches for the Treatment of the Ocular Morbidities in Patients with EEC Syndrome

 , , ,
, , ,
Abstract
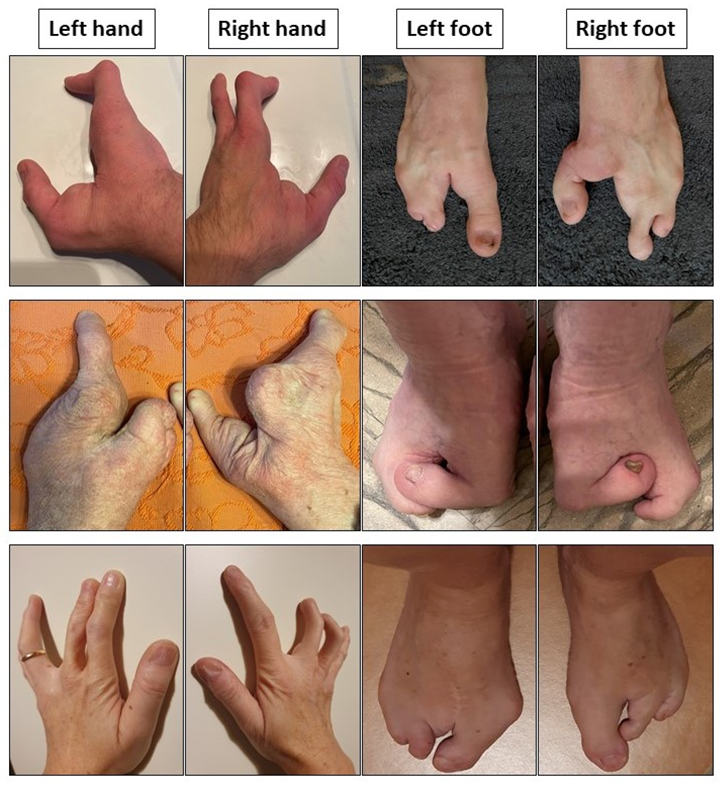
:1. Introduction
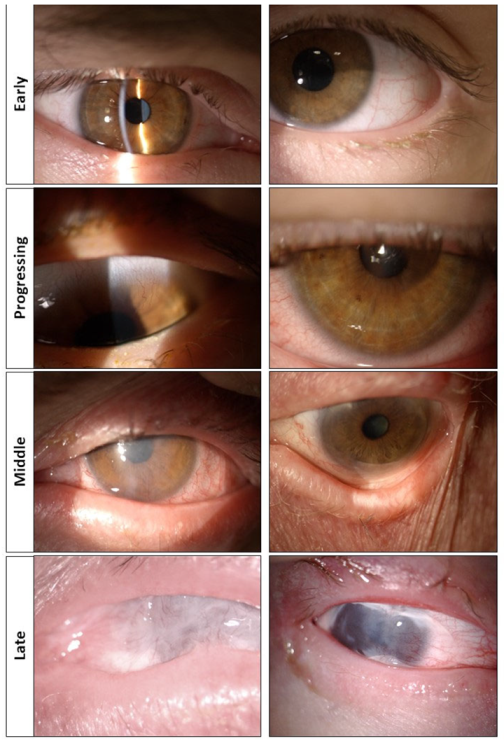
2. Corneal Transplantation Does Not Lead to Any Benefit
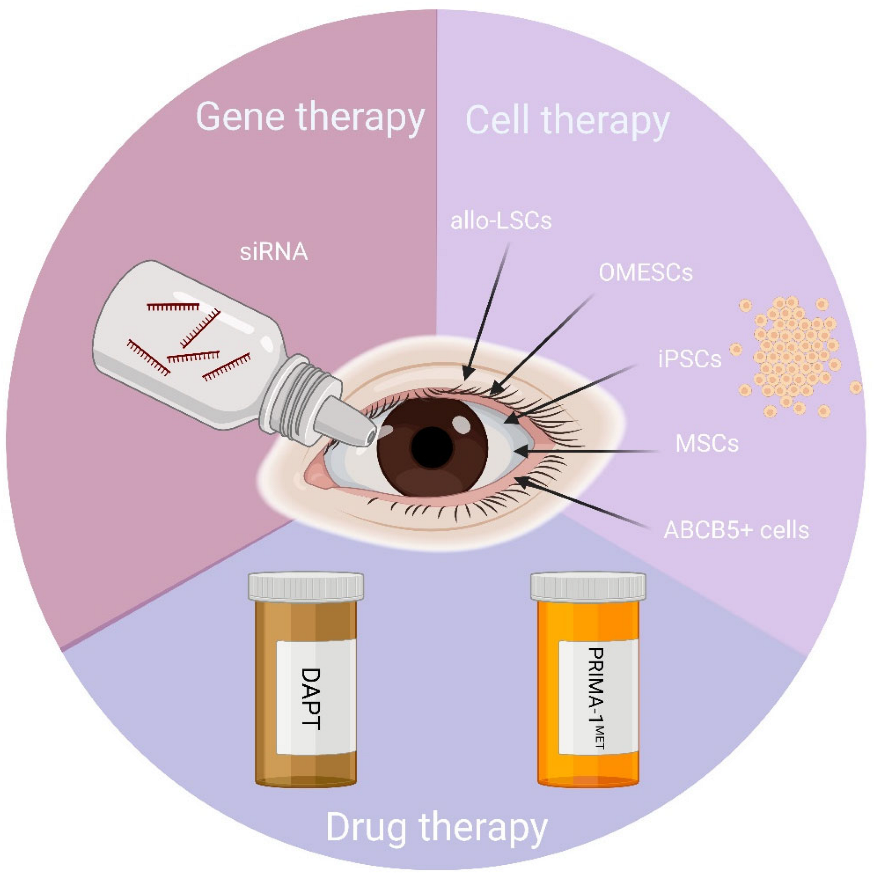
3. New Approaches for the Management of the Ocular Defects in Patients with EEC Syndrome
3.1. Allogeneic Limbal Stem Cells
3.2. Oral Mucosal Epithelial Stem Cells
3.3. Alternatives to Allogeneic Primary Limbal Stem Cells
3.3.1. Induced Pluripotent Stem Cells
3.3.2. ABCB5+ Cells and Mesenchymal Stem Cells
3.4. Gene Therapy-Based Approaches
3.5. Drug-Based Therapies
3.5.1. APR246/PRIMA-1MET
3.5.2. DAPT (N-[N-(3, 5-Difluorophenacetyl)-L-Alanyl]-S-Phenylglycine T-Butyl Ester)
4. Conclusions and Future Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Di Iorio, E.; Kaye, S.B.; Ponzin, D.; Barbaro, V.; Ferrari, S.; Böhm, E.; Nardiello, P.; Castaldo, G.; McGrath, J.A.; Willoughby, C.E. Limbal stem cell deficiency and ocular phenotype in ectrodactyly-ectodermal dysplasia-clefting syndrome caused by p63 mutations. Ophthalmology 2012, 119, 74–83. [Google Scholar] [CrossRef]
- Rinne, T.; Brunner, H.G.; Van Bokhoven, H. p63-associated disorders. Cell Cycle 2007, 6, 262–268. [Google Scholar] [CrossRef] [PubMed]
- Clements, S.E.; Techanukul, T.; Coman, D.; Mellerio, J.E.; McGrath, J.A. Molecular basis of EEC (ectrodactyly, ectodermal dysplasia, clefting) syndrome: Five new mutations in the DNA-binding domain of the TP63 gene and genotype–phenotype correlation. Br. J. Dermatol. 2010, 162, 201–207. [Google Scholar] [CrossRef] [PubMed]
- Novelli, F.; Ganini, C.; Melino, G.; Nucci, C.; Han, Y.; Shi, Y.; Wang, Y.; Candi, E. p63 in corneal and epidermal differentiation. Biochem. Biophys. Res. Commun. 2022, 610, 15–22. [Google Scholar] [CrossRef]
- Melino, G. p63 is a suppressor of tumorigenesis and metastasis interacting with mutant p53. Cell Death Differ. 2011, 18, 1487–1499. [Google Scholar] [CrossRef]
- Celli, J.; Duijf, P.; Hamel, B.C.J.; Bamshad, M.; Kramer, B.; Smits, A.P.T.; Newbury-Ecob, R.; Hennekam, R.C.M.; Van Buggenhout, G.; Van Haeringen, A.; et al. Heterozygous germline mutations in the p53 homolog p63 are the cause of EEC syndrome. Cell 1999, 99, 143–153. [Google Scholar] [CrossRef] [PubMed]
- Koster, M.I. p63 in skin development and ectodermal dysplasias. J. Investig. Dermatol. 2010, 130, 2352–2358. [Google Scholar] [CrossRef]
- Roelfsema, N.M.; Cobben, J.M. The EEC syndrome: A literature study. Clin. Dysmorphol. 1996, 5, 115–127. [Google Scholar] [CrossRef]
