
The Role of TP53 in Adaptation and Evolution



{kind=link}
{kind=link}
Abstract
:1. Introduction
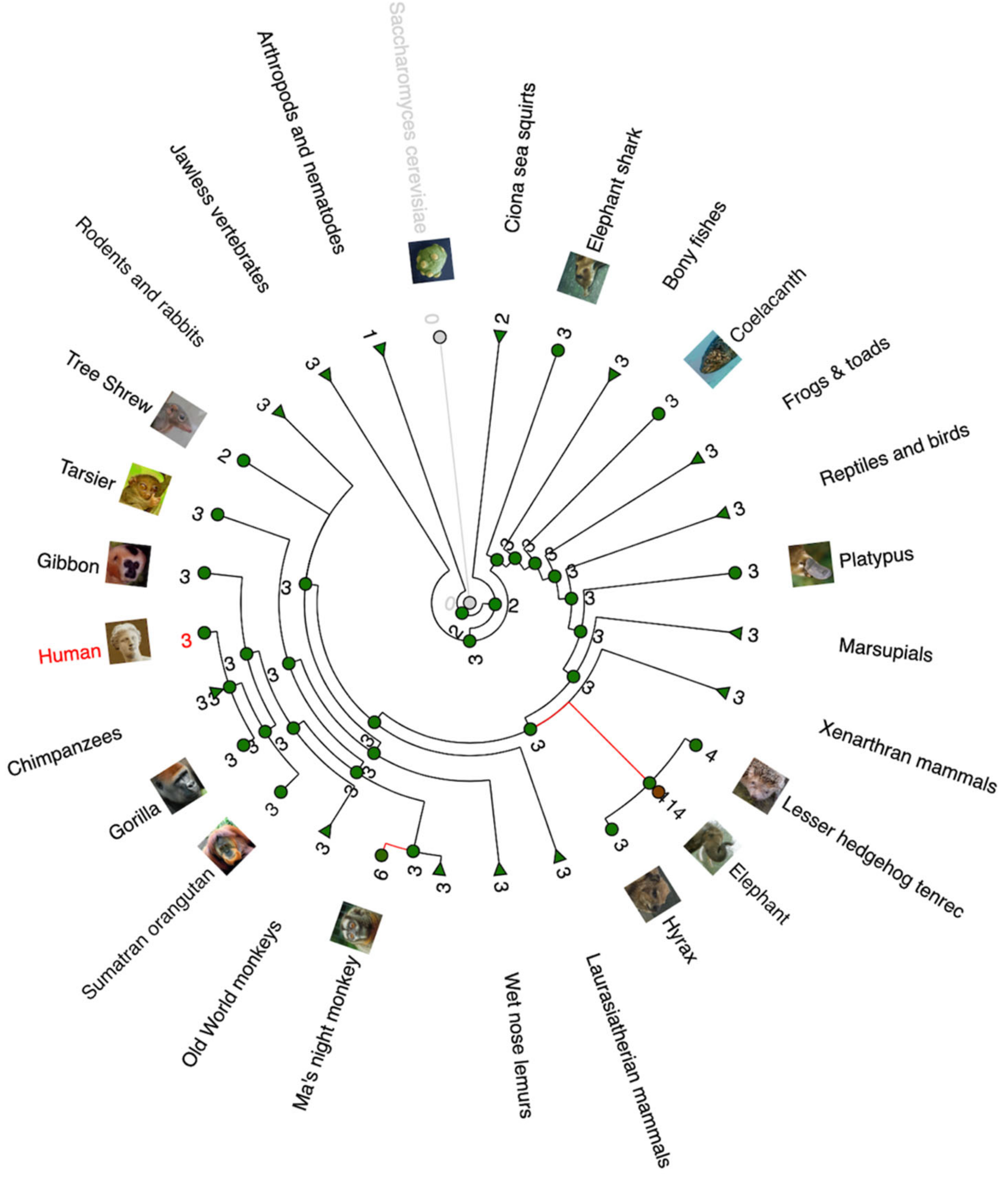
2. Evolution of p53 Family
3. Role of TP53 Variants in Increased Fertility and Longevity
3.1. Fertility and TP53
3.2. Longevity and TP53
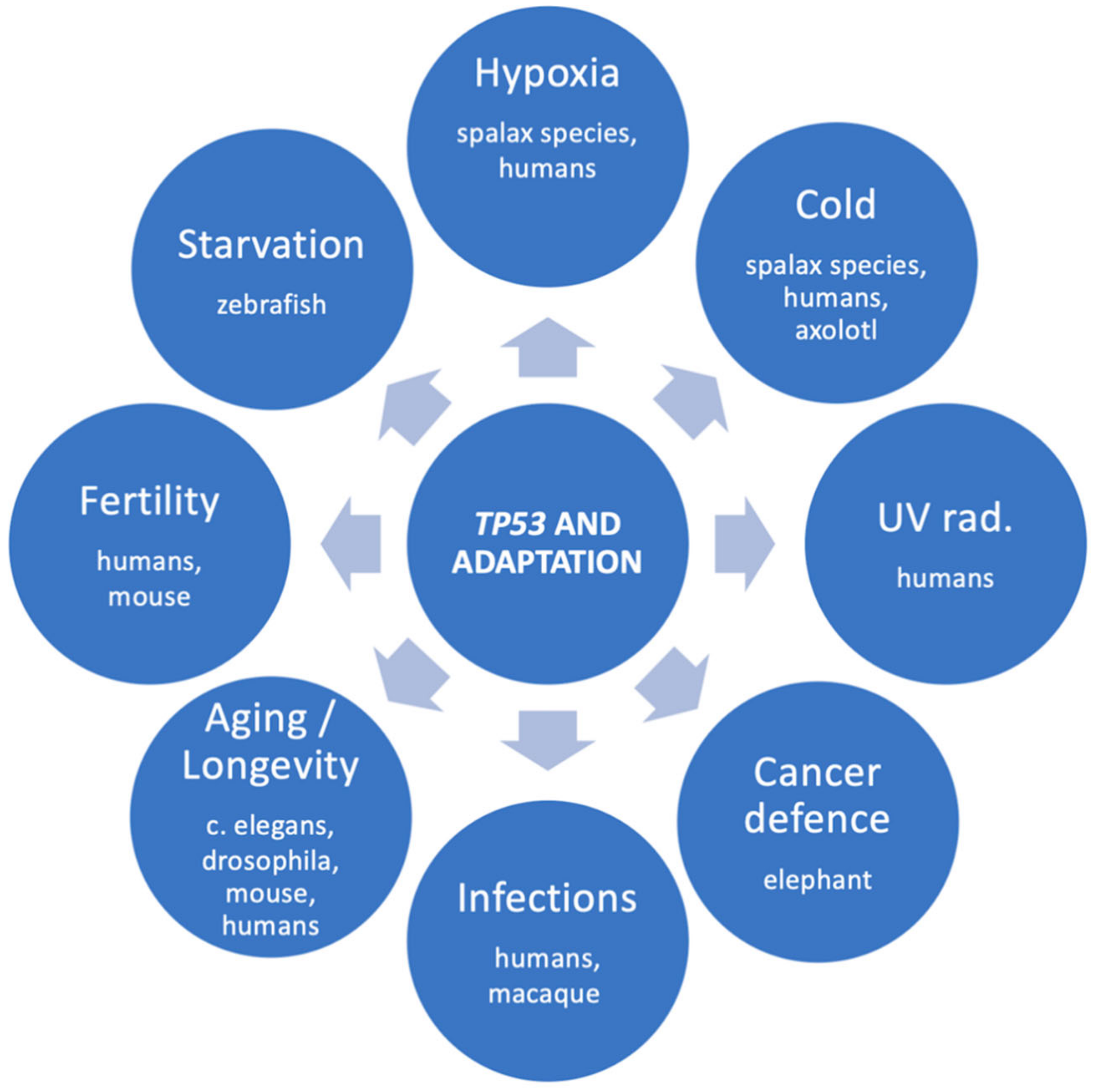
4. TP53 Is Crucial for Environmental Adaptation
5. Selection Pressures on Somatic TP53 Mutations
5.1. TP53 Mutations in Normal Tissues
5.2. TP53 Mutations in Malignant Tissues
6. TP53 Copy Numbers
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Capuozzo, M.; Santorsola, M.; Bocchetti, M.; Perri, F.; Cascella, M.; Granata, V.; Celotto, V.; Gualillo, O.; Cossu, A.M.; Nasti, G.; et al. P53: From Fundamental Biology to Clinical Applications in Cancer. Biology 2022, 11, 1325. [Google Scholar] [CrossRef]
- Kennedy, M.C.; Lowe, S.W. Mutant P53: It’s Not All One and the Same. Cell Death Differ. 2022, 29, 983–987. [Google Scholar] [CrossRef]
- Levine, A.J. P53: 800 Million Years of Evolution and 40 Years of Discovery. Nat. Rev. Cancer 2020, 20, 471–480. [Google Scholar] [CrossRef]
- Levine, A.J. Spontaneous and Inherited TP53 Genetic Alterations. Oncogene 2021, 40, 5975–5983. [Google Scholar] [CrossRef] [PubMed]
- Lane, D.P.; Cheok, C.F.; Brown, C.; Madhumalar, A.; Ghadessy, F.J.; Verma, C. Mdm2 and P53 Are Highly Conserved from Placozoans to Man. Cell Cycle 2010, 9, 540–547. [Google Scholar] [CrossRef] [PubMed]
- Hu, W.; Zheng, T.; Wang, J. Regulation of Fertility by the P53 Family Members. Genes Cancer 2011, 2, 420–430. [Google Scholar] [CrossRef] [PubMed]
- Beckman, G.; Birgander, R.; Sjalander, A.; Saha, N.; Holmberg, P.A.; Kivela, A.; Beckman, L. Is P53 Polymorphism Maintained by Natural Selection? Hum. Hered. 1994, 44, 266–270. [Google Scholar] [CrossRef] [PubMed]
- Dumont, P.; Leu, J.I.J.; Della Pietra, A.C.; George, D.L.; Murphy, M. The Codon 72 Polymorphic Variants of P53 Have Markedly Different Apoptotic Potential. Nat. Genet. 2003, 33, 357–365. [Google Scholar] [CrossRef]
- Hoyos, D.; Greenbaum, B.; Levine, A.J. The Genotypes and Phenotypes of Missense Mutations in the Proline Domain of the P53 Protein. Cell Death Differ. 2022, 29, 938–945. [Google Scholar] [CrossRef]
