
Cancer Stem Cell and Aggressiveness Traits Are Promoted by Stable Endothelin-Converting Enzyme-1c in Glioblastoma Cells

 , , , , ,
, , , , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. GlioVis Analysis
2.3. Lentiviral Cloning
2.4. Glioblastoma Stem-like Cell (GSC) Enrichment
2.5. Cell Viability
2.6. Cell Adhesion
2.7. Indirect Immunofluorescence (IIF)
2.8. Migration and Invasion
2.9. Vasculogenic Mimicry
2.10. Protein Stability
2.11. RT-PCR
2.12. Western Blot
2.13. Enzyme-Linked Immunosorbent Assay (ELISA)
2.14. Statistical Analysis
3. Result
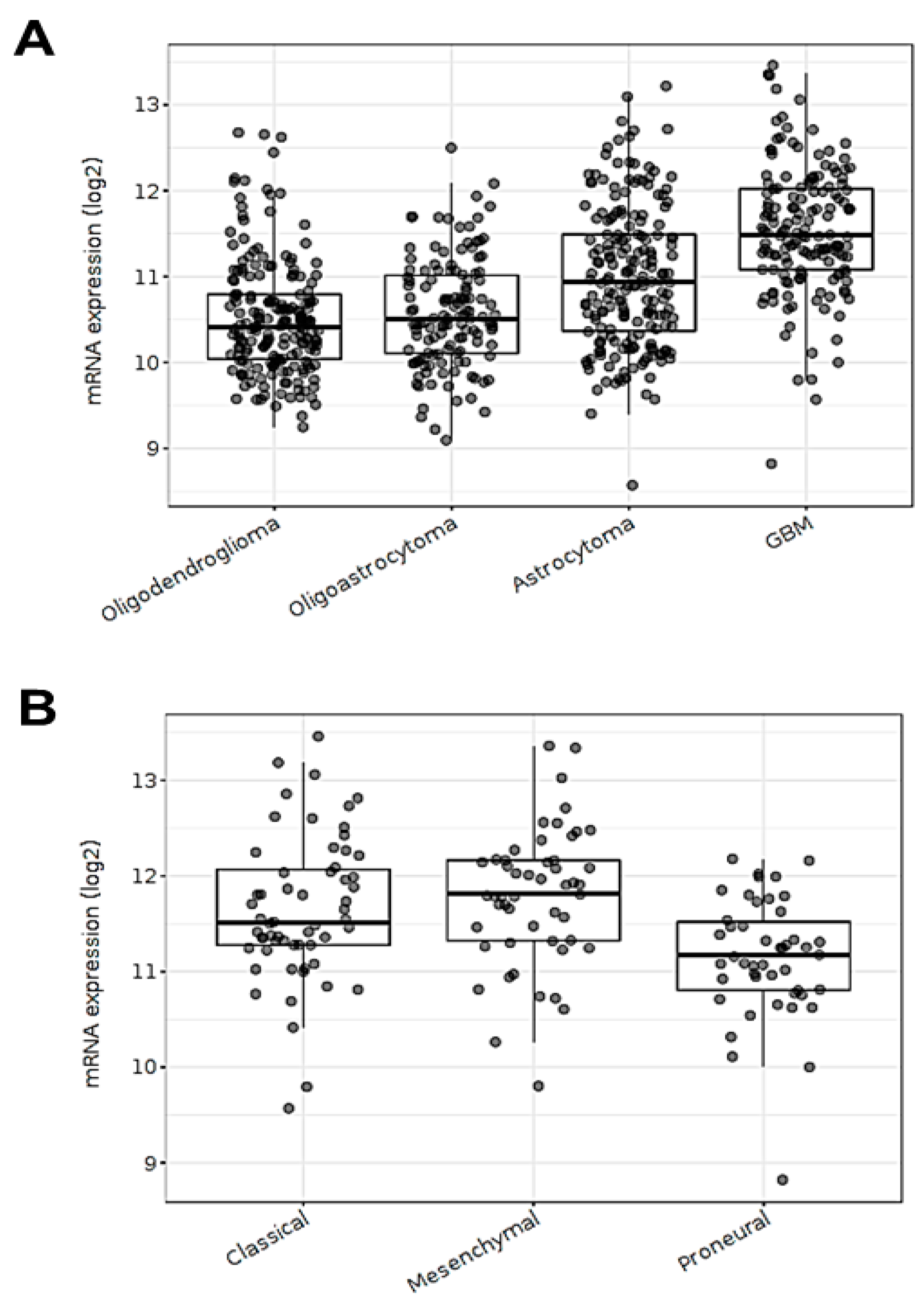
3.1. ECE1 Transcript Expression Is Related to Aggressive GBM Subtypes
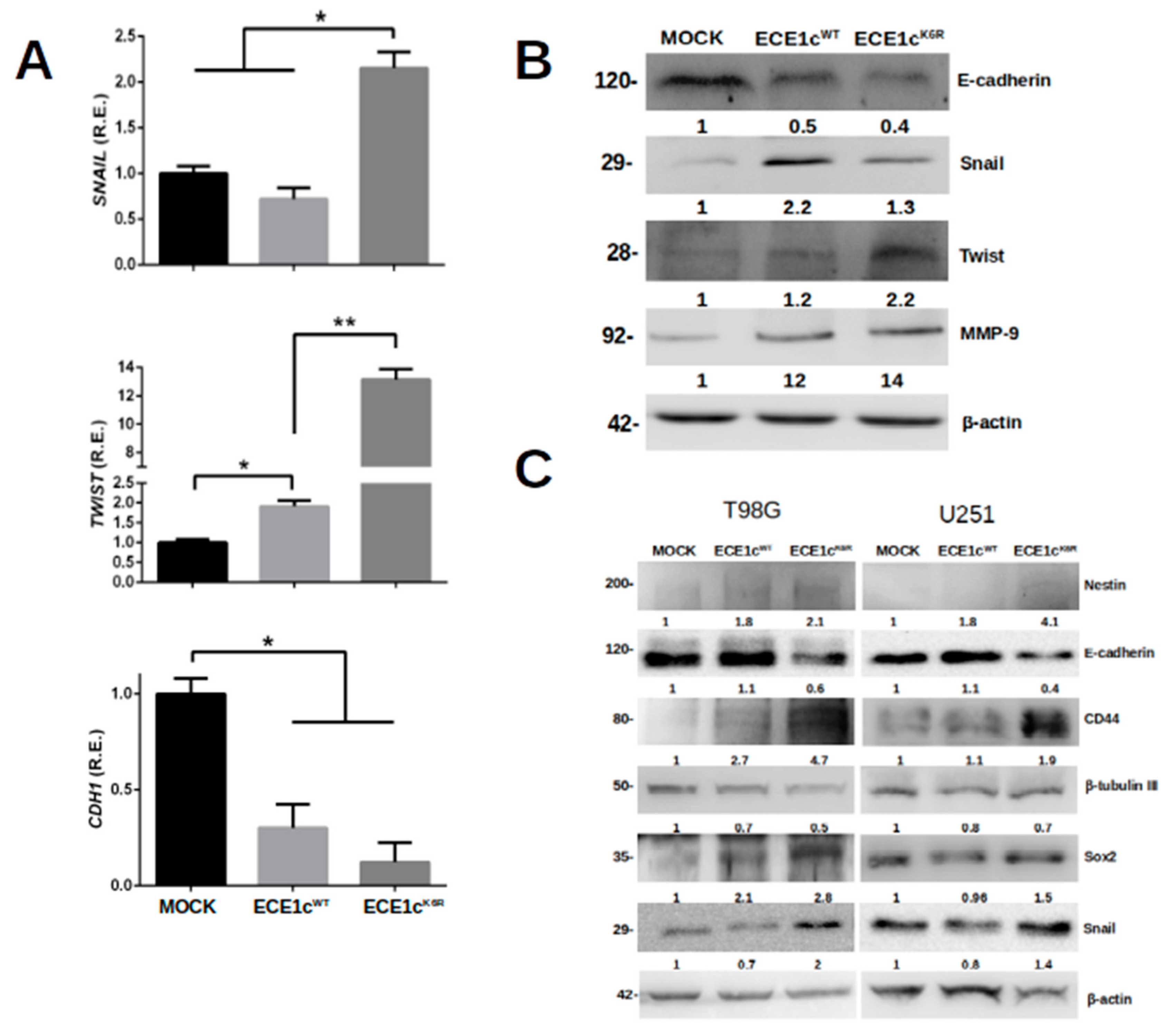
3.2. Lysine 6 Is Involved in CK2-Dependent ECE1c Stability
3.3. Highly Stable ECE1cK6R Promotes Stem-like Traits in GBM Cells
3.4. Chemoresistance Is Enhanced in ECE1cK6R-Expressing GBM Cells
3.5. ECE1cK6R Promotes Metastasis-Associated Traits in GBM Cells
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Wang, Y.; Jiang, T. Understanding high grade glioma: Molecular mechanism, therapy and comprehensive management. Cancer Lett. 2013, 331, 139–146. [Google Scholar] [CrossRef]
- Omuro, A. Glioblastoma and Other Malignant Gliomas. JAMA 2013, 310, 1842. [Google Scholar] [CrossRef] [PubMed]
- Wen, P.; Kesari, S. Malignant Gliomas in Adults. N. Engl. J. Med. 2008, 359, 492–507. [Google Scholar] [CrossRef]
- Stupp, R.; Mason, W.P.; van den Bent, M.J.; Weller, M.; Fisher, B.; Taphoorn, M.J.; Belanger, K.; Brandes, A.A.; Marosi, C.; Bogdahn, U.; et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N. Engl. J. Med. 2005, 352, 987–996. [Google Scholar] [CrossRef]
- Vtorushin, S.V.; Khristenko, K.Y.; Zavyalova, M.V.; Perelmuter, V.M.; Litviakov, N.V.; Denisov, E.V.; Dulesova, A.Y.; Cherdyntseva, N.V. The Phenomenon of Multi-Drug Resistance in the Treatment of Malignant Tumors. Exp. Oncol. 2014, 36, 144–156. [Google Scholar] [PubMed]
- Arévalo, Á.S.T.; Erices, J.I.; Uribe, D.A.; Howden, J.; Niechi, I.; Muñoz, S.; Martín, R.S.; Monrás, C.A.Q. Current Therapeutic Alternatives and New Perspectives in Glioblastoma Multiforme. Curr. Med. Chem. 2017, 24, 2781–2795. [Google Scholar] [CrossRef]
