Introduction to the Special Issue “Skeletal Muscle Atrophy: Mechanisms at a Cellular Level”

 ,
,  and
and 
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Skeletal Muscle
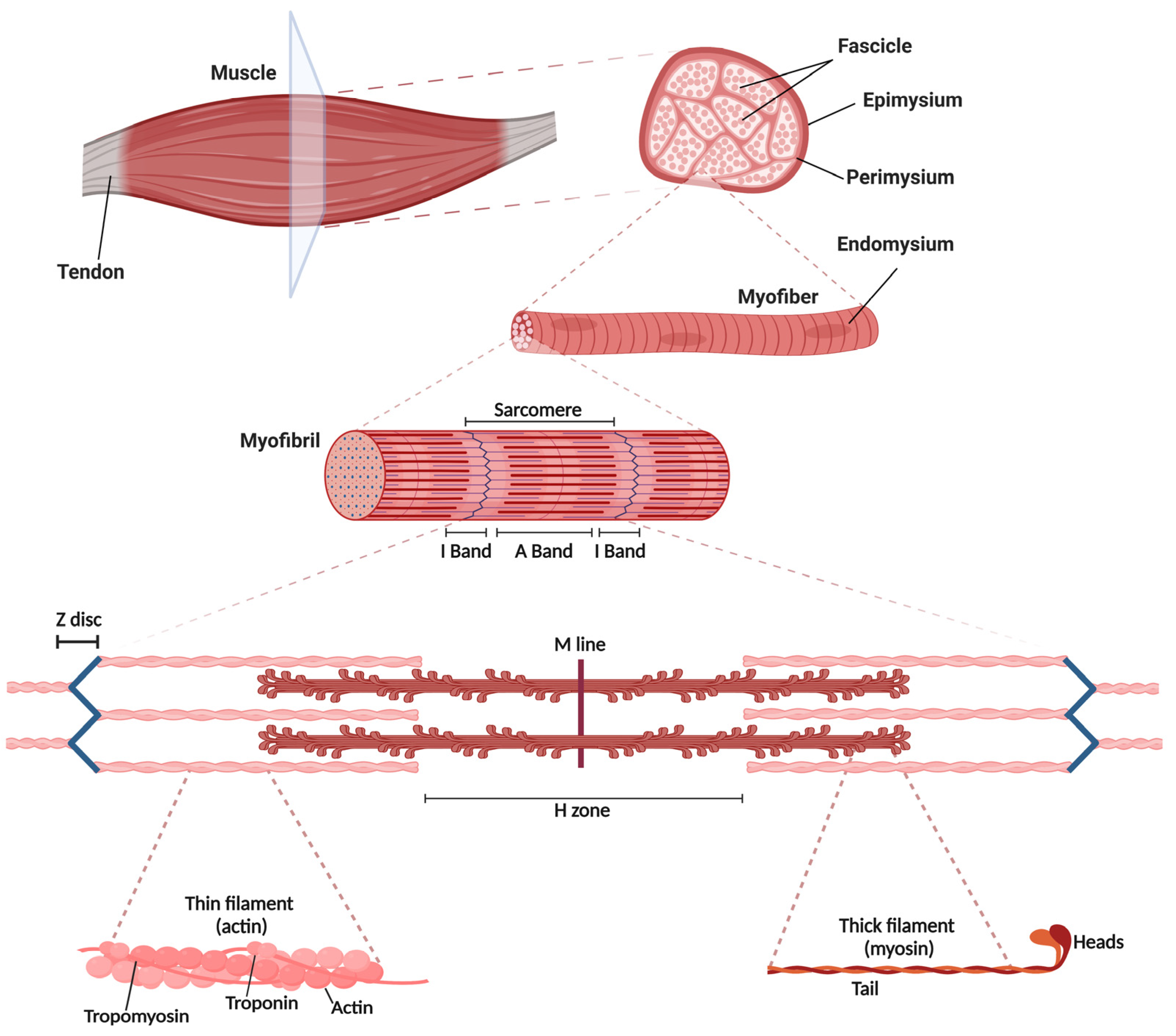
2.1. Skeletal Muscle Structure
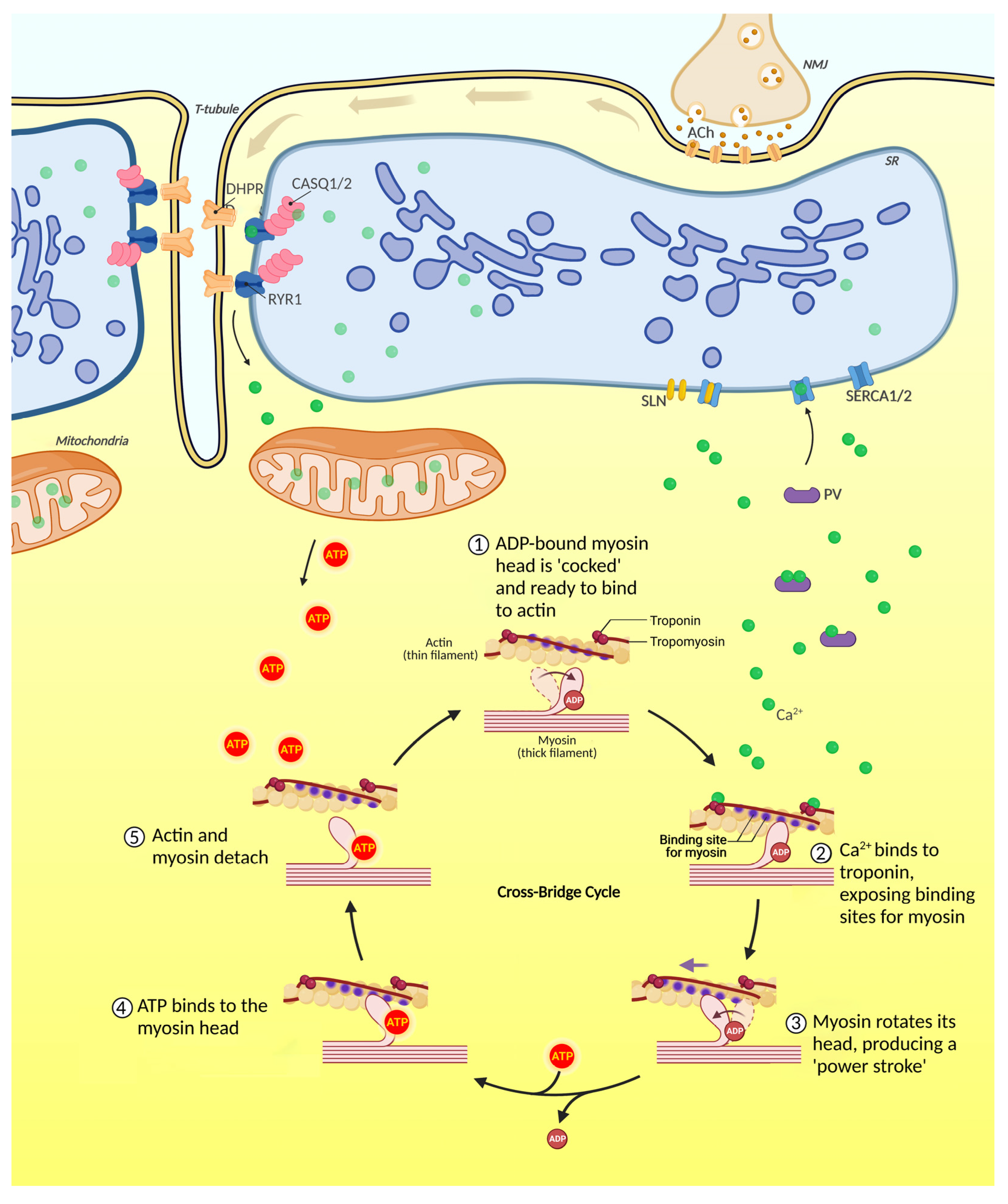
2.2. Skeletal Muscle Contraction
2.3. Fiber Types and Metabolism
3. Contribution to the Special Issue
3.1. Section 1
3.2. Section 2
3.3. Section 3
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Schiaffino, S.; Reggiani, C. Fiber Types in Mammalian Skeletal Muscles. Physiol. Rev. 2011, 91, 1447–1531. [Google Scholar] [CrossRef] [PubMed]
- Glass, D.J. Signalling pathways that mediate skeletal muscle hypertrophy and atrophy. Nature 2003, 5, 87–90. [Google Scholar] [CrossRef]
- Van Leeuwenhoek, A.I. A letter from Mr Anthony van Leeuwenhoek, F. R. S. containing his observations upon the seminal vessels, muscular fibres, and blood of whales. Philos. Trans. R. Soc. Lond. 1710, 27, 438–446. [Google Scholar] [CrossRef]
- Herzog, W. The multiple roles of titin in muscle contraction and force production. Biophys. Rev. 2018, 10, 1187–1199. [Google Scholar] [CrossRef] [PubMed]
- Barer, R. The structure of the striated muscle fibre. Biol. Rev. 1948, 23, 159–200. [Google Scholar] [CrossRef] [PubMed]
- Bennett, H.S. Modern Concepts of Structure of Striated Muscle. Available online: https://www.semanticscholar.org/paper/Modern-concepts-of-structure-of-striated-muscle.-Bennett/fb29f8162ac89b724323293cad64598ed8a412cc (accessed on 4 January 2023).
