
Prothymosin α Plays Role as a Brain Guardian through Ecto-F1 ATPase-P2Y12 Complex and TLR4/MD2

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
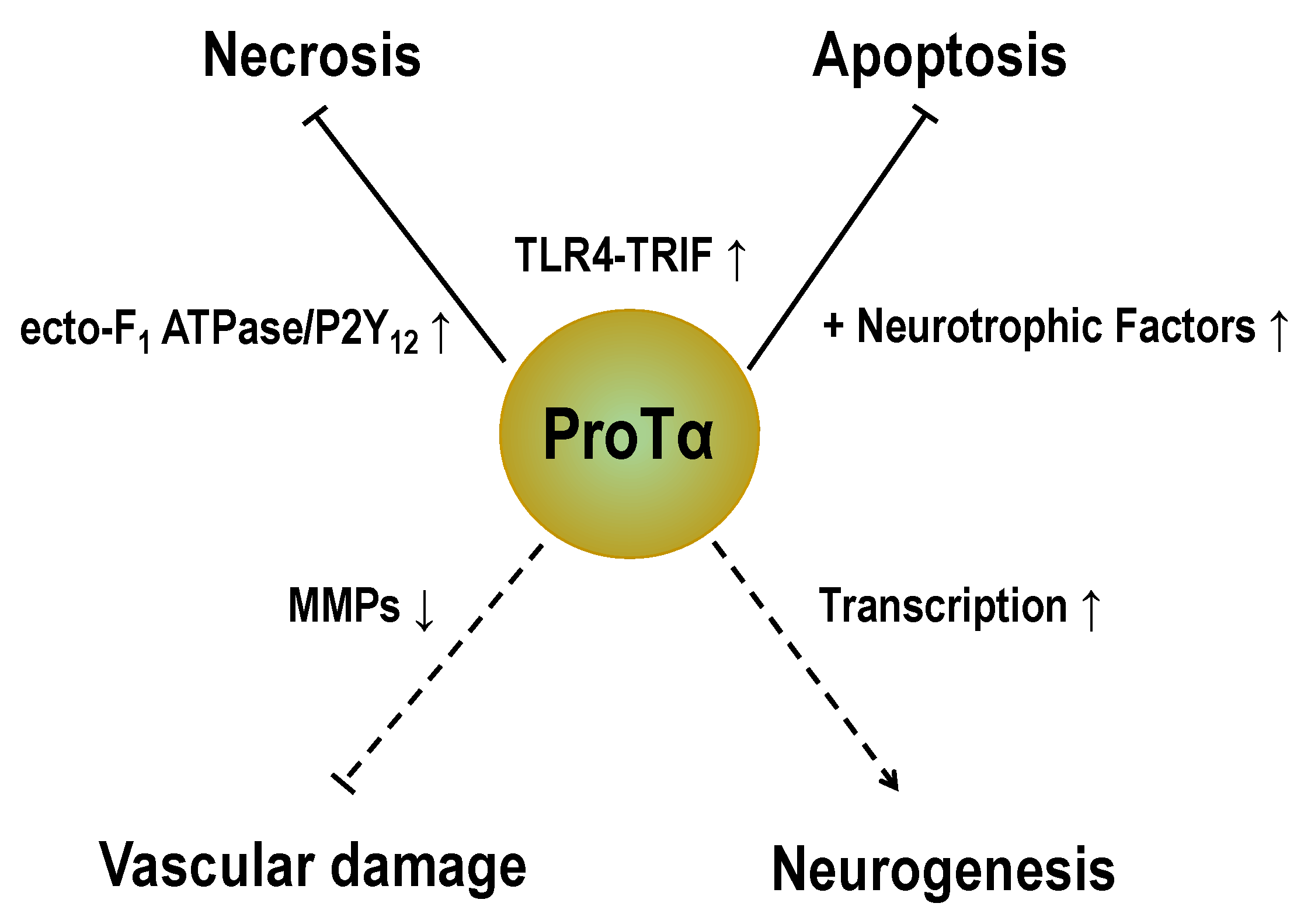
Abstract
:1. Introduction
2. Discovery of Necrosis-Inhibiting Factors
3. Cell-Death-Mode Switch by ProTα [5]
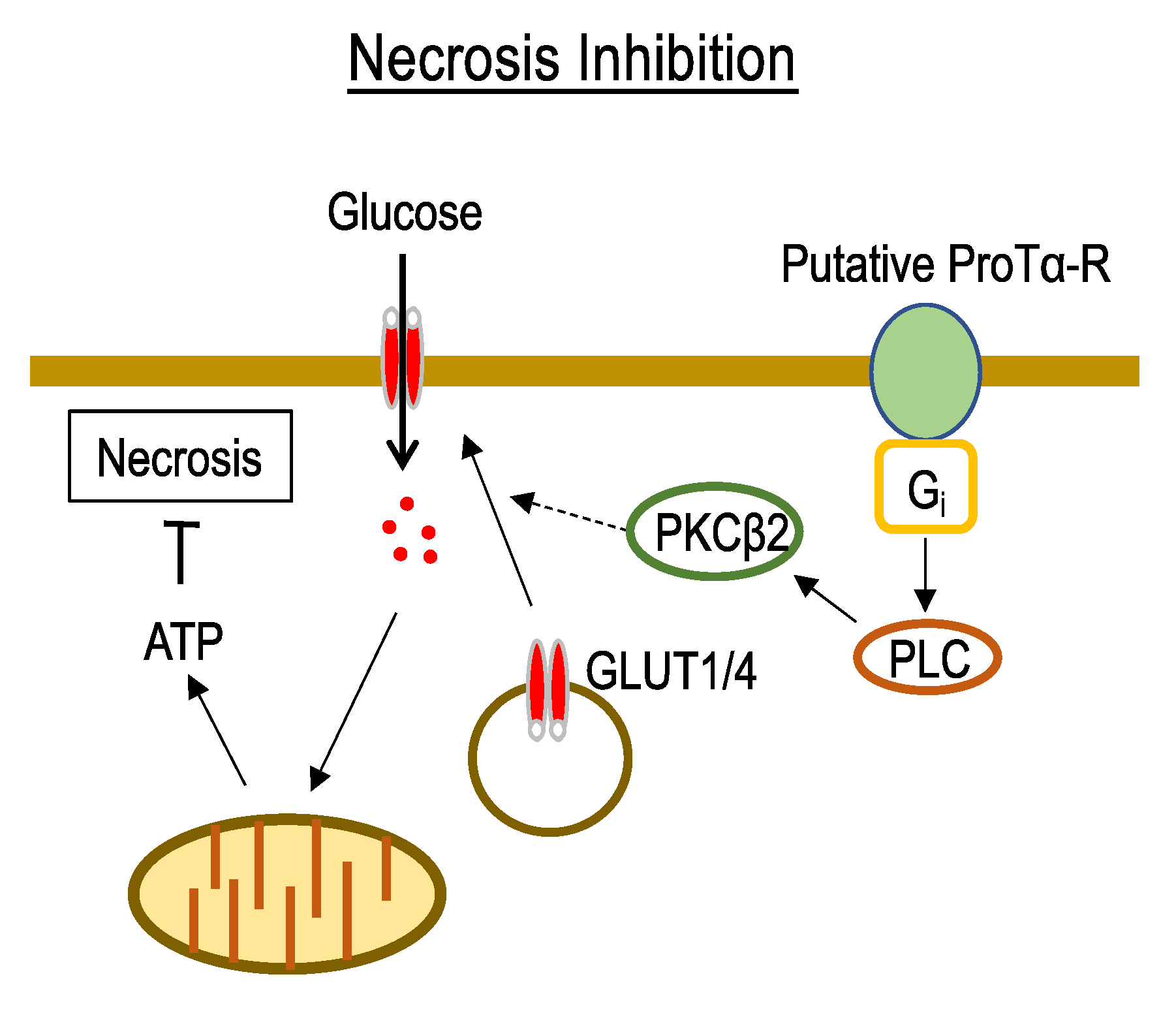
3.1. Mechanisms Underlying ProTα-Induced Inhibition of Neuronal Necrosis
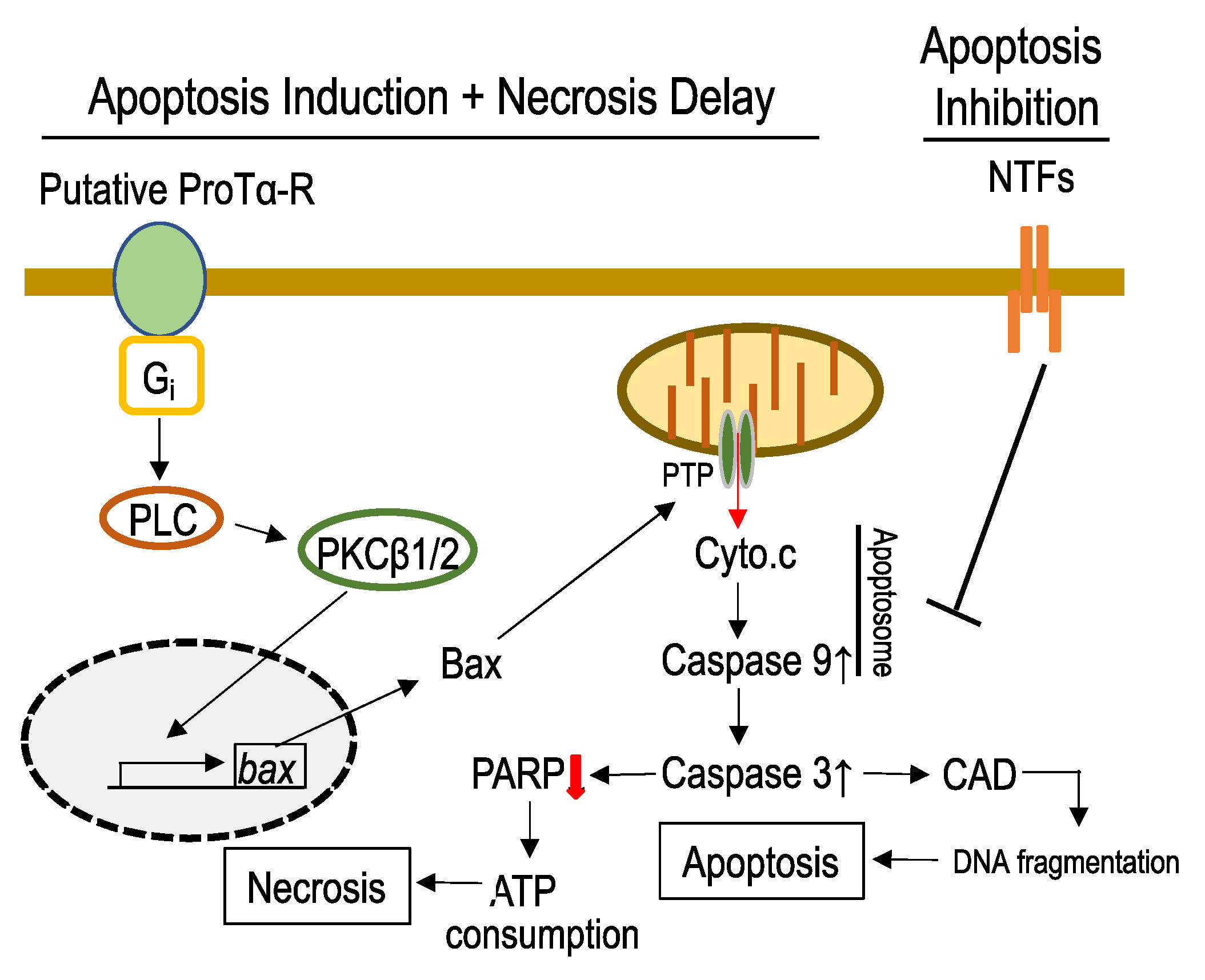
3.2. Mechanisms Underlying ProTα-Induced Apoptosis-Induction
4. In Vivo Beneficial Actions of ProTα
4.1. Cell-Death-Mode-Switch in Retinal Ischemia–Reperfusion Model
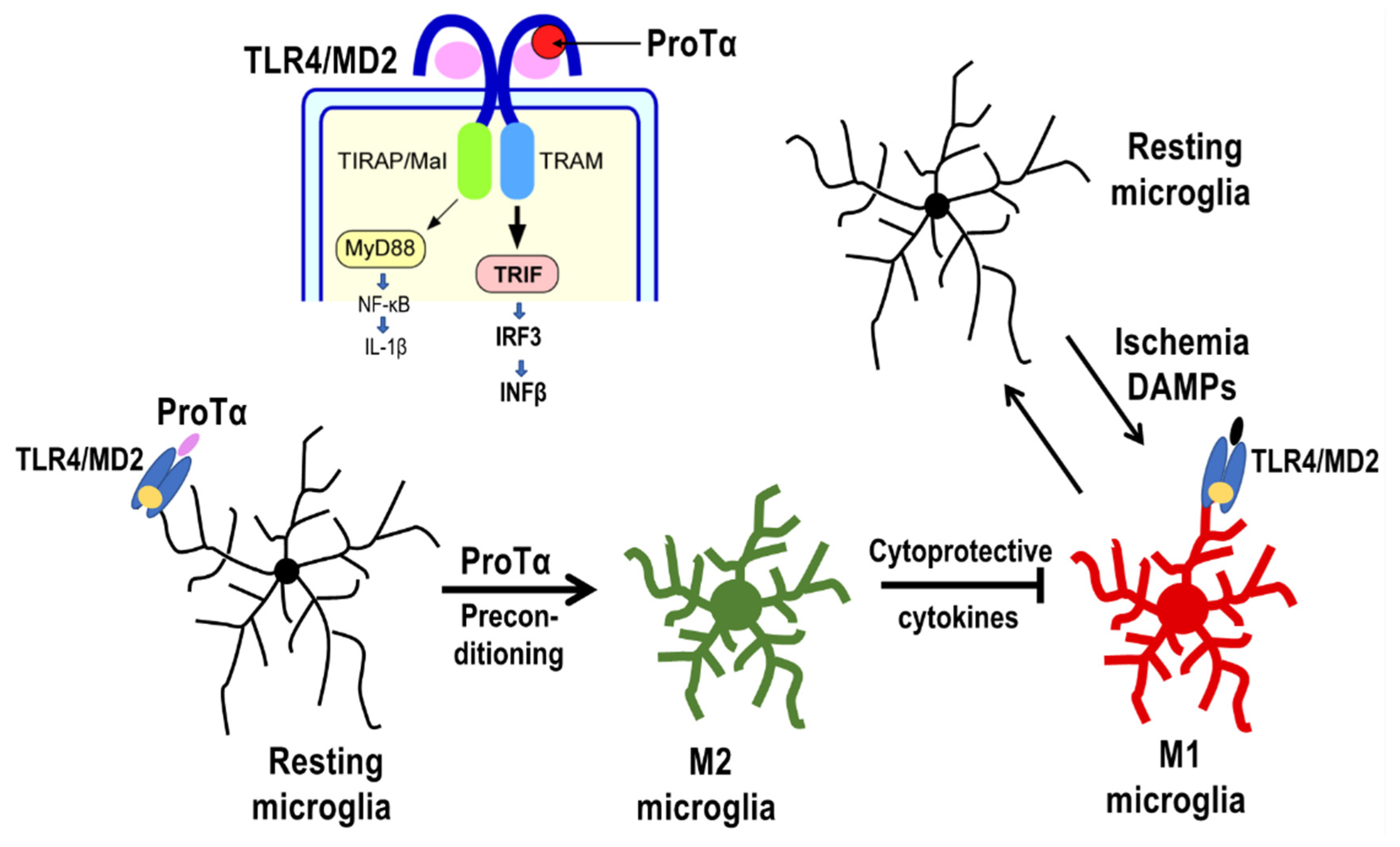
4.2. Preconditioned ProTα-Induced Retinal Protection via TLR4/MD2-TRIF Pathway
4.3. Possible ProTα Receptor Mechanisms via Ecto-F1 ATPase-P2Y12 Complex
5. ProTα Actions in the Cerebral Ischemia–Reperfusion Model
5.1. Inhibition of Cerebral Ischemia-Induced Brain Damage
5.2. Inhibition of Late tPA-Induced Hemorrhage
6. In Vivo Role of ProTα Using Transgenic Mice [49]
6.1. Gross Behavioral Activities of ProTα+/− Mice
6.2. Enhanced Anxiety-like Behaviors of ProTα+/− Mice
6.3. Impaired Learning and Memory in ProTα+/− Mice
6.4. Declined LTP Induction in ProTα+/− Mice
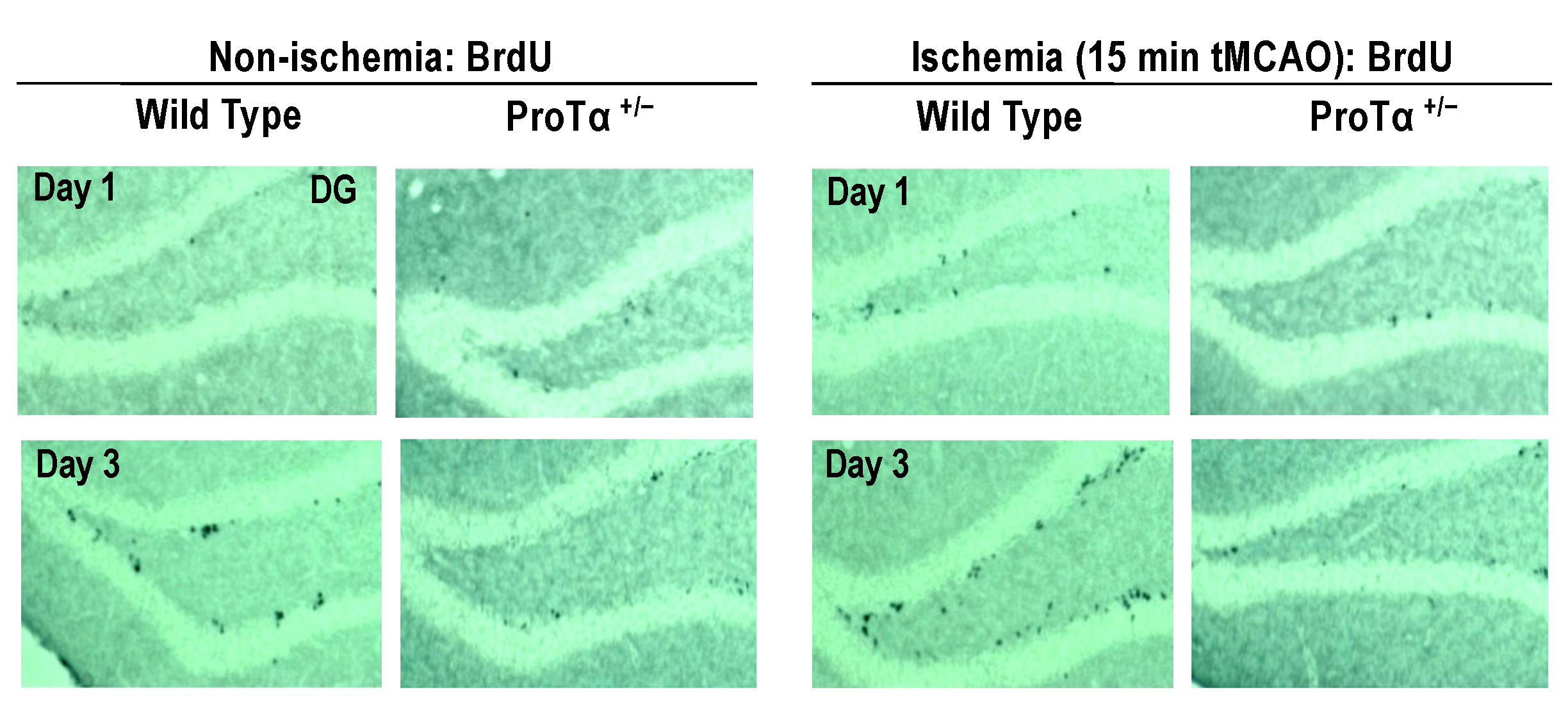
6.5. Decreased Adult Hippocampal Neurogenesis in ProTα+/− Mice
6.6. Conditional ProTα Knock-Out Mice
7. Multiple Intracellular Functions
8. Conclusions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
Abbreviations
| ProTα | Prothymosin alpha |
| NTFs | Neurotrophic factors |
| TLR4 | Toll-like receptor 4 |
| TRIF | Toll–IL-1 receptor domain TLR4 containing adaptor inducing IFN-β |
| RIPK | Receptor-interacting serine/threonine protein kinase |
| LD | Low density |
| HD | High density |
| GFAP | Glial fibrillary acidic protein |
| GLUT | Glucose transporter |
| PLC | Phospholipase C |
| PKC | Protein kinase C |
| AS-ODN | Antisense oligodeoxynucleotide |
| PTP | Permeability transition pore |
| Cyto.c | Cytochrome c |
| BIP-V5 | Bax inhibitor peptide V5 |
| CAD | Caspase-activated end-DNase |
| PARP | Poly (ADP-ribose) polymerase |
| BDNF | Brain-derived neuron AS-ODN-ophic factor |
| H&E | Hematoxylin and eosin |
| ERG | Electroretinography |
| i.vt. | Intravitreous injection |
| GCL | Ganglion cell layer |
| INL | Inner nuclear cell layer |
| ONL | Outer nuclear cell layer |
| EPO | Erythropoietin |
| QCM | Quartz crystal microbalance |
| DAMPs | Damage-associated molecular patterns |
| E-NTDPase | Ectonucleoside triphosphate diphosphohydrolase |
| TTC | 2,3,5-Triphenyltetrazolium chloride |
| BBCAO | Bilateral common carotid arteries occlusion |
| MMPs | Matrix metalloproteases |
| ZO-1 | Zonula occludens |
| ROI | Region of interest |
| MED system | Planar multielectrode array and electronics |