- Kang, H.J.; Feng, Z.; Sun, Y.; Atwal, G.; Murphy, M.E.; Rebbeck, T.R.; Rosenwaks, Z.; Levine, A.J.; Hu, W. Single-Nucleotide Polymorphisms in the P53 Pathway Regulate Fertility in Humans. Proc. Natl. Acad. Sci. USA 2009, 106, 9761–9766. [Google Scholar] [CrossRef] [Green Version]
- Hu, W.; Feng, Z.; Teresky, A.K.; Levine, A.J. P53 Regulates Maternal Reproduction through LIF. Nature 2007, 450, 721–724. [Google Scholar] [CrossRef] [PubMed]
- Lledo, B.; Turienzo, A.; Ortiz, J.A.; Morales, R.; Ten, J.; Llácer, J.; Bernabeu, R. Negative Effect of P72 Polymorphism on P53 Gene in IVF Outcome in Patients with Repeated Implantation Failure and Pregnancy Loss. J. Assist. Reprod. Genet. 2014, 31, 169–172. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Yang, X.; Wang, Z. Association between P53 Arg72Pro Polymorphism and Recurrent Pregnancy Loss: An Updated Systematic Review and Meta-Analysis. Reprod. Biomed. Online 2015, 31, 149–153. [Google Scholar] [CrossRef] [PubMed]
- Tagliani-Ribeiro, A.; Paskulin, D.D.; Oliveira, M.; Zagonel-Oliveira, M.; Longo, D.; Ramallo, V.; Ashton-Prolla, P.; Saraiva-Pereira, M.L.; Fagundes, N.J.R.; Schuler-Faccini, L.; et al. High Twinning Rate in Cândido Godói: A New Role for P53 in Human Fertility. Hum. Reprod. 2012, 27, 2866–2871. [Google Scholar] [CrossRef] [PubMed]
- Li, A.; Ganeshan, L.; O’Neill, C. The Effect of Trp53 Gene-Dosage and Parent-of-Origin of Inheritance on Mouse Gamete and Embryo Function in Vitro. Biol. Reprod. 2012, 86, 1–6. [Google Scholar] [CrossRef]
- Byars, S.G.; Voskarides, K. Antagonistic Pleiotropy in Human Disease. J. Mol. Evol. 2020, 88, 12–25. [Google Scholar] [CrossRef]
- Bauer, J.H.; Poon, P.C.; Glatt-Deeley, H.; Abrams, J.M.; Helfand, S.L. Neuronal Expression of P53 Dominant-Negative Proteins in Adult Drosophila Melanogaster Extends Life Span. Curr. Biol. 2005, 15, 2063–2068. [Google Scholar] [CrossRef]
- Ingaramo, M.C.; Sánchez, J.A.; Dekanty, A. Regulation and Function of P53: A Perspective from Drosophila Studies. Mech. Dev. 2018, 154, 82–90. [Google Scholar] [CrossRef]
- Waskar, M.; Landis, G.N.; Shen, J.; Curtis, C.; Tozer, K.; Abdueva, D.; Skvortsov, D.; Tavaré, S.; Tower, J. Drosophila Melanogaster P53 Has Developmental Stage-Specific and Sex-Specific Effects on Adult Life Span Indicative of Sexual Antagonistic Pleiotropy. Aging 2009, 1, 903–936. [Google Scholar] [CrossRef]