- Bastiancich, C.; Bastiat, G.; Lagarce, F. Gemcitabine and glioblastoma: Challenges and current perspectives. Drug Discov. Today 2018, 23, 416–423. [Google Scholar] [CrossRef]
- Bambury, R.M.; Morris, P.G. The search for novel therapeutic strategies in the treatment of recurrent glioblastoma multiforme. Expert Rev. Anticancer Ther. 2014, 14, 955–964. [Google Scholar] [CrossRef]
- Campos, B.; Olsen, L.R.; Urup, T.; Poulsen, H.S. A comprehensive profile of recurrent glioblastoma. Oncogene 2016, 35, 5819–5825. [Google Scholar] [CrossRef] [PubMed]
- Torres, A.; Vargas, Y.; Uribe, D.; Jaramillo, C.; Gleisner, A.; Salazar-Onfray, F.; López, M.N.; Melo, R.; Oyarzún, C.; San Martín, R.; et al. Adenosine A3 receptor elicits chemoresistance mediated by multiple resistance-associated protein-1 in human glioblastoma stem-like cells. Oncotarget 2016, 7, 67373–67386. [Google Scholar] [CrossRef] [Green Version]
- Uribe, D.; Torres, Á.; Rocha, J.D.; Niechi, I.; Oyarzún, C.; Sobrevia, L.; San Martín, R.; Quezada, C. Multidrug resistance in glioblastoma stem-like cells: Role of the hypoxic microenvironment and adenosine signaling. Mol. Aspects Med. 2017, 55, 140–151. [Google Scholar] [CrossRef]
- Chen, Z.; Shi, T.; Zhang, L.; Zhu, P.; Deng, M.; Huang, C.; Hu, T.; Jiang, L.; Li, J. Mammalian drug efflux transporters of the ATP binding cassette (ABC) family in multidrug resistance: A review of the past decade. Cancer Lett. 2016, 370, 153–164. [Google Scholar] [CrossRef]
- Torres, Á.; Erices, J.I.; Sanchez, F.; Ehrenfeld, P.; Turchi, L.; Virolle, T.; Uribe, D.; Niechi, I.; Spichiger, C.; Rocha, J.D.; et al. Extracellular adenosine promotes cell migration/invasion of Glioblastoma Stem-like Cells through A3 Adenosine Receptor activation under hypoxia. Cancer Lett. 2019, 446, 112–122. [Google Scholar] [CrossRef]
- Torres, Á.; Arriagada, V.; Erices, J.I.; Toro, M.D.L.Á.; Rocha, J.D.; Niechi, I.; Carrasco, C.; Oyarzún, C.; Quezada, C. FK506 Attenuates the MRP1-Mediated Chemoresistant Phenotype in Glioblastoma Stem-Like Cells. Int. J. Mol. Sci. 2018, 19, 2697. [Google Scholar] [CrossRef]
- Bonnans, C.; Chou, J.; Werb, Z. Remodelling the extracellular matrix in development and disease. Nat. Rev. Mol. Cell Biol. 2014, 15, 786–801. [Google Scholar] [CrossRef]