- van Bokhoven, H.; Hamel, B.C.J.; Bamshad, M.; Sangiorgi, E.; Gurrieri, F.; Duijf, P.H.G.; Vanmolkot, K.R.J.; van Beusekom, E.; van Beersum, S.E.C.; Celli, J.; et al. p63 Gene Mutations in EEC Syndrome, Limb-Mammary Syndrome, and Isolated Split Hand–Split Foot Malformation Suggest a Genotype-Phenotype Correlation. Am. J. Hum. Genet. 2001, 69, 481–492. [Google Scholar] [CrossRef]
- Candi, E.; Rufini, A.; Terrinoni, A.; Dinsdale, D.; Ranalli, M.; Paradisi, A.; De Laurenzi, V.; Spagnoli, L.G.; Catani, M.V.; Ramadan, S.; et al. Differential roles of p63 isoforms in epidermal development: Selective genetic complementation in p63 null mice. Cell Death Differ. 2006, 13, 1037–1047. [Google Scholar] [CrossRef] [Green Version]
- Barbaro, V.; Nasti, A.A.; Raffa, P.; Migliorati, A.; Nespeca, P.; Ferrari, S.; Palumbo, E.; Bertolin, M.; Breda, C.; Miceli, F.; et al. Personalized Stem Cell Therapy to Correct Corneal Defects Due to a Unique Homozygous-Heterozygous Mosaicism of Ectrodactyly-Ectodermal Dysplasia-Clefting Syndrome. Stem Cells Transl. Med. 2016, 5, 1098–1105. [Google Scholar] [CrossRef] [PubMed]
- Mawhorter, L.G.; Ruttum, M.S.; Koenig, S.B. Keratopathy in a Family with the Ectrodactyly-Ectodermal Dysplasia-clefting Syndrome. Ophthalmology 1985, 92, 1427–1431. [Google Scholar] [CrossRef]
- McNab, A.A.; Potts, M.J.; Welham, R.A. The EEC syndrome and its ocular manifestations. Br. J. Ophthalmol. 1989, 73, 261–264. [Google Scholar] [CrossRef]
- Rodini, E.S.O.; Richieri-Costa, A. EEC syndrome: Report on 20 new patients, clinical and genetic considerations. Am. J. Med. Genet. 1990, 37, 42–53. [Google Scholar] [CrossRef]
- Buss, P.W.; Hughes, H.E.; Clarke, A. Twenty-four cases of the EEC syndrome: Clinical presentation and management. J. Med. Genet. 1995, 32, 716–723. [Google Scholar] [CrossRef] [PubMed]
- Bonnar, E.; Logan, P.; Eustace, P. Absent meibomian glands: A marker for EEC syndrome. Eye 1996, 10, 355–361. [Google Scholar] [CrossRef] [PubMed]
- Elmann, S.; Hanson, S.A.; Bunce, C.N.; Shinder, R. Ectrodactyly Ectodermal Dysplasia Clefting (EEC) syndrome: A rare cause of congenital lacrimal anomalies. Ophthalmic Plast. Reconstr. Surg. 2015, 31, e35–e37. [Google Scholar] [CrossRef] [PubMed]
- Fried, K. Ectrodactyly-ectodermal dysplasia-clefting (EEC) syndrome. Clin. Genet. 1972, 3, 396–400. [Google Scholar] [CrossRef] [PubMed]
- Baum, J.L.; Bull, M.J. Ocular Manifestations of the Ectrodactyly, Ectodermal Dysplasia, Cleft Lip-Palate Syndrome. Am. J. Ophthalmol. 1974, 78, 211–216. [Google Scholar] [CrossRef]
- Nobe, J.R. Results of Penetrating Keratoplasty for the Treatment of Corneal Perforations. Arch. Ophthalmol. 1990, 108, 939. [Google Scholar] [CrossRef]
- Mader, T.H.; Stulting, R.D. Penetrating keratoplasty in ectodermal dysplasia. Am. J. Ophthalmol. 1990, 110, 319–320. [Google Scholar] [CrossRef] [PubMed]