| LTP | Long-term potentiation |
| fEPSP | Field excitatory post-synaptic potential |
| DG | Dentate gyrus |
| ZEB2 | Zinc finger E-box binding homeobox2 |
| Keap1 | Kelch-like ECH-associated protein 1 |
| ANT | Adenine nucleotide translocase |
References
- Ueda, H. Prothymosin alpha and cell death mode switch, a novel target for the prevention of cerebral ischemia-induced damage. Pharmacol. Ther. 2009, 123, 323–333. [Google Scholar] [CrossRef] [PubMed]
- Saleem, S. Apoptosis, Autophagy, Necrosis and Their Multi Galore Crosstalk in Neurodegeneration. Neuroscience 2021, 469, 162–174. [Google Scholar] [CrossRef] [PubMed]
- Mompean, M.; Li, W.; Li, J.; Laage, S.; Siemer, A.B.; Bozkurt, G.; Wu, H.; McDermott, A.E. The Structure of the Necrosome RIPK1-RIPK3 Core, a Human Hetero-Amyloid Signaling Complex. Cell 2018, 173, 1244–1253. [Google Scholar] [CrossRef]
- Moriwaki, K.; Bertin, J.; Gough, P.J.; Orlowski, G.M.; Chan, F.K. Differential roles of RIPK1 and RIPK3 in TNF-induced necroptosis and chemotherapeutic agent-induced cell death. Cell Death Dis. 2015, 6, e1636. [Google Scholar] [CrossRef] [PubMed]
- Ueda, H.; Fujita, R.; Yoshida, A.; Matsunaga, H.; Ueda, M. Identification of prothymosin-alpha1, the necrosis-apoptosis switch molecule in cortical neuronal cultures. J. Cell Biol. 2007, 176, 853–862. [Google Scholar] [CrossRef] [PubMed]
- Galluzzi, L.; Kepp, O.; Krautwald, S.; Kroemer, G.; Linkermann, A. Molecular mechanisms of regulated necrosis. Semin. Cell Dev. Biol. 2014, 35, 24–32. [Google Scholar] [CrossRef]
- Vandenabeele, P.; Galluzzi, L.; Vanden Berghe, T.; Kroemer, G. Molecular mechanisms of necroptosis: An ordered cellular explosion. Nat. Rev. Mol. Cell. Biol. 2010, 11, 700–714. [Google Scholar] [CrossRef]
- Kijogi, C.M.; Khayeka-Wandabwa, C.; Sasaki, K.; Tanaka, Y.; Kurosu, H.; Matsunaga, H.; Ueda, H. Subcellular dissemination of prothymosin alpha at normal physiology: Immunohistochemical vis-a-vis western blotting perspective. BMC Physiol. 2016, 16, 2. [Google Scholar] [CrossRef]
- Matsunaga, H.; Ueda, H. Stress-induced non-vesicular release of prothymosin-alpha initiated by an interaction with S100A13, and its blockade by caspase-3 cleavage. Cell Death Differ. 2010, 17, 1760–1772. [Google Scholar] [CrossRef]
- Ueda, H.; Matsunaga, H.; Halder, S.K. Prothymosin alpha plays multifunctional cell robustness roles in genomic, epigenetic, and nongenomic mechanisms. Ann. N. Y. Acad. Sci. 2012, 1269, 34–43. [Google Scholar] [CrossRef]
- Laster, S.M.; Wood, J.G.; Gooding, L.R. Tumor necrosis factor can induce both apoptic and necrotic forms of cell lysis. J. Immunol. 1988, 141, 2629–2634. [Google Scholar] [CrossRef]