- Maugeri-Saccà, M.; Di Martino, S.; De Maria, R. Biological and clinical implications of cancer stem cells in primary brain tumors. Front. Oncol. 2013, 3, 6. [Google Scholar] [CrossRef]
- Rocha, R.; Torres, Á.; Ojeda, K.; Uribe, D.; Rocha, D.; Erices, J.; Niechi, I.; Ehrenfeld, P.; San Martín, R.; Quezada, C. The Adenosine A3 Receptor Regulates Differentiation of Glioblastoma Stem-Like Cells to Endothelial Cells under Hypoxia. Int. J. Mol. Sci. 2018, 19, 1228. [Google Scholar] [CrossRef] [PubMed]
- Rosanò, L.; Bagnato, A. Endothelin therapeutics in cancer: Where are we? Am. J. Physiol. Regul. Integr. Comp. Physiol. 2016, 310, R469–R475. [Google Scholar] [CrossRef] [PubMed]
- Rosanò, L.; Spinella, F.; Bagnato, A. Endothelin 1 in cancer: Biological implications and therapeutic opportunities. Nat. Rev. Cancer 2013, 13, 637–651. [Google Scholar] [CrossRef] [PubMed]
- Tapia, J.C.; Niechi, I. Endothelin-converting enzyme-1 in cancer aggressiveness. Cancer Lett. 2019, 452, 152–157. [Google Scholar] [CrossRef]
- Niechi, I.; Silva, E.; Cabello, P.; Huerta, H.; Carrasco, V.; Villar, P.; Cataldo, L.R.; Marcelain, K.; Armisen, R.; Varas-Godoy, M.; et al. Colon cancer cell invasion is promoted by protein kinase CK2 through increase of endothelin-converting enzyme-1c protein stability. Oncotarget 2015, 6, 42749–42760. [Google Scholar] [CrossRef] [PubMed]
- Kuruppu, S.; Tochon-Danguy, N.; Smith, A.I. Protein Kinase C recognition sites in the cytoplasmic domain of Endothelin Converting Enzyme-1c. Biochem. Biophys. Res. Commun. 2012, 427, 606–610. [Google Scholar] [CrossRef] [PubMed]
- Pérez-Moreno, P.; Quezada-Meza, C.; Chavez-Almarza, C.; Niechi, I.; Silva-Pavez, E.; Trigo-Hidalgo, C.; Aguayo, F.; Jara, L.; Cáceres-Verschae, A.; Varas-Godoy, M.; et al. Phosphorylation of Endothelin-Converting Enzyme-1c at Serines 18 and 20 by CK2 Promotes Aggressiveness Traits in Colorectal Cancer Cells. Front. Oncol. 2020, 10, 1004. [Google Scholar] [CrossRef] [PubMed]
- Pérez-Moreno, P.; Indo, S.; Niechi, I.; Huerta, H.; Cabello, P.; Jara, L.; Aguayo, F.; Varas-Godoy, M.; Burzio, V.A.; Tapia, J.C. Endothelin-converting enzyme-1c promotes stem cell traits and aggressiveness in colorectal cancer cells. Mol. Oncol. 2020, 14, 347–362. [Google Scholar] [CrossRef]