- Anderson, N.J.; Hardten, D.R.; McCarty, T.M. Penetrating Keratoplasty and Keratolimbal Allograft Transplantation for Corneal Perforations Associated with the Ectodermal Dysplasia Syndrome. Cornea 2003, 22, 385–388. [Google Scholar] [CrossRef]
- Bigatà, X.; Bielsa, I.; Artigas, M.; Azón, A.; Ribera, M.; Ferrándiz, C. The ectrodactyly-ectodermal dysplasia-clefting syndrome (EEC): Report of five cases. Pediatr. Dermatol. 2003, 20, 113–118. [Google Scholar] [CrossRef] [PubMed]
- Daya, S.M.; Ilari, F.A.C.S.L. Living related conjunctival limbal allograft for the treatment of stem cell deficiency. Ophthalmology 2001, 108, 126–133. [Google Scholar] [CrossRef] [PubMed]
- Rama, P.; Matuska, S.; Paganoni, G.; Spinelli, A.; De Luca, M.; Pellegrini, G. Limbal stem-cell therapy and long-term corneal regeneration. N. Engl. J. Med. 2010, 363, 147–155. [Google Scholar] [CrossRef]
- Shortt, A.J.; Secker, G.A.; Rajan, M.S.; Meligonis, G.; Dart, J.K.; Tuft, S.J.; Daniels, J.T. Ex vivo expansion and transplantation of limbal epithelial stem cells. Ophthalmology 2008, 115, 1989–1997. [Google Scholar] [CrossRef]
- Daya, S.M.; Watson, A.; Sharpe, J.R.; Giledi, O.; Rowe, A.; Martin, R.; James, S.E. Outcomes and DNA analysis of ex vivo expanded stem cell allograft for ocular surface reconstruction. Ophthalmology 2005, 112, 470–477. [Google Scholar] [CrossRef] [PubMed]
- Cheung, A.Y.; Sarnicola, E.; Kurji, K.H.; Govil, A.; Mogilishetty, G.; Eslani, M.; Wright, E.; Brailey, P.; Holland, E.J. Cincinnati Protocol for Preoperative Screening and Donor Selection for Ocular Surface Stem Cell Transplantation. Cornea 2018, 37, 1192–1197. [Google Scholar] [CrossRef] [PubMed]
- Behaegel, J.; Tassignon, M.J.; Lagali, N.; Consejo, A.; Koppen, C.; Dhubhghaill, S.N. Outcomes of Human Leukocyte Antigen-Matched Allogeneic Cultivated Limbal Epithelial Transplantation in Aniridia-Associated Keratopathy-A Single-Center Retrospective Analysis. Cornea 2022, 41, 69–77. [Google Scholar] [CrossRef] [PubMed]
- Inatomi, T.; Nakamura, T.; Kojyo, M.; Koizumi, N.; Sotozono, C.; Kinoshita, S. Ocular surface reconstruction with combination of cultivated autologous oral mucosal epithelial transplantation and penetrating keratoplasty. Am. J. Ophthalmol. 2006, 142, 757–764.e1. [Google Scholar] [CrossRef]
- Nakamura, T.; Inatomi, T.; Sotozono, C.; Amemiya, T.; Kanamura, N.; Kinoshita, S. Transplantation of cultivated autologous oral mucosal epithelial cells in patients with severe ocular surface disorders. Br. J. Ophthalmol. 2004, 88, 1280–1284. [Google Scholar] [CrossRef] [PubMed]
- Cyranoski, D. Woman is first to receive cornea made from “reprogrammed” stem cells. Nature 2019, 534, 310–312. [Google Scholar] [CrossRef] [PubMed]