- Pfeil, E.M.; Brands, J.; Merten, N.; Vogtle, T.; Vescovo, M.; Rick, U.; Albrecht, I.M.; Heycke, N.; Kawakami, K.; Ono, Y.; et al. Heterotrimeric G Protein Subunit Galphaq Is a Master Switch for Gbetagamma-Mediated Calcium Mobilization by Gi-Coupled GPCRs. Mol. Cell 2020, 80, 940–954. [Google Scholar] [CrossRef]
- Ueda, H.; Miyamae, T.; Fukushima, N.; Takeshima, H.; Fukuda, K.; Sasaki, Y.; Misu, Y. Opioid mu- and kappa-receptor mediate phospholipase C activation through Gi1 in Xenopus oocytes. Brain Res. Mol. Brain Res. 1995, 32, 166–170. [Google Scholar] [CrossRef]
- Ueda, H.; Tamura, S.; Fukushima, N.; Katada, T.; Ui, M.; Satoh, M. Inositol 1,4,5-trisphosphate-gated calcium transport through plasma membranes in nerve terminals. J. Neurosci. 1996, 16, 2891–2900. [Google Scholar] [CrossRef]
- Ueda, H.; Yoshihara, Y.; Misawa, H.; Fukushima, N.; Katada, T.; Ui, M.; Takagi, H.; Satoh, M. The kyotorphin (tyrosine-arginine) receptor and a selective reconstitution with purified Gi, measured with GTPase and phospholipase C assays. J. Biol. Chem. 1989, 264, 3732–3741. [Google Scholar] [CrossRef]
- Chandra, D.; Tang, D.G. Mitochondrially localized active caspase-9 and caspase-3 result mostly from translocation from the cytosol and partly from caspase-mediated activation in the organelle. Lack of evidence for Apaf-1-mediated procaspase-9 activation in the mitochondria. J. Biol. Chem. 2003, 278, 17408–17420. [Google Scholar] [CrossRef]
- Gnesutta, N.; Minden, A. Death receptor-induced activation of initiator caspase 8 is antagonized by serine/threonine kinase PAK4. Mol. Cell. Biol. 2003, 23, 7838–7848. [Google Scholar] [CrossRef]
- Yoneda, T.; Imaizumi, K.; Oono, K.; Yui, D.; Gomi, F.; Katayama, T.; Tohyama, M. Activation of caspase-12, an endoplastic reticulum (ER) resident caspase, through tumor necrosis factor receptor-associated factor 2-dependent mechanism in response to the ER stress. J. Biol. Chem. 2001, 276, 13935–13940. [Google Scholar] [CrossRef]
- Smaili, S.S.; Hsu, Y.T.; Sanders, K.M.; Russell, J.T.; Youle, R.J. Bax translocation to mitochondria subsequent to a rapid loss of mitochondrial membrane potential. Cell Death Differ. 2001, 8, 909–920. [Google Scholar] [CrossRef]
- Rivas-Carrillo, J.D.; Soto-Gutierrez, A.; Navarro-Alvarez, N.; Noguchi, H.; Okitsu, T.; Chen, Y.; Yuasa, T.; Tanaka, K.; Narushima, M.; Miki, A.; et al. Cell-permeable pentapeptide V5 inhibits apoptosis and enhances insulin secretion, allowing experimental single-donor islet transplantation in mice. Diabetes 2007, 56, 1259–1267. [Google Scholar] [CrossRef] [Green Version]
- Fadeel, B.; Ottosson, A.; Pervaiz, S. Big wheel keeps on turning: Apoptosome regulation and its role in chemoresistance. Cell Death Differ. 2008, 15, 443–452. [Google Scholar] [CrossRef] [PubMed]
- Lopez, K.E.; Bouchier-Hayes, L. Lethal and Non-Lethal Functions of Caspases in the DNA Damage Response. Cells 2022, 11, 1887. [Google Scholar] [CrossRef] [PubMed]