- Arum, O.; Johnson, T.E. Reduced Expression of the Caenorhabditis Elegans P53 Ortholog Cep-1 Results in Increased Longevity. J. Gerontol. Ser. A Biol. Sci. Med. Sci. 2007, 62, 951–959. [Google Scholar] [CrossRef] [Green Version]
- Yanase, S.; Suda, H.; Yasuda, K.; Ishii, N. Impaired P53/CEP-1 Is Associated with Lifespan Extension through an Age-Related Imbalance in the Energy Metabolism of C. Elegans. Genes Cells 2017, 22, 1004–1010. [Google Scholar] [CrossRef]
- Van Heemst, D.; Mooijaart, S.P.; Beekman, M.; Schreuder, J.; De Craen, A.J.M.; Brandt, B.W.; Eline Slagboom, P.; Westendorp, R.G.J. Variation in the Human TP53 Gene Affects Old Age Survival and Cancer Mortality. Exp. Gerontol. 2005, 40, 11–15. [Google Scholar] [CrossRef]
- Bojesen, S.E.; Nordestgaard, B.G. The Common Germline Arg72Pro Polymorphism of P53 and Increased Longevity in Humans. Cell Cycle 2008, 7, 158–163. [Google Scholar] [CrossRef]
- Zhao, Y.; Wu, L.; Yue, X.; Zhang, C.; Wang, J.; Li, J.; Sun, X.; Zhu, Y.; Feng, Z.; Hu, W. A Polymorphism in the Tumor Suppressor P53 Affects Aging and Longevity in Mouse Models. Elife 2018, 7, e34701. [Google Scholar] [CrossRef]
- Groß, S.; Immel, U.-D.; Klintschar, M.; Bartel, F. Germline Genetics of the P53 Pathway Affect Longevity in a Gender Specific Manner. Curr. Aging Sci. 2014, 7, 91–100. [Google Scholar] [CrossRef]
- Gencel-Augusto, J.; Lozano, G. P53 Tetramerization: At the Center of the Dominant-Negative Effect of Mutant P53. Genes Dev. 2020, 34, 1128–1146. [Google Scholar] [CrossRef]
- Mehta, S.; Campbell, H.; Drummond, C.J.; Li, K.; Murray, K.; Slatter, T.; Bourdon, J.; Braithwaite, A.W. Adaptive Homeostasis and the P53 Isoform Network. EMBO Rep. 2021, 22, e53085. [Google Scholar] [CrossRef]
- Ashur-Fabian, O.; Avivi, A.; Trakhtenbrot, L.; Adamsky, K.; Cohen, M.; Kajakaro, G.; Joel, A.; Amariglio, N.; Nevo, E.; Rechavi, G. Evolution of P53 in Hypoxia-Stressed Spalax Mimics Human Tumor Mutation. Proc. Natl. Acad. Sci. USA 2004, 101, 12236–12241. [Google Scholar] [CrossRef]
- Ellis, M.; Stern, O.; Ashur-Fabian, O. The Double Benefit of Spalax P53: Surviving Underground Hypoxia While Defying Lung Cancer Cells in Vitro via Autophagy and Caspase-Dependent Cell Death. Oncotarget 2016, 7, 63242–63251. [Google Scholar] [CrossRef]