- Rall, J.A. What makes skeletal muscle striated? Discoveries in the endosarcomeric and exosarcomeric cytoskeleton. Adv. Physiol. Educ. 2018, 42, 672–684. [Google Scholar] [CrossRef] [PubMed]
- Grabarek, Z.; Grabarek, J.; Leavis, P.C.; Gergely, J. Cooperative binding to the Ca2+-specific sites of troponin C in regulated actin and actomyosin. J. Biol. Chem. 1983, 258, 14098–14102. [Google Scholar] [CrossRef]
- Franzini-Armstrong, C. The relationship between form and function throughout the history of excitation–contraction coupling. J. Gen. Physiol. 2018, 150, 189–210. [Google Scholar] [CrossRef]
- Al-Qusairi, L.; Laporte, J. T-tubule biogenesis and triad formation in skeletal muscle and implication in human diseases. Skelet. Muscle 2011, 1, 26. [Google Scholar] [CrossRef]
- Wei, L.; Hanna, A.D.; Beard, N.A.; Dulhunty, A.F. Unique isoform-specific properties of calsequestrin in the heart and skeletal muscle. Cell Calcium 2009, 45, 474–484. [Google Scholar] [CrossRef]
- Novák, P.; Soukup, T. Calsequestrin Distribution, Structure and Function, Its Role in Normal and Pathological Situations and the Effect of Thyroid Hormones. Physiol. Res. 2011, 60, 439–452. [Google Scholar] [CrossRef]
- Kuo, I.; Ehrlich, B.E. Signaling in Muscle Contraction. Cold Spring Harb. Perspect. Biol. 2015, 7, a006023. [Google Scholar] [CrossRef] [PubMed]
- Marty, I.; Fauré, J. Excitation-Contraction Coupling Alterations in Myopathies. J. Neuromuscul. Dis. 2016, 3, 443–453. [Google Scholar] [CrossRef]
- Scott, W.; Stevens, J.; Binder–Macleod, S.A. Human Skeletal Muscle Fiber Type Classifications. Phys. Ther. 2001, 81, 1810–1816. [Google Scholar] [CrossRef]
- Ørtenblad, N.; Nielsen, J.; Boushel, R.; Söderlund, K.; Saltin, B.; Holmberg, H.-C. The Muscle Fiber Profiles, Mitochondrial Content, and Enzyme Activities of the Exceptionally Well-Trained Arm and Leg Muscles of Elite Cross-Country Skiers. Front. Physiol. 2018, 9, 1031. [Google Scholar] [CrossRef] [PubMed]
- Harridge, S.; Bottinelli, R.; Canepari, M.; Pellegrino, M.A.; Reggiani, C.; Esbjörnsson, M.; Saltin, B. Whole-muscle and single-fibre contractile properties and myosin heavy chain isoforms in humans. Pflugers Arch. 1996, 432, 913–920. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, J.; Gejl, K.D.; Hey-Mogensen, M.; Holmberg, H.-C.; Suetta, C.; Krustrup, P.; Elemans, C.P.H.; Ørtenblad, N. Plasticity in mitochondrial cristae density allows metabolic capacity modulation in human skeletal muscle. J. Physiol. 2017, 595, 2839–2847. [Google Scholar] [CrossRef]
- Gorza, L.; Sorge, M.; Seclì, L.; Brancaccio, M. Master Regulators of Muscle Atrophy: Role of Costamere Components. Cells 2021, 10, 61. [Google Scholar] [CrossRef]
- Canfora, I.; Tarantino, N.; Pierno, S. Metabolic Pathways and Ion Channels Involved in Skeletal Muscle Atrophy: A Starting Point for Potential Therapeutic Strategies. Cells 2022, 11, 2566. [Google Scholar] [CrossRef]
- Marchioretti, C.; Zuccaro, E.; Pandey, U.B.; Rosati, J.; Basso, M.; Pennuto, M. Skeletal Muscle Pathogenesis in Polyglutamine Diseases. Cells 2022, 11, 2105. [Google Scholar] [CrossRef]
- Aquila, G.; Cecconi, A.D.R.; Brault, J.J.; Corli, O.; Piccirillo, R. Nutraceuticals and Exercise against Muscle Wasting during Cancer Cachexia. Cells 2020, 9, 2536. [Google Scholar] [CrossRef]