- Pronk, G.J.; Ramer, K.; Amiri, P.; Williams, L.T. Requirement of an ICE-like protease for induction of apoptosis and ceramide generation by REAPER. Science 1996, 271, 808–810. [Google Scholar] [CrossRef]
- Weaver, A.N.; Yang, E.S. Beyond DNA Repair: Additional Functions of PARP-1 in Cancer. Front. Oncol. 2013, 3, 290. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.V.; Chen, H.C.; Walsh, C.M. Necroptotic signaling in adaptive and innate immunity. Semin. Cell Dev. Biol. 2014, 35, 33–39. [Google Scholar] [CrossRef]
- Fujita, R.; Ueda, M.; Fujiwara, K.; Ueda, H. Prothymosin-alpha plays a defensive role in retinal ischemia through necrosis and apoptosis inhibition. Cell Death Differ. 2009, 16, 349–358. [Google Scholar] [CrossRef]
- Mosoian, A.; Teixeira, A.; Burns, C.S.; Sander, L.E.; Gusella, G.L.; He, C.; Blander, J.M.; Klotman, P.; Klotman, M.E. Prothymosin-alpha inhibits HIV-1 via Toll-like receptor 4-mediated type I interferon induction. Proc. Natl. Acad. Sci. USA 2010, 107, 10178–10183. [Google Scholar] [CrossRef]
- Skopeliti, M.; Iconomidou, V.A.; Derhovanessian, E.; Pawelec, G.; Voelter, W.; Kalbacher, H.; Hamodrakas, S.J.; Tsitsilonis, O.E. Prothymosin alpha immunoactive carboxyl-terminal peptide TKKQKTDEDD stimulates lymphocyte reactions, induces dendritic cell maturation and adopts a beta-sheet conformation in a sequence-specific manner. Mol. Immunol. 2009, 46, 784–792. [Google Scholar] [CrossRef]
- Vartanian, K.B.; Stevens, S.L.; Marsh, B.J.; Williams-Karnesky, R.; Lessov, N.S.; Stenzel-Poore, M.P. LPS preconditioning redirects TLR signaling following stroke: TRIF-IRF3 plays a seminal role in mediating tolerance to ischemic injury. J. Neuroinflamm. 2011, 8, 140. [Google Scholar] [CrossRef]
- Omotuyi, O.; Matsunaga, H.; Ueda, H. Evidence for ProTalpha-TLR4/MD-2 binding: Molecular dynamics and gravimetric assay studies. Expert Opin. Biol. Ther. 2015, 15 (Suppl. S1), S223–S229. [Google Scholar] [CrossRef]
- Halder, S.K.; Matsunaga, H.; Ishii, K.J.; Akira, S.; Miyake, K.; Ueda, H. Retinal cell type-specific prevention of ischemia-induced damages by LPS-TLR4 signaling through microglia. J. Neurochem. 2013, 126, 243–260. [Google Scholar] [CrossRef]
- Halder, S.K.; Matsunaga, H.; Ishii, K.J.; Ueda, H. Prothymosin-alpha preconditioning activates TLR4-TRIF signaling to induce protection of ischemic retina. J. Neurochem. 2015, 135, 1161–1177. [Google Scholar] [CrossRef]
- Halder, S.K.; Matsunaga, H.; Ueda, H. Experimental evidence for the involvement of F(0)/F(1) ATPase and subsequent P2Y(12) receptor activation in prothymosin alpha-induced protection of retinal ischemic damage. J. Pharmacol. Sci. 2020, 143, 127–131. [Google Scholar] [CrossRef]