- Shams, I.; Malik, A.; Manov, I.; Joel, A.; Band, M.; Avivi, A. Transcription Pattern of P53-Targeted DNA Repair Genes in the Hypoxia-Tolerant Subterranean Mole Rat Spalax. J. Mol. Biol. 2013, 425, 1111–1118. [Google Scholar] [CrossRef]
- Band, M.; Ashur-Fabian, O.; Avivi, A. The Expression of P53-Target Genes in the Hypoxia-Tolerant Subterranean Mole-Rat Is Hypoxia-Dependent and Similar to Expression Patterns in Solid Tumors. Cell Cycle 2010, 9, 3367–3372. [Google Scholar] [CrossRef]
- Fang, X.; Nevo, E.; Han, L.; Levanon, E.Y.; Zhao, J.; Avivi, A.; Larkin, D.; Jiang, X.; Feranchuk, S.; Zhu, Y.; et al. Genome-Wide Adaptive Complexes to Underground Stresses in Blind Mole Rats Spalax. Nat. Commun. 2014, 5, 3966. [Google Scholar] [CrossRef]
- Zhao, Y.; Ren, J.L.; Wang, M.Y.; Zhang, S.T.; Liua, Y.; Li, M.; Cao, Y.B.; Zu, H.Y.; Chen, X.C.; Wu, C.I.; et al. Codon 104 Variation of P53 Gene Provides Adaptive Apoptotic Responses to Extreme Environments in Mammals of the Tibet Plateau. Proc. Natl. Acad. Sci. USA 2013, 110, 20639–20644. [Google Scholar] [CrossRef]
- Kimura, K.; Shinmura, K.; Hasegawa, T.; Beppu, Y.; Yokoyama, R.; Yokota, J. Germline P53 Mutation in a Patient with Multiple Primary Cancers. Jpn. J. Clin. Oncol. 2001, 31, 349–351. [Google Scholar] [CrossRef]
- Zhao, Y.; Tang, J.W.; Yang, Z.; Cao, Y.B.; Ren, J.L.; Ben-Abu, Y.; Li, K.; Chen, X.Q.; Du, J.Z.; Nevo, E. Adaptive Methylation Regulation of P53 Pathway in Sympatric Speciation of Blind Mole Rats, Spalax. Proc. Natl. Acad. Sci. USA 2016, 113, 2146–2151. [Google Scholar] [CrossRef]
- Villiard, É.; Brinkmann, H.; Moiseeva, O.; Mallette, F.A.; Ferbeyre, G.; Roy, S. Urodele P53 Tolerates Amino Acid Changes Found in P53 Variants Linked to Human Cancer. BMC Evol. Biol. 2007, 7, 180. [Google Scholar] [CrossRef]
- Jacovas, V.C.; Couto-Silva, C.M.; Nunes, K.; Lemes, R.B.; de Oliveira, M.Z.; Salzano, F.M.; Bortolini, M.C.; Hünemeier, T. Selection Scan Reveals Three New Loci Related to High Altitude Adaptation in Native Andeans. Sci. Rep. 2018, 8, 12733. [Google Scholar] [CrossRef]
- Jacovas, V.C.; Rovaris, D.L.; Peréz, O.; De Azevedo, S.; Macedo, G.S.; Sandoval, J.R.; Salazar-Granara, A.; Villena, M.; Dugoujon, J.M.; Bisso-Machado, R.; et al. Genetic Variations in the TP53 Pathway in Native Americans Strongly Suggest Adaptation to the High Altitudes of the Andes. PLoS ONE 2015, 10, e0137823. [Google Scholar] [CrossRef]