- Liang, L.; Qu, L.; Dingm, Y. Protein and mRNA characterization in human colorectal carcinoma cell lines with different metastatic potentials. Cancer Investig. 2007, 25, 427–434. [Google Scholar] [CrossRef]
- Lin, K.; Tai, C.; Hsu, J.; Li, C.; Fang, C.; Lai, H.; Hseu, C.; Lin, Y.; Uen, Y. Overexpression of Nuclear Protein Kinase CK2 a Catalytic Subunit (CK2a) as a Poor Prognosticator in Human Colorectal Cancer. PLoS ONE 2011, 6, e17193. [Google Scholar]
- Rowse, A.L.; Gibson, S.A.; Meares, G.P.; Rajbhandari, R.; Nozell, S.E.; Dees, K.J.; Hjelmeland, A.B.; McFarland, B.C.; Benveniste, E.N. Protein kinase CK2 is important for the function of glioblastoma brain tumor initiating cells. J. Neurooncol. 2017, 132, 219–229. [Google Scholar] [CrossRef]
- Agarwal, M.; Nitta, R.T.; Li, G. Casein Kinase 2: A novel player in glioblastoma therapy and cancer stem cells. J. Mol. Genet. Med. 2013, 8, 1000094. [Google Scholar]
- Ruzzene, M.; Pinna, L. Addiction to protein kinase CK2: A common denominator of diverse cancer cells? Biochim. Biophys. Acta 2010, 1804, 499–504. [Google Scholar] [CrossRef]
- Ji, H.; Lu, Z. The role of protein kinase CK2 in glioblastoma development. Clin. Cancer Res. 2013, 19, 6335–6337. [Google Scholar] [CrossRef]
- Silva-Pavez, E.; Villar, P.; Trigo, C.; Caamaño, E.; Niechi, I.; Pérez, P.; Muñoz, J.P.; Aguayo, F.; Burzio, V.A.; Varas-Godoy, M.; et al. CK2 inhibition with silmitasertib promotes methuosis-like cell death associated to catastrophic massive vacuolization of colorectal cancer cells. Cell Death Dis. 2019, 10, 73. [Google Scholar] [CrossRef] [PubMed]
- Ponce, D.P.; Yefi, R.; Cabello, P.; Maturana, J.L.; Niechi, I.; Silva, E.; Galindo, M.; Antonelli, M.; Marcelain, K.; Armisen, R.; et al. CK2 functionally interacts with AKT/PKB to promote the β-catenin-dependent expression of survivin and enhance cell survival. Mol. Cell. Biochem. 2011, 356, 127–132. [Google Scholar] [CrossRef]
- Tapia, J.C.; Torres, V.; Rodriguez, D.; Leyton, L.; Quest, A. Casein kinase 2 (CK2) increases survivin expression via enhanced beta-catenin-T cell factor/lymphoid enhancer binding factor-dependent transcription. Proc. Natl. Acad. Sci. USA 2006, 103, 15079–15084. [Google Scholar] [CrossRef]