- Ueda, H.; Matsunaga, H.; Matsushita, Y.; Maeda, S.; Iwamoto, R.; Yokoyama, S.; Shirouzu, M. Ecto-F(0)/F(1) ATPase as a novel candidate of prothymosin alpha receptor. Expert Opin. Biol. Ther. 2018, 18, 89–94. [Google Scholar] [CrossRef]
- Freeman, K.W.; Bowman, B.R.; Zetter, B.R. Regenerative protein thymosin beta-4 is a novel regulator of purinergic signaling. FASEB J. 2011, 25, 907–915. [Google Scholar] [CrossRef]
- Martinez, L.O.; Jacquet, S.; Esteve, J.P.; Rolland, C.; Cabezon, E.; Champagne, E.; Pineau, T.; Georgeaud, V.; Walker, J.E.; Terce, F.; et al. Ectopic beta-chain of ATP synthase is an apolipoprotein A-I receptor in hepatic HDL endocytosis. Nature 2003, 421, 75–79. [Google Scholar] [CrossRef]
- Yegutkin, G.G. Nucleotide- and nucleoside-converting ectoenzymes: Important modulators of purinergic signalling cascade. Biochim. Biophys. Acta 2008, 1783, 673–694. [Google Scholar] [CrossRef]
- Cardouat, G.; Duparc, T.; Fried, S.; Perret, B.; Najib, S.; Martinez, L.O. Ectopic adenine nucleotide translocase activity controls extracellular ADP levels and regulates the F(1)-ATPase-mediated HDL endocytosis pathway on hepatocytes. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2017, 1862, 832–841. [Google Scholar] [CrossRef]
- Martinez, L.O.; Najib, S.; Perret, B.; Cabou, C.; Lichtenstein, L. Ecto-F1-ATPase/P2Y pathways in metabolic and vascular functions of high density lipoproteins. Atherosclerosis 2015, 238, 89–100. [Google Scholar] [CrossRef]
- Cabou, C.; Honorato, P.; Briceno, L.; Ghezali, L.; Duparc, T.; Leon, M.; Combes, G.; Frayssinhes, L.; Fournel, A.; Abot, A.; et al. Pharmacological inhibition of the F(1) -ATPase/P2Y(1) pathway suppresses the effect of apolipoprotein A1 on endothelial nitric oxide synthesis and vasorelaxation. Acta Physiol. 2019, 226, e13268. [Google Scholar] [CrossRef] [Green Version]
- Fujita, R.; Ma, Y.; Ueda, H. Lysophosphatidic acid-induced membrane ruffling and brain-derived neurotrophic factor gene expression are mediated by ATP release in primary microglia. J. Neurochem. 2008, 107, 152–160. [Google Scholar] [CrossRef] [PubMed]
- Fujita, R.; Ueda, H. Prothymosin-alpha1 prevents necrosis and apoptosis following stroke. Cell Death Differ. 2007, 14, 1839–1842. [Google Scholar] [CrossRef] [PubMed]
- Balami, J.S.; Hadley, G.; Sutherland, B.A.; Karbalai, H.; Buchan, A.M. The exact science of stroke thrombolysis and the quiet art of patient selection. Brain 2013, 136, 3528–3553. [Google Scholar] [CrossRef] [PubMed]
- Cheng, N.T.; Kim, A.S. Intravenous Thrombolysis for Acute Ischemic Stroke Within 3 Hours Versus Between 3 and 4.5 Hours of Symptom Onset. Neurohospitalist 2015, 5, 101–109. [Google Scholar] [CrossRef] [PubMed]
- Davis, S.M.; Donnan, G.A. 4.5 hours: The new time window for tissue plasminogen activator in stroke. Stroke 2009, 40, 2266–2267. [Google Scholar] [CrossRef]
