
Implications of Senescent Cell Burden and NRF2 Pathway in Uremic Calcification: A Translational Study

 , , , ,
, , , ,  , , , , , and
, , , , , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Animal Model of Uremic VC: Tissue Sampling and Blood Parameters
2.2. Observational Clinical Study and In Vitro Assay: Patients and Sampling
2.3. Histology
2.4. mRNA Expression
2.5. In Vitro Assay
2.6. Statistics
2.7. Study Approval
3. Results
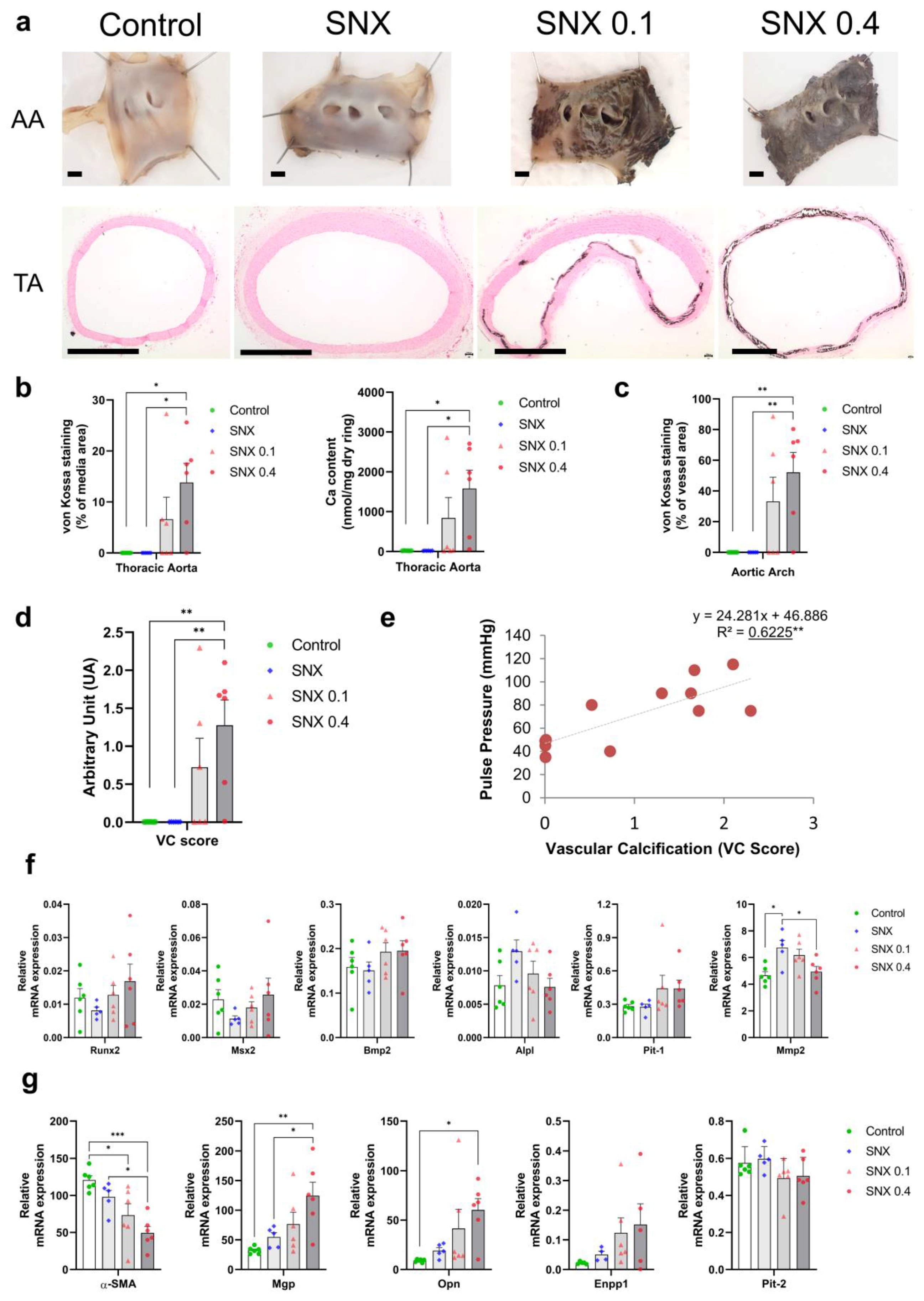
3.1. Validation of the Pre-Clinical Uremic VC Model