- Yefi, R.; Ponce, D.P.; Niechi, I.; Silva, E.; Cabello, P.; Rodriguez, D.A.; Marcelain, K.; Armisen, R.; Quest, A.F.G.; Tapia, J.C. Protein kinase CK2 promotes cancer cell viability via up-regulation of cyclooxygenase-2 expression and enhanced prostaglandin E2 production. J. Cell Biochem. 2011, 112, 3167–3175. [Google Scholar] [CrossRef] [PubMed]
- Berger, Y.; Dehmlow, H.; Blum-Kaelin, D.; Kitas, E.A.; Löffler, B.M.; Aebi, J.D.; Juillerat-Jeanneret, L. Endothelin-converting enzyme-1 inhibition and growth of human glioblastoma cells. J. Med. Chem. 2005, 48, 483–498. [Google Scholar] [CrossRef]
- Bowman, R.L.; Wang, Q.; Carro, A.; Verhaak, R.G.; Squatrito, M. GlioVis data portal for visualization and analysis of brain tumor expression datasets. Neuro Oncol. 2017, 19, 139–141. [Google Scholar] [CrossRef]
- Varas-Godoy, M.; Lladser, A.; Farfan, N.; Villota, C.; Villegas, J.; Tapia, J.C.; Burzio, L.O.; Burzio, V.A.; Valenzuela, P.D.T. In vivo knockdown of antisense non-coding mitochondrial RNAs by a lentiviral-encoded shRNA inhibits melanoma tumor growth and lung colonization. Pigment Cell Melanoma Res. 2018, 31, 64–72. [Google Scholar] [CrossRef] [PubMed]
- Racordon, D.; Valdivia, A.; Mingo, G.; Erices, R.; Aravena, R.; Santoro, F.; Bravo, M.L.; Ramirez, C.; Gonzalez, P.; Sandoval, A.; et al. Structural and functional identification of vasculogenic mimicry in vitro. Sci. Rep. 2017, 7, 6985. [Google Scholar] [CrossRef]
- Ajani, J.A.; Song, S.; Hochster, H.S.; Steinberg, I.B. Cancer stem cells: The promise and the potential. Semin. Oncol. 2015, 42 (Suppl. 1), S3–S17. [Google Scholar] [CrossRef]
- Fanali, C.; Lucchetti, D.; Farina, M.; Corbi, M.; Cufino, V.; Cittadini, A.; Sgambato, A. Cancer stem cells in colorectal cancer from pathogenesis to therapy: Controversies and perspectives. World J. Gastroenterol. 2014, 20, 923–942. [Google Scholar] [CrossRef]
- Hadjimichael, C.; Chanoumidou, K.; Papadopoulou, N.; Arampatzi, P.; Papamatheakis, J.; Kretsovali, A. Common stemness regulators of embryonic and cancer stem cells. World J. Stem Cells 2015, 7, 1150–1184. [Google Scholar]
- Hsieh, W.T.; Yeh, W.L.; Cheng, R.Y.; Lin, C.; Tsai, C.F.; Huang, B.R.; Wu, C.Y.J.; Lin, H.Y.; Huang, S.S.; Lu, D.Y. Exogenous endothelin-1 induces cell migration and matrix metalloproteinase expression in U251 human glioblastoma multiforme. J. Neurooncol. 2014, 118, 257–269. [Google Scholar] [CrossRef]