- Shi, H.; Tan, S.-J.; Zhong, H.; Hu, W.; Levine, A.; Xiao, C.-J.; Peng, Y.; Qi, X.-B.; Shou, W.-H.; Ma, R.-L.Z.; et al. Winter Temperature and UV Are Tightly Linked to Genetic Changes in the P53 Tumor Suppressor Pathway in Eastern Asia. Am. J. Hum. Genet. 2009, 84, 534–541. [Google Scholar] [CrossRef]
- Matoba, S.; Kang, J.G.; Patino, W.D.; Wragg, A.; Boehm, M.; Gavrilova, O.; Hurley, P.J.; Bunz, F.; Hwang, P.M. P53 Regulates Mitochondrial Respiration. Science 2006, 312, 1650–1653. [Google Scholar] [CrossRef]
- Voskarides, K.; Koutsofti, C.; Pozova, M. TP53 Mutant Versus Wild-Type Zebrafish Larvae Under Starvation Stress: Larvae Can Live Up to 17 Days Post-Fertilization Without Food. Zebrafish 2022, 19, 49–55. [Google Scholar] [CrossRef]
- Thomas, M.; David, P.; Banks, L. The Role of the E6-P53 Interaction in the Molecular Pathogenesis of HPV. Oncogene 1999, 18, 7690–7700. [Google Scholar] [CrossRef]
- Long, T.; Burk, R.D.; Chan, P.K.S.; Chen, Z. Non-Human Primate Papillomavirus E6-Mediated P53 Degradation Reveals Ancient Evolutionary Adaptation of Carcinogenic Phenotype to Host Niche. PLoS Pathog. 2022, 18, e1010444. [Google Scholar] [CrossRef]
- Tomasetti, C. Mutated Clones Are the New Normal: Measuring and Understanding the Dynamics of Clonal Cell Populations Is Key for Cancer Prevention. Science 2019, 364, 938–939. [Google Scholar] [CrossRef]
- Luijts, T.; Elliott, K.; Siaw, J.T.; Van de Velde, J.; Beyls, E.; Claeys, A.; Lammens, T.; Larsson, E.; Willaert, W.; Vral, A.; et al. A Clinically Annotated Post-Mortem Approach to Study Multi-Organ Somatic Mutational Clonality in Normal Tissues. Sci. Rep. 2022, 12, 10322. [Google Scholar] [CrossRef]
- Martincorena, I.; Roshan, A.; Gerstung, M.; Ellis, P.; Van Loo, P.; McLaren, S.; Wedge, D.C.; Fullam, A.; Alexandrov, L.B.; Tubio, J.M.; et al. High Burden and Pervasive Positive Selection of Somatic Mutations in Normal Human Skin. Science 2015, 348, 880–886. [Google Scholar] [CrossRef]
- Martincorena, I.; Fowler, J.C.; Wabik, A.; Lawson, A.R.J.; Abascal, F.; Hall, M.W.J.; Cagan, A.; Murai, K.; Mahbubani, K.; Stratton, M.R.; et al. Somatic Mutant Clones Colonize the Human Esophagus with Age. Science 2018, 362, 911–917. [Google Scholar] [CrossRef]