- Trevisan, M.; Alvisi, G.; Barbaro, V.; Barzon, L.; Raffa, P.; Migliorati, A.; Desole, G.; Ruzittu, S.; Masi, G.; Di Iorio, E.; et al. Oral Mucosa-Derived Induced Pluripotent Stem Cells from Patients with Ectrodactyly-Ectodermal Dysplasia-Clefting Syndrome. Cell. Reprogram. 2018, 20, 215–224. [Google Scholar] [CrossRef] [PubMed]
- Alvisi, G.; Trevisan, M.; Masi, G.; Canel, V.; Caenazzo, L.; Nespeca, P.; Barzon, L.; Di Iorio, E.; Barbaro, V.; Palù, G. Generation of a transgene-free human induced pluripotent stem cell line (UNIPDi001-A) from oral mucosa epithelial stem cells. Stem Cell Res. 2018, 28, 177–180. [Google Scholar] [CrossRef] [PubMed]
- Trevisan, M.; Di Iorio, E.; Masi, G.; Riccetti, S.; Barzon, L.; Alvisi, G.; Caenazzo, L.; Barbaro, V.; Palù, G. Induced pluripotent stem cells line (UNIPDi003-A) from a patient affected by EEC syndrome carrying the R279H mutation in TP63 gene. Stem Cell Res. 2018, 28, 141–144. [Google Scholar] [CrossRef] [PubMed]
- Trevisan, M.; Barbaro, V.; Riccetti, S.; Masi, G.; Barzon, L.; Nespeca, P.; Alvisi, G.; Di Iorio, E.; Palù, G. Generation of a transgene-free induced pluripotent stem cells line (UNIPDi002-A) from oral mucosa epithelial stem cells carrying the R304Q mutation in TP63 gene. Stem Cell Res. 2018, 28, 149–152. [Google Scholar] [CrossRef] [PubMed]
- Shalom-Feuerstein, R.; Serror, L.; Aberdam, E.; Müller, F.J.; Van Bokhoven, H.; Wiman, K.G.; Zhou, H.; Aberdam, D.; Petit, I. Impaired epithelial differentiation of induced pluripotent stem cells from ectodermal dysplasia-related patients is rescued by the small compound APR-246/PRIMA-1MET. Proc. Natl. Acad. Sci. USA 2013, 110, 2152–2156. [Google Scholar] [CrossRef]
- Ksander, B.R.; Kolovou, P.E.; Wilson, B.J.; Saab, K.R.; Guo, Q.; Ma, J.; McGuire, S.P.; Gregory, M.S.; Vincent, W.J.B.; Perez, V.L.; et al. ABCB5 is a limbal stem cell gene required for corneal development and repair. Nature 2014, 511, 353–357. [Google Scholar] [CrossRef]
- Norrick, A.; Esterlechner, J.; Niebergall-Roth, E.; Dehio, U.; Sadeghi, S.; Schröder, H.M.; Ballikaya, S.; Stemler, N.; Ganss, C.; Dieter, K.; et al. Process development and safety evaluation of ABCB5+ limbal stem cells as advanced-therapy medicinal product to treat limbal stem cell deficiency. Stem Cell Res. Ther. 2021, 12, 194. [Google Scholar] [CrossRef]
- Griffin, M.D.; Ryan, A.E.; Alagesan, S.; Lohan, P.; Treacy, O.; Ritter, T. Anti-donor immune responses elicited by allogeneic mesenchymal stem cells: What have we learned so far? Immunol. Cell Biol. 2013, 91, 40–51. [Google Scholar] [CrossRef]
- Calonge, M.; Pérez, I.; Galindo, S.; Nieto-Miguel, T.; López-Paniagua, M.; Fernández, I.; Alberca, M.; García-Sancho, J.; Sánchez, A.; Herreras, J.M. A proof-of-concept clinical trial using mesenchymal stem cells for the treatment of corneal epithelial stem cell deficiency. Transl. Res. 2019, 206, 18–40. [Google Scholar] [CrossRef] [PubMed]