- De Los Rios la Rosa, F.; Khoury, J.; Kissela, B.M.; Flaherty, M.L.; Alwell, K.; Moomaw, C.J.; Khatri, P.; Adeoye, O.; Woo, D.; Ferioli, S.; et al. Eligibility for Intravenous Recombinant Tissue-Type Plasminogen Activator Within a Population: The Effect of the European Cooperative Acute Stroke Study (ECASS) III Trial. Stroke 2012, 43, 1591–1595. [Google Scholar] [CrossRef]
- Suzuki, Y.; Nagai, N.; Umemura, K. A Review of the Mechanisms of Blood-Brain Barrier Permeability by Tissue-Type Plasminogen Activator Treatment for Cerebral Ischemia. Front. Cell. Neurosci. 2016, 10, 2. [Google Scholar] [CrossRef]
- Halder, S.K.; Matsunaga, H.; Ueda, H. Prothymosin alpha and its mimetic hexapeptide improve delayed tissue plasminogen activator-induced brain damage following cerebral ischemia. J. Neurochem. 2020, 153, 772–789. [Google Scholar] [CrossRef]
- Ueda, H.; Sasaki, K.; Halder, S.K.; Deguchi, Y.; Takao, K.; Miyakawa, T.; Tajima, A. Prothymosin alpha-deficiency enhances anxiety-like behaviors and impairs learning/memory functions and neurogenesis. J. Neurochem. 2017, 141, 124–136. [Google Scholar] [CrossRef]
- Bianco, N.R.; Montano, M.M. Regulation of prothymosin alpha by estrogen receptor alpha: Molecular mechanisms and relevance in estrogen-mediated breast cell growth. Oncogene 2002, 21, 5233–5244. [Google Scholar] [CrossRef] [Green Version]
- Ha, S.Y.; Song, D.H.; Hwang, S.H.; Cho, S.Y.; Park, C.K. Expression of prothymosin alpha predicts early recurrence and poor prognosis of hepatocellular carcinoma. Hepatobiliary Pancreat. Dis. Int. 2015, 14, 171–177. [Google Scholar] [CrossRef]
- Karapetian, R.N.; Evstafieva, A.G.; Abaeva, I.S.; Chichkova, N.V.; Filonov, G.S.; Rubtsov, Y.P.; Sukhacheva, E.A.; Melnikov, S.V.; Schneider, U.; Wanker, E.E.; et al. Nuclear oncoprotein prothymosin alpha is a partner of Keap1: Implications for expression of oxidative stress-protecting genes. Mol. Cell. Biol. 2005, 25, 1089–1099. [Google Scholar] [CrossRef]
- Su, B.H.; Tseng, Y.L.; Shieh, G.S.; Chen, Y.C.; Shiang, Y.C.; Wu, P.; Li, K.J.; Yen, T.H.; Shiau, A.L.; Wu, C.L. Prothymosin alpha overexpression contributes to the development of pulmonary emphysema. Nat. Commun. 2013, 4, 1906. [Google Scholar] [CrossRef]
- Mosoian, A. Intracellular and extracellular cytokine-like functions of prothymosin alpha: Implications for the development of immunotherapies. Future Med. Chem. 2011, 3, 1199–1208. [Google Scholar] [CrossRef]
- Ioannou, K.; Derhovanessian, E.; Tsakiri, E.; Samara, P.; Kalbacher, H.; Voelter, W.; Trougakos, I.P.; Pawelec, G.; Tsitsilonis, O.E. Prothymosin alpha and a prothymosin alpha-derived peptide enhance T(H)1-type immune responses against defined HER-2/neu epitopes. BMC Immunol. 2013, 14, 43. [Google Scholar] [CrossRef]