- Colom, B.; Alcolea, M.P.; Piedrafita, G.; Hall, M.W.J.; Wabik, A.; Dentro, S.C.; Fowler, J.C.; Herms, A.; King, C.; Ong, S.H.; et al. Spatial Competition Shapes the Dynamic Mutational Landscape of Normal Esophageal Epithelium. Nat. Genet. 2020, 52, 604–614. [Google Scholar] [CrossRef]
- Yokoyama, A.; Kakiuchi, N.; Yoshizato, T.; Nannya, Y.; Suzuki, H.; Takeuchi, Y.; Shiozawa, Y.; Sato, Y.; Aoki, K.; Kim, S.K.; et al. Age-Related Remodelling of Oesophageal Epithelia by Mutated Cancer Drivers. Nature 2019, 565, 312–317. [Google Scholar] [CrossRef]
- Lee-Six, H.; Olafsson, S.; Ellis, P.; Osborne, R.J.; Sanders, M.A.; Moore, L.; Georgakopoulos, N.; Torrente, F.; Noorani, A.; Goddard, M.; et al. The Landscape of Somatic Mutation in Normal Colorectal Epithelial Cells. Nature 2019, 574, 532–537. [Google Scholar] [CrossRef]
- Yoshida, K.; Gowers, K.H.C.; Lee-Six, H.; Chandrasekharan, D.P.; Coorens, T.; Maughan, E.F.; Beal, K.; Menzies, A.; Millar, F.R.; Anderson, E.; et al. Tobacco Smoking and Somatic Mutations in Human Bronchial Epithelium. Nature 2020, 578, 266–272. [Google Scholar] [CrossRef]
- Li, R.; Du, Y.; Chen, Z.; Xu, D.; Lin, T.; Jin, S.; Wang, G.; Liu, Z.; Lu, M.; Chen, X.; et al. Macroscopic Somatic Clonal Expansion in Morphologically Normal Human Urothelium. Science 2020, 370, 82–89. [Google Scholar] [CrossRef] [PubMed]
- Colom, B.; Herms, A.; Hall, M.W.J.; Dentro, S.C.; King, C.; Sood, R.K.; Alcolea, M.P.; Piedrafita, G.; Fernandez-Antoran, D.; Ong, S.H.; et al. Mutant Clones in Normal Epithelium Outcompete and Eliminate Emerging Tumours. Nature 2021, 598, 510–514. [Google Scholar] [CrossRef] [PubMed]
- Weinstein, J.N.; Collisson, E.A.; Mills, G.B.; Shaw, K.R.M.; Ozenberger, B.A.; Ellrott, K.; Sander, C.; Stuart, J.M.; Chang, K.; Creighton, C.J.; et al. The Cancer Genome Atlas Pan-Cancer Analysis Project. Nat. Genet. 2013, 45, 1113–1120. [Google Scholar] [CrossRef]
- Muzny, D.M.; Bainbridge, M.N.; Chang, K.; Dinh, H.H.; Drummond, J.A.; Fowler, G.; Kovar, C.L.; Lewis, L.R.; Morgan, M.B.; Newsham, I.F.; et al. Comprehensive Molecular Characterization of Human Colon and Rectal Cancer. Nature 2012, 487, 330–337. [Google Scholar] [CrossRef]
- Serrano, M.; Lin, A.W.; McCurrach, M.E.; Beach, D.; Lowe, S.W. Oncogenic Ras Provokes Premature Cell Senescence Associated with Accumulation of P53 and P16(INK4a). Cell 1997, 88, 593–602. [Google Scholar] [CrossRef] [PubMed]