- Adams, V.; Gußen, V.; Zozulya, S.; Cruz, A.; Moriscot, A.; Linke, A.; Labeit, S. Small-Molecule Chemical Knockdown of MuRF1 in Melanoma Bearing Mice Attenuates Tumor Cachexia Associated Myopathy. Cells 2020, 9, 2272. [Google Scholar] [CrossRef]
- Viana, L.R.; Chiocchetti, G.D.M.E.; Oroy, L.; Vieira, W.F.; Busanello, E.N.B.; Marques, A.C.; Salgado, C.D.M.; de Oliveira, A.L.R.; Vieira, A.S.; Suarez, P.S.; et al. Leucine-Rich Diet Improved Muscle Function in Cachectic Walker 256 Tumour-Bearing Wistar Rats. Cells 2021, 10, 3272. [Google Scholar] [CrossRef] [PubMed]
- Alves, P.K.N.; Cruz, A.; Silva, W.J.; Labeit, S.; Moriscot, A.S. Leucine Supplementation Decreases HDAC4 Expression and Nuclear Localization in Skeletal Muscle Fiber of Rats Submitted to Hindlimb Immobilization. Cells 2020, 9, 2582. [Google Scholar] [CrossRef]
- Marmolejo-Martínez-Artesero, S.; Romeo-Guitart, D.; García, L.M.; Barreiro, E.; Casas, C. NeuroHeal Reduces Muscle Atrophy and Modulates Associated Autophagy. Cells 2020, 9, 1575. [Google Scholar] [CrossRef] [PubMed]
- Marmolejo-Martínez-Artesero, S.; Romeo-Guitart, D.; Venegas, V.; Marotta, M.; Casas, C. NeuroHeal Improves Muscle Regeneration after Injury. Cells 2020, 10, 22. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; An, S.; Lee, S.-J.; Kang, J.-S. Protein Arginine Methyltransferases in Neuromuscular Function and Diseases. Cells 2022, 11, 364. [Google Scholar] [CrossRef] [PubMed]
- Cussonneau, L.; Boyer, C.; Brun, C.; Deval, C.; Loizon, E.; Meugnier, E.; Gueret, E.; Dubois, E.; Taillandier, D.; Polge, C.; et al. Concurrent BMP Signaling Maintenance and TGF-β Signaling Inhibition Is a Hallmark of Natural Resistance to Muscle Atrophy in the Hibernating Bear. Cells 2021, 10, 1873. [Google Scholar] [CrossRef] [PubMed]
- Choi, H.-J.; Yeon, M.-H.; Jun, H.-S. Schisandrae chinensis Fructus Extract Ameliorates Muscle Atrophy in Streptozotocin-Induced Diabetic Mice by Downregulation of the CREB-KLF15 and Autophagy–Lysosomal Pathways. Cells 2021, 10, 2283. [Google Scholar] [CrossRef]
- Liu, Y.; Zhou, Z.; Li, K.; Wang, P.; Chen, Y.; Deng, S.; Li, W.; Yu, K.; Wang, K. VMP1 Regulated by chi-miR-124a Effects Goat Myoblast Proliferation, Autophagy, and Apoptosis through the PI3K/ULK1/mTOR Signaling Pathway. Cells 2022, 11, 2227. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zuccaro, E.; Marchioretti, C.; Pirazzini, M.; Pennuto, M. Introduction to the Special Issue “Skeletal Muscle Atrophy: Mechanisms at a Cellular Level”. Cells 2023, 12, 502. https://doi.org/10.3390/cells12030502
Zuccaro E, Marchioretti C, Pirazzini M, Pennuto M. Introduction to the Special Issue “Skeletal Muscle Atrophy: Mechanisms at a Cellular Level”. Cells. 2023; 12(3):502. https://doi.org/10.3390/cells12030502
Chicago/Turabian StyleZuccaro, Emanuela, Caterina Marchioretti, Marco Pirazzini, and Maria Pennuto. 2023. "Introduction to the Special Issue “Skeletal Muscle Atrophy: Mechanisms at a Cellular Level”" Cells 12, no. 3: 502. https://doi.org/10.3390/cells12030502
APA StyleZuccaro, E., Marchioretti, C., Pirazzini, M., & Pennuto, M. (2023). Introduction to the Special Issue “Skeletal Muscle Atrophy: Mechanisms at a Cellular Level”. Cells, 12(3), 502. https://doi.org/10.3390/cells12030502