- Halder, S.K.; Sasaki, K.; Ueda, H. Ggamma7-specific prothymosin alpha deletion causes stress- and age-dependent motor dysfunction and anxiety. Biochem. Biophys. Res. Commun. 2020, 522, 264–269. [Google Scholar] [CrossRef]
- Deacon, R.M. Digging and marble burying in mice: Simple methods for in vivo identification of biological impacts. Nat. Protoc. 2006, 1, 122–124. [Google Scholar] [CrossRef]
- Thomas, A.; Burant, A.; Bui, N.; Graham, D.; Yuva-Paylor, L.A.; Paylor, R. Marble burying reflects a repetitive and perseverative behavior more than novelty-induced anxiety. Psychopharmacology 2009, 204, 361–373. [Google Scholar] [CrossRef]
- Nagai, J.; Kurokawa, M.; Takeshima, H.; Kieffer, B.L.; Ueda, H. Circadian-dependent learning and memory enhancement in nociceptin receptor-deficient mice with a novel KUROBOX apparatus using stress-free positive cue task. J. Pharmacol. Exp. Ther. 2007, 321, 195–201. [Google Scholar] [CrossRef]
- Gruart, A.; Munoz, M.D.; Delgado-Garcia, J.M. Involvement of the CA3-CA1 synapse in the acquisition of associative learning in behaving mice. J. Neurosci. 2006, 26, 1077–1087. [Google Scholar] [CrossRef]
- Oka, H.; Shimono, K.; Ogawa, R.; Sugihara, H.; Taketani, M. A new planar multielectrode array for extracellular recording: Application to hippocampal acute slice. J. Neurosci. Methods 1999, 93, 61–67. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, K.; Omotuyi, O.I.; Ueda, M.; Shinohara, K.; Ueda, H. NMDA receptor agonists reverse impaired psychomotor and cognitive functions associated with hippocampal Hbegf-deficiency in mice. Mol. Brain 2015, 8, 83. [Google Scholar] [CrossRef] [PubMed]
- Shimono, K.; Kubota, D.; Brucher, F.; Taketani, M.; Lynch, G. Asymmetrical distribution of the Schaffer projections within the apical dendrites of hippocampal field CA1. Brain Res. 2002, 950, 279–287. [Google Scholar] [CrossRef] [PubMed]
- Whitlock, J.R.; Heynen, A.J.; Shuler, M.G.; Bear, M.F. Learning induces long-term potentiation in the hippocampus. Science 2006, 313, 1093–1097. [Google Scholar] [CrossRef] [PubMed]
- Kitamura, T.; Saitoh, Y.; Takashima, N.; Murayama, A.; Niibori, Y.; Ageta, H.; Sekiguchi, M.; Sugiyama, H.; Inokuchi, K. Adult neurogenesis modulates the hippocampus-dependent period of associative fear memory. Cell 2009, 139, 814–827. [Google Scholar] [CrossRef]
- Revest, J.M.; Dupret, D.; Koehl, M.; Funk-Reiter, C.; Grosjean, N.; Piazza, P.V.; Abrous, D.N. Adult hippocampal neurogenesis is involved in anxiety-related behaviors. Mol. Psychiatry 2009, 14, 959–967. [Google Scholar] [CrossRef]
- Snyder, J.S.; Soumier, A.; Brewer, M.; Pickel, J.; Cameron, H.A. Adult hippocampal neurogenesis buffers stress responses and depressive behaviour. Nature 2011, 476, 458–461. [Google Scholar] [CrossRef]