- Tollis, M.; Boddy, A.M.; Maley, C.C. Peto’s Paradox: How Has Evolution Solved the Problem of Cancer Prevention? BMC Biol. 2017, 15, 60. [Google Scholar] [CrossRef]
- Peto, H.; Roe, F.J.C.; Lee, P.N.; Levy, L.; Clack, J. Cancer and Ageing in Mice and Men. Br. J. Cancer 1975, 32, 411–426. [Google Scholar] [CrossRef]
- Nunney, L. The Real War on Cancer: The Evolutionary Dynamics of Cancer Suppression. Evol. Appl. 2013, 6, 11–19. [Google Scholar] [CrossRef]
- Abegglen, L.M.; Caulin, A.F.; Chan, A.; Lee, K.; Robinson, R.; Campbell, M.S.; Kiso, W.K.; Schmitt, D.L.; Waddell, P.J.; Bhaskara, S.; et al. Potential Mechanisms for Cancer Resistance in Elephants and Comparative Cellular Response to DNA Damage in Humans. JAMA J. Am. Med. Assoc. 2015, 314, 1850–1860. [Google Scholar] [CrossRef]
- García-Cao, I.; García-Cao, M.; Martín-Caballero, J.; Criado, L.M.; Klatt, P.; Flores, J.M.; Weill, J.C.; Blasco, M.A.; Serrano, M. “Super P53” Mice Exhibit Enhanced DNA Damage Response, Are Tumor Resistant and Age Normally. EMBO J. 2002, 21, 6225–6235. [Google Scholar] [CrossRef] [PubMed]
- Sulak, M.; Fong, L.; Mika, K.; Chigurupati, S.; Yon, L.; Mongan, N.P.; Emes, R.D.; Lynch, V.J. TP53 Copy Number Expansion Is Associated with the Evolution of Increased Body Size and an Enhanced DNA Damage Response in Elephants. eLife 2016, 5, e11994. [Google Scholar] [CrossRef] [PubMed]
- Padariya, M.; Jooste, M.L.; Hupp, T.; Fåhraeus, R.; Vojtesek, B.; Vollrath, F.; Kalathiya, U.; Karakostis, K. The Elephant Evolved P53 Isoforms That Escape MDM2-Mediated Repression and Cancer. Mol. Biol. Evol. 2022, 39, msac149. [Google Scholar] [CrossRef] [PubMed]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Voskarides, K.; Giannopoulou, N. The Role of TP53 in Adaptation and Evolution. Cells 2023, 12, 512. https://doi.org/10.3390/cells12030512
Voskarides K, Giannopoulou N. The Role of TP53 in Adaptation and Evolution. Cells. 2023; 12(3):512. https://doi.org/10.3390/cells12030512
Chicago/Turabian StyleVoskarides, Konstantinos, and Nefeli Giannopoulou. 2023. "The Role of TP53 in Adaptation and Evolution" Cells 12, no. 3: 512. https://doi.org/10.3390/cells12030512
APA StyleVoskarides, K., & Giannopoulou, N. (2023). The Role of TP53 in Adaptation and Evolution. Cells, 12(3), 512. https://doi.org/10.3390/cells12030512