- Allen, E.H.A.; Atkinson, S.D.; Liao, H.; Moore, J.E.; Leslie Pedrioli, D.M.; Smith, F.J.D.; McLean, W.H.I.; Moore, C.B.T. Allele-specific siRNA silencing for the common keratin 12 founder mutation in Meesmann epithelial corneal dystrophy. Investig. Ophthalmol. Vis. Sci. 2013, 54, 494–502. [Google Scholar] [CrossRef] [PubMed]
- Courtney, D.G.; Atkinson, S.D.; Allen, E.H.A.; Moore, J.E.; Walsh, C.P.; Pedrioli, D.M.L.; MacEwen, C.J.; Pellegrini, G.; Maurizi, E.; Serafini, C.; et al. siRNA silencing of the mutant keratin 12 allele in corneal limbal epithelial cells grown from patients with Meesmann’s epithelial corneal dystrophy. Investig. Ophthalmol. Vis. Sci. 2014, 55, 3352–3360. [Google Scholar] [CrossRef] [PubMed]
- Courtney, D.G.; Atkinson, S.D.; Moore, J.E.; Maurizi, E.; Serafini, C.; Pellegrini, G.; Black, G.C.; Manson, F.D.; Yam, G.H.F.; MacEwen, C.J.; et al. Development of allele-specific gene-silencing siRNAs for TGFBI Arg124Cys in lattice corneal dystrophy type I. Investig. Ophthalmol. Vis. Sci. 2014, 55, 977–985. [Google Scholar] [CrossRef] [PubMed]
- Atkinson, S.D.; McGilligan, V.E.; Liao, H.; Szeverenyi, I.; Smith, F.J.D.; Moore, C.B.T.; McLean, W.H.I. Development of allele-specific therapeutic siRNA for keratin 5 mutations in epidermolysis bullosa simplex. J. Investig. Dermatol. 2011, 131, 2079–2086. [Google Scholar] [CrossRef]
- Leslie Pedrioli, D.M.; Fu, D.J.; Gonzalez-Gonzalez, E.; Contag, C.H.; Kaspar, R.L.; Smith, F.J.D.; McLean, W.H.I. Generic and personalized RNAi-based therapeutics for a dominant-negative epidermal fragility disorder. J. Investig. Dermatol. 2012, 132, 1627–1635. [Google Scholar] [CrossRef]
- Barbaro, V.; Nasti, A.A.; Del Vecchio, C.; Ferrari, S.; Migliorati, A.; Raffa, P.; Lariccia, V.; Nespeca, P.; Biasolo, M.; Willoughby, C.E.; et al. Correction of Mutant p63 in EEC Syndrome Using siRNA Mediated Allele-Specific Silencing Restores Defective Stem Cell Function. Stem Cells 2016, 34, 1588–1600. [Google Scholar] [CrossRef]
- Barbaro, V.; Confalonieri, L.; Vallini, I.; Ferrari, S.; Ponzin, D.; Mantero, G.; Willoughby, C.E.; Parekh, M.; Di Iorio, E. Development of an allele-specific real-time PCR assay for discrimination and quantification of p63 R279H mutation in EEC syndrome. J. Mol. Diagn. 2012, 14, 38–45. [Google Scholar] [CrossRef]
- Novelli, F.; Lena, A.M.; Panatta, E.; Nasser, W.; Shalom-Feuerstein, R.; Candi, E.; Melino, G. Allele-specific silencing of EEC p63 mutant R304W restores p63 transcriptional activity. Cell Death Dis. 2016, 7, e2227. [Google Scholar] [CrossRef]
- Käsmann, B.; Ruprecht, K.W. Ocular manifestations in a father and son with EEC syndrome. Graefes Arch. Clin. Exp. Ophthalmol. 1997, 235, 512–516. [Google Scholar] [CrossRef]