- Swatek, K.N.; Komander, D. Ubiquitin modifications. Cell Res. 2016, 26, 399–422. [Google Scholar] [CrossRef] [Green Version]
- Dikic, I. Proteasomal and Autophagic Degradation Systems. Annu. Rev. Biochem. 2017, 86, 193–224. [Google Scholar] [CrossRef]
- Herhaus, L.; Perez-Oliva, A.B.; Cozza, G.; Gourlay, R.; Weidlich, S.; Campbell, D.G.; Pinna, L.A.; Sapkota, G.P. Casein kinase 2 (CK2) phosphorylates the deubiquitylase OTUB1 at Ser16 to trigger its nuclear localization. Sci. Signal. 2015, 8, ra35. [Google Scholar] [CrossRef]
- Duncan, J.S.; Litchfield, D.W. Too much of a good thing: The role of protein kinase CK2 in tumorigenesis and prospects for therapeutic inhibition of CK2. Biochim. Biophys. Acta 2008, 1784, 33–47. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.W.; Hasanuzzaman, M.; Cho, M.; Kim, N.H.; Choi, H.Y.; Han, J.W.; Park, H.J.; Oh, J.W.; Shin, J.G. Role of 14-3-3 sigma in over-expression of P-gp by rifampin and paclitaxel stimulation through interaction with PXR. Cell. Signal. 2017, 31, 124–134. [Google Scholar] [CrossRef]
- Coffman, L.; Mooney, C.; Lim, J.; Bai, S.; Silva, I.; Gong, Y.; Yang, K.; Buckanovich, R.J. Endothelin receptor-A is required for the recruitment of antitumor T cells and modulates chemotherapy induction of cancer stem cells. Cancer Biol. Ther. 2013, 14, 184–192. [Google Scholar] [CrossRef] [PubMed]
- Emery, I.F.; Gopalan, A.; Wood, S.; Chow, K.H.; Battelli, C.; George, J.; Blaszyk, H.; Florman, J.; Yun, K. Expression and function of ABCG2 and XIAP in glioblastomas. J. Neurooncol. 2017, 133, 47–57. [Google Scholar] [CrossRef] [PubMed]
- Yi, G.Z.; Liu, Y.W.; Xiang, W.; Wang, H.; Chen, Z.Y.; Xie, S.; Qi, S.T. Akt and β-catenin contribute to TMZ resistance and EMT of MGMT negative malignant glioma cell line. J. Neurol. Sci. 2016, 367, 101–106. [Google Scholar] [CrossRef]
- Rosanò, L.; Bagnato, A. Disrupting the endothelin and Wnt relationship to overcome chemoresistance. Mol. Cell. Oncol. 2015, 2, e995025. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.J.; Lee, H.J.; Kim, M.S.; Choi, H.J.; He, J.; Wu, Q.; Aldape, K.; Weinberg, J.S.; Yung, W.K.A.; Conrad, C.A.; et al. Macitentan, a Dual Endothelin Receptor Antagonist, in Combination with Temozolomide Leads to Glioblastoma Regression and Long-term Survival in Mice. Clin. Cancer Res. 2015, 21, 4630–4641. [Google Scholar] [CrossRef] [PubMed]