3.2. VSMC Osteoblastic Transition and Thoracic Calcification in Uremic Rats
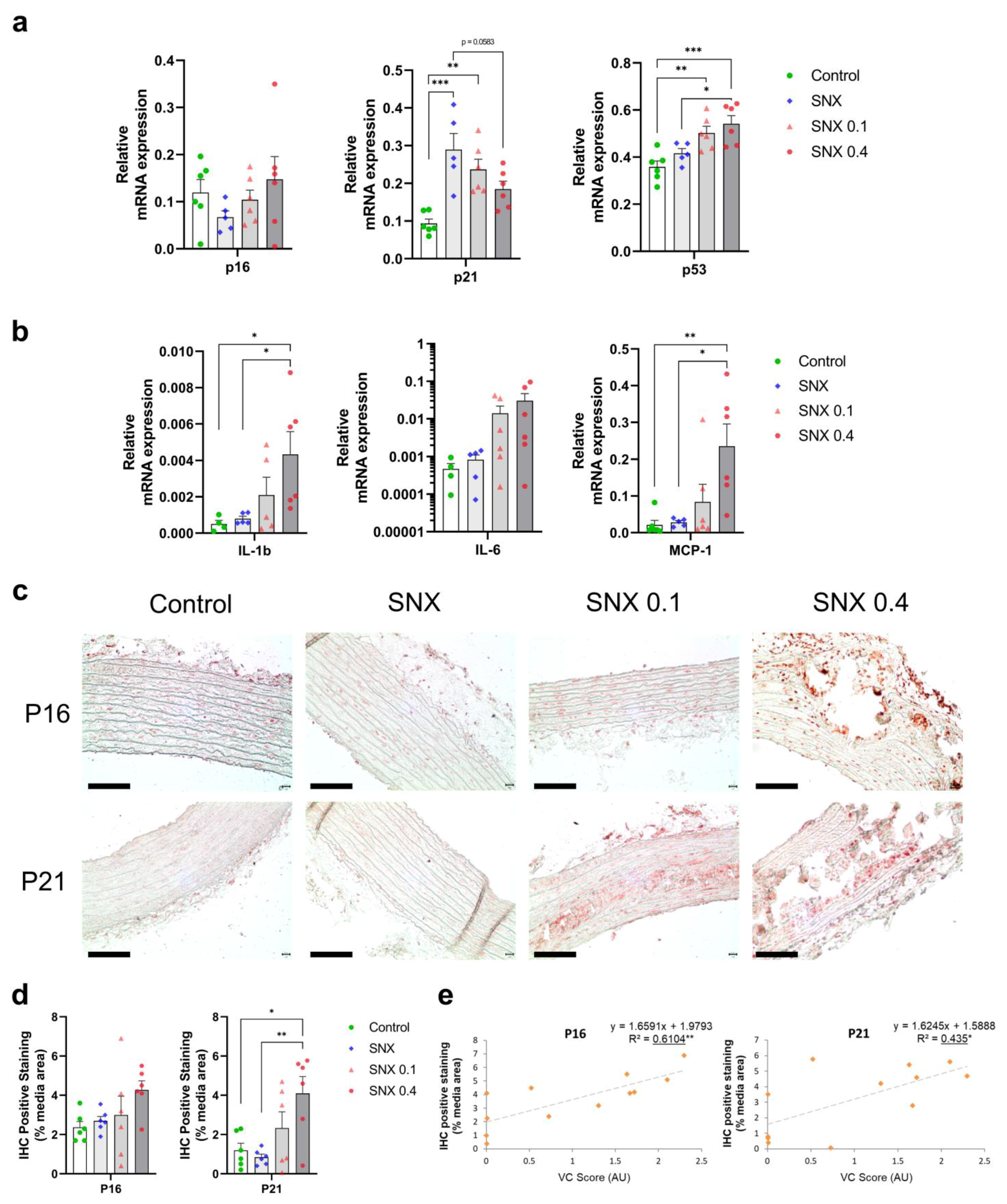
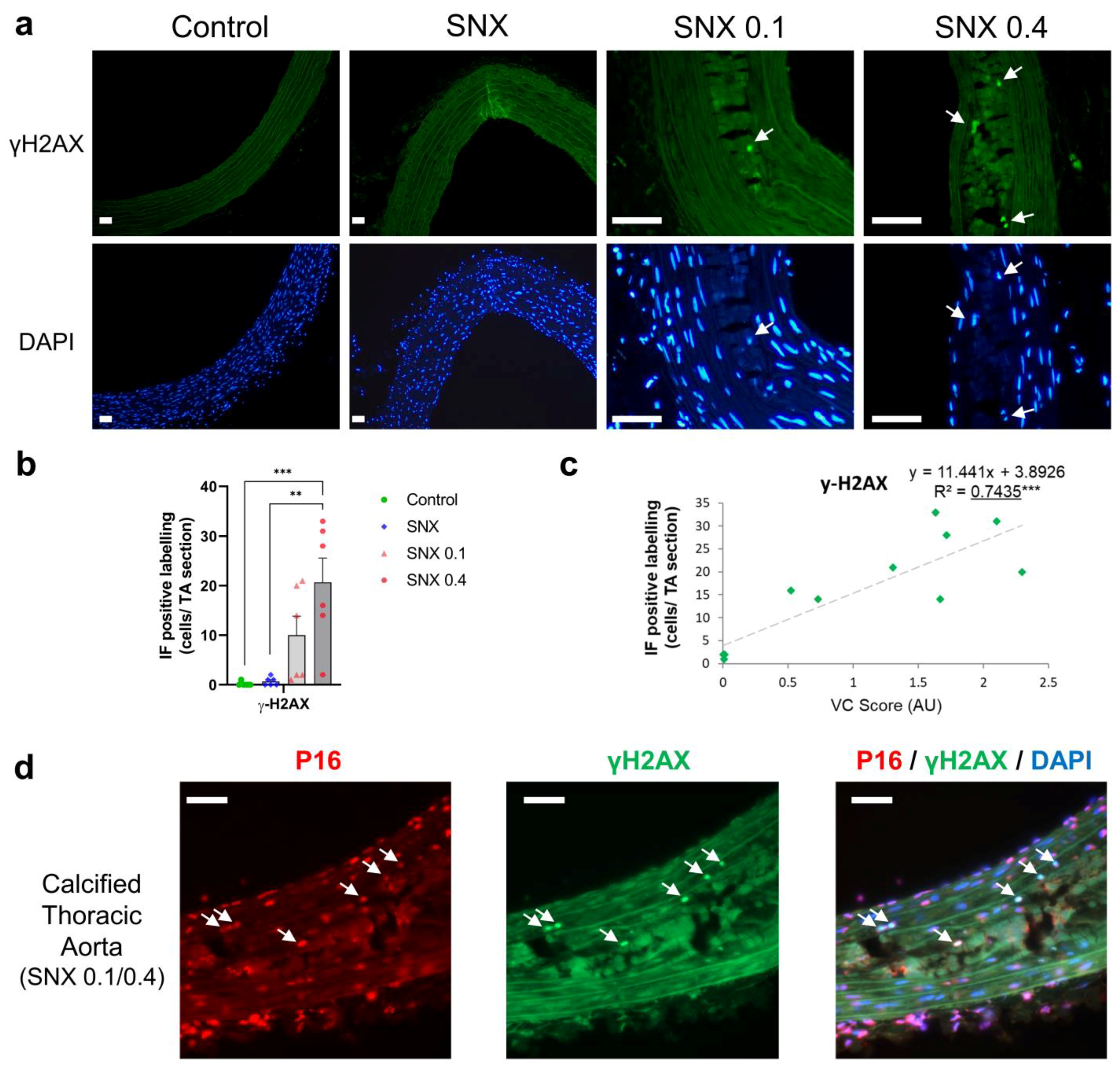
3.3. Increased Senescent Cell Burden in Calcified Rats