- Lehmann, S.; Bykov, V.J.N.; Ali, D.; Andreń, O.; Cherif, H.; Tidefelt, U.; Uggla, B.; Yachnin, J.; Juliusson, G.; Moshfegh, A.; et al. Targeting p53 in vivo: A first-in-human study with p53-targeting compound APR-246 in refractory hematologic malignancies and prostate cancer. J. Clin. Oncol. 2012, 30, 3633–3639. [Google Scholar] [CrossRef]
- Shen, J.; Van Den Bogaard, E.H.; Kouwenhoven, E.N.; Bykov, V.J.N.; Rinne, T.; Zhang, Q.; Tjabringa, G.S.; Gilissen, C.; Van Heeringen, S.J.; Schalkwijk, J.; et al. APR-246/PRIMA-1(MET) rescues epidermal differentiation in skin keratinocytes derived from EEC syndrome patients with p63 mutations. Proc. Natl. Acad. Sci. USA 2013, 110, 2157–2162. [Google Scholar] [CrossRef]
- Aberdam, E.; Roux, L.N.; Secrétan, P.H.; Boralevi, F.; Schlatter, J.; Morice-Picard, F.; Sol, S.; Bodemer, C.; Missero, C.; Cisternino, S.; et al. Improvement of epidermal covering on AEC patients with severe skin erosions by PRIMA-1MET/APR-246. Cell Death Dis. 2020, 11, 30. [Google Scholar] [CrossRef] [PubMed]
- Balistreri, C.R.; Madonna, R.; Melino, G.; Caruso, C. The emerging role of Notch pathway in ageing: Focus on the related mechanisms in age-related diseases. Ageing Res. Rev. 2016, 29, 50–65. [Google Scholar] [CrossRef] [PubMed]
- Totaro, A.; Castellan, M.; Battilana, G.; Zanconato, F.; Azzolin, L.; Giulitti, S.; Cordenonsi, M.; Piccolo, S. YAP/TAZ link cell mechanics to Notch signalling to control epidermal stem cell fate. Nat. Commun. 2017, 8, 15206. [Google Scholar] [CrossRef] [PubMed]
- Djalilian, A.R.; Namavari, A.; Ito, A.; Balali, S.; Afshar, A.; Lavker, R.M.; Yue, B.Y.J.T. Down-regulation of Notch signaling during corneal epithelial proliferation. Mol. Vis. 2008, 14, 1041–1049. [Google Scholar]
- Movahedan, A.; Majdi, M.; Afsharkhamseh, N.; Sagha, H.M.; Saadat, N.S.; Shalileh, K.; Milani, B.Y.; Ying, H.; Djalilian, A.R. Notch inhibition during corneal epithelial wound healing promotes migration. Investig. Ophthalmol. Vis. Sci. 2012, 53, 7476–7483. [Google Scholar] [CrossRef]
- Nickoloff, B.J.; Osborne, B.A.; Miele, L. Notch signaling as a therapeutic target in cancer: A new approach to the development of cell fate modifying agents. Oncogene 2003, 22, 6598–6608. [Google Scholar] [CrossRef]
- Pannuti, A.; Foreman, K.; Rizzo, P.; Osipo, C.; Golde, T.; Osborne, B.; Miele, L. Targeting Notch to target cancer stem cells. Clin. Cancer Res. 2010, 16, 3141–3152. [Google Scholar] [CrossRef]
- Barbaro, V.; Orvieto, A.; Alvisi, G.; Bertolin, M.; Bonelli, F.; Liehr, T.; Harutyunyan, T.; Kankel, S.; Joksic, G.; Ferrari, S.; et al. Analysis and pharmacological modulation of senescence in human epithelial stem cells. J. Cell. Mol. Med. 2022, 26, 3977–3994. [Google Scholar] [CrossRef]