- Rayhman, O.; Klipper, E.; Muller, L.; Davidson, B.; Reich, R.; Meidan, R. Small interfering RNA molecules targeting endothelin-converting enzyme-1 inhibit endothelin-1 synthesis and the invasive phenotype of ovarian carcinoma cells. Cancer Res. 2008, 68, 9265–9273. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lambert, L.A.; Whyteside, A.R.; Turner, A.J.; Usmani, B.A. Isoforms of endothelin-converting enzyme-1 (ECE-1) have opposing effects on prostate cancer cell invasion. Br. J. Cancer 2008, 99, 1114–1120. [Google Scholar] [CrossRef] [Green Version]









Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Niechi, I.; Erices, J.I.; Carrillo-Beltrán, D.; Uribe-Ojeda, A.; Torres, Á.; Rocha, J.D.; Uribe, D.; Toro, M.A.; Villalobos-Nova, K.; Gaete-Ramírez, B.; et al. Cancer Stem Cell and Aggressiveness Traits Are Promoted by Stable Endothelin-Converting Enzyme-1c in Glioblastoma Cells. Cells 2023, 12, 506. https://doi.org/10.3390/cells12030506
Niechi I, Erices JI, Carrillo-Beltrán D, Uribe-Ojeda A, Torres Á, Rocha JD, Uribe D, Toro MA, Villalobos-Nova K, Gaete-Ramírez B, et al. Cancer Stem Cell and Aggressiveness Traits Are Promoted by Stable Endothelin-Converting Enzyme-1c in Glioblastoma Cells. Cells. 2023; 12(3):506. https://doi.org/10.3390/cells12030506
Chicago/Turabian StyleNiechi, Ignacio, José I. Erices, Diego Carrillo-Beltrán, Atenea Uribe-Ojeda, Ángelo Torres, José Dellis Rocha, Daniel Uribe, María A. Toro, Karla Villalobos-Nova, Belén Gaete-Ramírez, and et al. 2023. "Cancer Stem Cell and Aggressiveness Traits Are Promoted by Stable Endothelin-Converting Enzyme-1c in Glioblastoma Cells" Cells 12, no. 3: 506. https://doi.org/10.3390/cells12030506
APA StyleNiechi, I., Erices, J. I., Carrillo-Beltrán, D., Uribe-Ojeda, A., Torres, Á., Rocha, J. D., Uribe, D., Toro, M. A., Villalobos-Nova, K., Gaete-Ramírez, B., Mingo, G., Owen, G. I., Varas-Godoy, M., Jara, L., Aguayo, F., Burzio, V. A., Quezada-Monrás, C., & Tapia, J. C. (2023). Cancer Stem Cell and Aggressiveness Traits Are Promoted by Stable Endothelin-Converting Enzyme-1c in Glioblastoma Cells. Cells, 12(3), 506. https://doi.org/10.3390/cells12030506