- Dhakshinamoorthy, S.; Long, D.J., 2nd; Jaiswal, A.K. Antioxidant regulation of genes encoding enzymes that detoxify xenobiotics and carcinogens. Curr. Top. Cell. Regul. 2000, 36, 201–216. [Google Scholar] [CrossRef]
- Jaiswal, A.K. Regulation of genes encoding NAD(P)H:quinone oxidoreductases. Free Radic. Biol. Med. 2000, 29, 254–262. [Google Scholar] [CrossRef]
- Kobayashi, M.; Yamamoto, M. Nrf2-Keap1 regulation of cellular defense mechanisms against electrophiles and reactive oxygen species. Adv. Enzyme Regul. 2006, 46, 113–140. [Google Scholar] [CrossRef]
- McMahon, M.; Thomas, N.; Itoh, K.; Yamamoto, M.; Hayes, J.D. Dimerization of substrate adaptors can facilitate cullin-mediated ubiquitylation of proteins by a “tethering” mechanism: A two-site interaction model for the Nrf2-Keap1 complex. J. Biol. Chem. 2006, 281, 24756–24768. [Google Scholar] [CrossRef]
- Gomez-Marquez, J. Function of prothymosin alpha in chromatin decondensation and expression of thymosin beta-4 linked to angiogenesis and synaptic plasticity. Ann. N. Y. Acad. Sci. 2007, 1112, 201–209. [Google Scholar] [CrossRef]
- Subramanian, C.; Hasan, S.; Rowe, M.; Hottiger, M.; Orre, R.; Robertson, E.S. Epstein-Barr virus nuclear antigen 3C and prothymosin alpha interact with the p300 transcriptional coactivator at the CH1 and CH3/HAT domains and cooperate in regulation of transcription and histone acetylation. J. Virol. 2002, 76, 4699–4708. [Google Scholar] [CrossRef]
- Karetsou, Z.; Kretsovali, A.; Murphy, C.; Tsolas, O.; Papamarcaki, T. Prothymosin alpha interacts with the CREB-binding protein and potentiates transcription. EMBO Rep. 2002, 3, 361–366. [Google Scholar] [CrossRef]
- Happel, N.; Doenecke, D. Histone H1 and its isoforms: Contribution to chromatin structure and function. Gene 2009, 431, 1–12. [Google Scholar] [CrossRef]
- Gladka, M.M.; Kohela, A.; Molenaar, B.; Versteeg, D.; Kooijman, L.; Monshouwer-Kloots, J.; Kremer, V.; Vos, H.R.; Huibers, M.M.H.; Haigh, J.J.; et al. Cardiomyocytes stimulate angiogenesis after ischemic injury in a ZEB2-dependent manner. Nat. Commun. 2021, 12, 84. [Google Scholar] [CrossRef]






Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ueda, H. Prothymosin α Plays Role as a Brain Guardian through Ecto-F1 ATPase-P2Y12 Complex and TLR4/MD2. Cells 2023, 12, 496. https://doi.org/10.3390/cells12030496
Ueda H. Prothymosin α Plays Role as a Brain Guardian through Ecto-F1 ATPase-P2Y12 Complex and TLR4/MD2. Cells. 2023; 12(3):496. https://doi.org/10.3390/cells12030496
Chicago/Turabian StyleUeda, Hiroshi. 2023. "Prothymosin α Plays Role as a Brain Guardian through Ecto-F1 ATPase-P2Y12 Complex and TLR4/MD2" Cells 12, no. 3: 496. https://doi.org/10.3390/cells12030496
APA StyleUeda, H. (2023). Prothymosin α Plays Role as a Brain Guardian through Ecto-F1 ATPase-P2Y12 Complex and TLR4/MD2. Cells, 12(3), 496. https://doi.org/10.3390/cells12030496