- Qiu, J.; Gjini, J.; Arif, T.; Moore, K.; Lin, M.; Ghaffari, S. Using mitochondrial activity to select for potent human hematopoietic stem cells. Blood Adv. 2021, 5, 1605–1616. [Google Scholar] [CrossRef]
- Shanbhag, S.S.; Patel, C.N.; Goyal, R.; Donthineni, P.R.; Singh, V.; Basu, S. Simple limbal epithelial transplantation (SLET): Review of indications, surgical technique, mechanism, outcomes, limitations, and impact. Indian J. Ophthalmol. 2019, 67, 1265–1277. [Google Scholar] [CrossRef]
- Skeens, H.M.; Brooks, B.P.; Holland, E.J. Congenital Aniridia Variant: Minimally Abnormal Irides with Severe Limbal Stem Cell Deficiency. Ophthalmology 2011, 118, 1260–1264. [Google Scholar] [CrossRef]
- Ilari, L.; Daya, S.M. Long-term outcomes of keratolimbal allograft for the treatment of severe ocular surface disorders. Ophthalmology 2002, 109, 1278–1284. [Google Scholar] [CrossRef] [PubMed]
- Nishida, K.; Yamato, M.; Hayashida, Y.; Watanabe, K.; Yamamoto, K.; Adachi, E.; Nagai, S.; Kikuchi, A.; Maeda, N.; Watanabe, H.; et al. Corneal Reconstruction with Tissue-Engineered Cell Sheets Composed of Autologous Oral Mucosal Epithelium. N. Engl. J. Med. 2004, 351, 1187–1196. [Google Scholar] [CrossRef] [PubMed]
- Inatomi, T.; Nakamura, T.; Koizumi, N.; Sotozono, C.; Yokoi, N.; Kinoshita, S. Midterm Results on Ocular Surface Reconstruction Using Cultivated Autologous Oral Mucosal Epithelial Transplantation. Am. J. Ophthalmol. 2006, 141, 267–275.e1. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Sheha, H.; Fu, Y.; Giegengack, M.; Tseng, S.C.G. Oral Mucosal Graft with Amniotic Membrane Transplantation for Total Limbal Stem Cell Deficiency. Am. J. Ophthalmol. 2011, 152, 739–747.e1. [Google Scholar] [CrossRef]
- Burillon, C.; Huot, L.; Justin, V.; Nataf, S.; Chapuis, F.; Decullier, E.; Damour, O. Cultured Autologous Oral Mucosal Epithelial Cell Sheet (CAOMECS) Transplantation for the Treatment of Corneal Limbal Epithelial Stem Cell Deficiency. Investig. Ophthalmol. Vis. Sci. 2012, 53, 1325. [Google Scholar] [CrossRef]
- Mozafari, M.; Lauschke, M.; Institutet, K.; Lagali, S.N.; Samoila, O.; Gocan, D. Clinical Outcomes From Cultivated Allogenic Stem Cells vs. Oral Mucosa Epithelial Transplants in Total Bilateral Stem Cells Deficiency. Front. Med. 2020, 7, 43. [Google Scholar] [CrossRef]
- Wu, W.; Tang, L.; D’Amore, P.A.; Lei, H. Application of CRISPR-Cas9 in eye disease. Exp. Eye Res. 2017, 161, 116–123. [Google Scholar] [CrossRef]
- Roux, L.N.; Petit, I.; Domart, R.; Concordet, J.-P.; Qu, J.; Zhou, H.; Joliot, A.; Ferrigno, O.; Aberdam, D. Modeling of Aniridia-Related Keratopathy by CRISPR/Cas9 Genome Editing of Human Limbal Epithelial Cells and Rescue by Recombinant PAX6 Protein. Stem Cells 2018, 36, 1421–1429. [Google Scholar] [CrossRef] [PubMed] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Reference | Cases | Age | Gender | Follow-Up Time | Outcome |
|---|---|---|---|---|---|
| Corneal transplantation | |||||
| [18] | 1 patient | 13 | F | 9 years | Graft opacification, neovascularisation, partial blindness |
| [19] | 1 patient | 5 | F | 1 year | Graft opacification, re-epithelialisation, neovascularisation, partial blindness |
| [20] | 1 patient | 33 | F | unavailable | Corneal melting and perforation |
| [21] | 2 patients | 45 23 | F M | 10 months 22 months | Staphylococcal ulcerative keratitis followed by corneal perforation, secondary penetrating keratoplasty, clear vision; Mild epithelial erosion, marginal scarring and neovascularisation |
| [22] | 2 patients | 25 56 | F F | 14 months 6 months | Penetrating keratoplasty was repeated 5 times in left eye and 2 times in right Eye due to corneal perforations, clear vision and mild peripheral neovasularisation; Stromal scarring, loss of normal lamellar architecture, discontinuous Bowman’s membrane and hypercellualr stroma with a few chronic Inflammatory cells |
| [23] | 1 patient | 28 | F | unavailable | Unavailable |
| unpublished | 1 patient | 62 | M | 7 years | Symblepharon/ankyloblepharon |
| Reference | Age and Gender | Pre-Operative Visual Acuity | Post-Operative Visual Acuity | Dosage of Cyclosporin a | Follow-Up Time | Clinical Outcome | Further Information |
|---|---|---|---|---|---|---|---|
| [26] | 32, female | PL | PL | 3.5 mg/kg for 6 months | 6 months | failure | |
| [27] | 3, female | unknown | 20/160, amblyopia | nil | 27 months | success | DNA from host only at months 1 and 6 |
| [27] | 31, female | 4/200 | 4/200 | 3 mg/kg tapered to 2 mg/kg after 2 weeks, indefinitely | 27 months | failure | DNA from host only at month 7 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Barbaro, V.; Bonelli, F.; Ferrari, S.; La Vella, G.; Di Iorio, E. Innovative Therapeutic Approaches for the Treatment of the Ocular Morbidities in Patients with EEC Syndrome. Cells 2023, 12, 495. https://doi.org/10.3390/cells12030495
Barbaro V, Bonelli F, Ferrari S, La Vella G, Di Iorio E. Innovative Therapeutic Approaches for the Treatment of the Ocular Morbidities in Patients with EEC Syndrome. Cells. 2023; 12(3):495. https://doi.org/10.3390/cells12030495
Chicago/Turabian StyleBarbaro, Vanessa, Filippo Bonelli, Stefano Ferrari, Giulia La Vella, and Enzo Di Iorio. 2023. "Innovative Therapeutic Approaches for the Treatment of the Ocular Morbidities in Patients with EEC Syndrome" Cells 12, no. 3: 495. https://doi.org/10.3390/cells12030495
APA StyleBarbaro, V., Bonelli, F., Ferrari, S., La Vella, G., & Di Iorio, E. (2023). Innovative Therapeutic Approaches for the Treatment of the Ocular Morbidities in Patients with EEC Syndrome. Cells, 12(3), 495. https://doi.org/10.3390/cells12030495