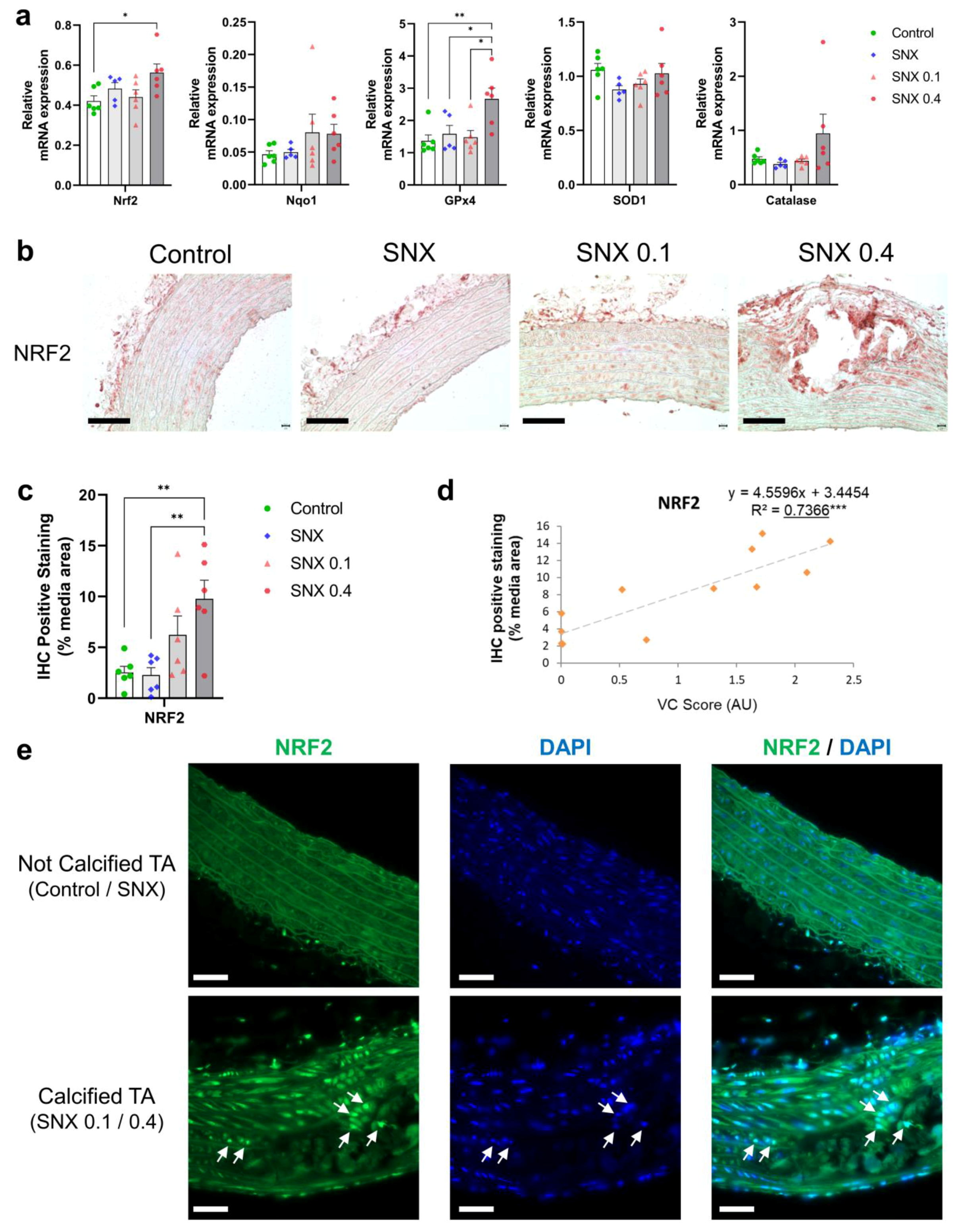
3.4. Oxidative Stress Regulation through NRF2 Pathway in Calcified Rats
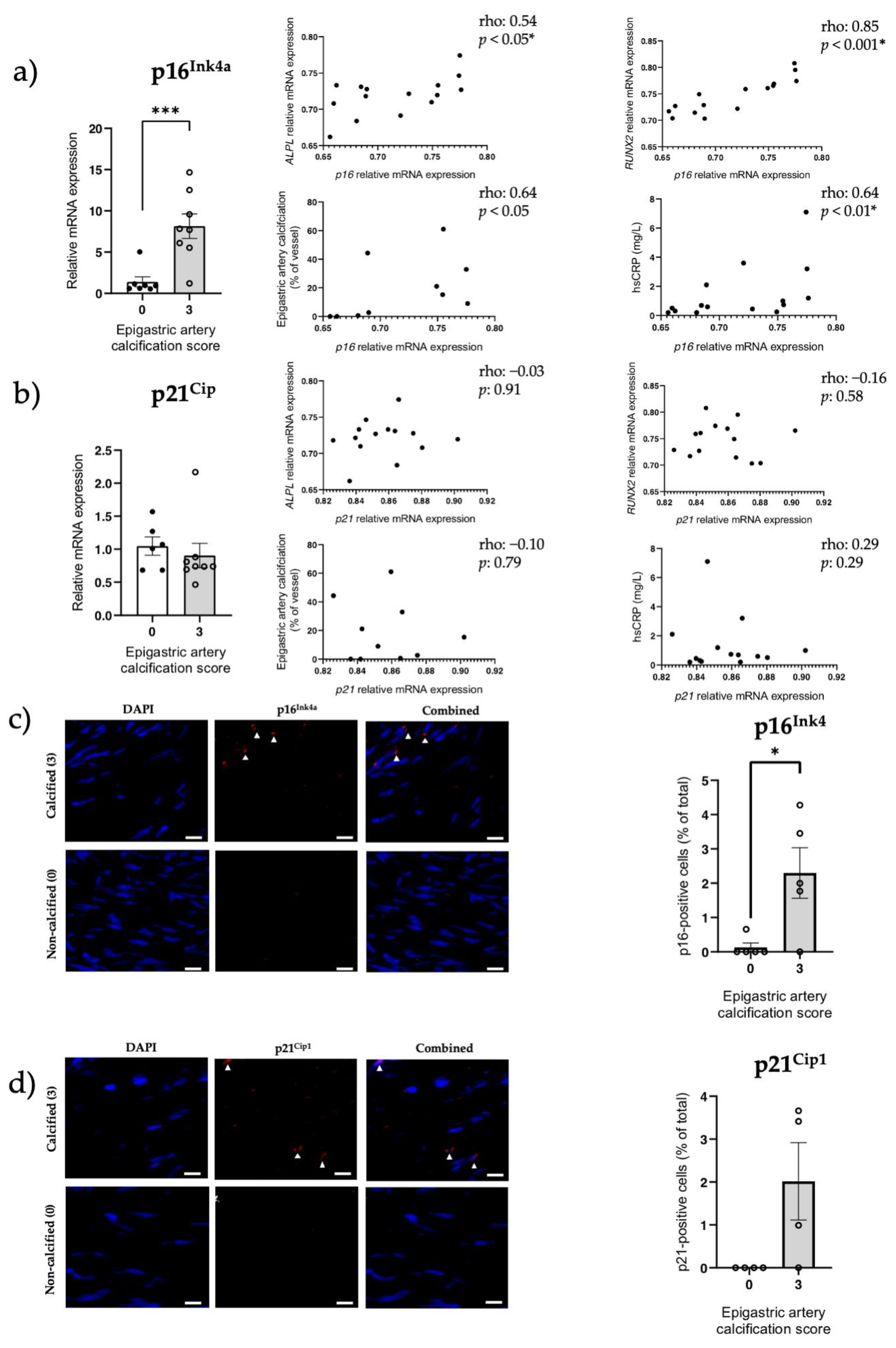
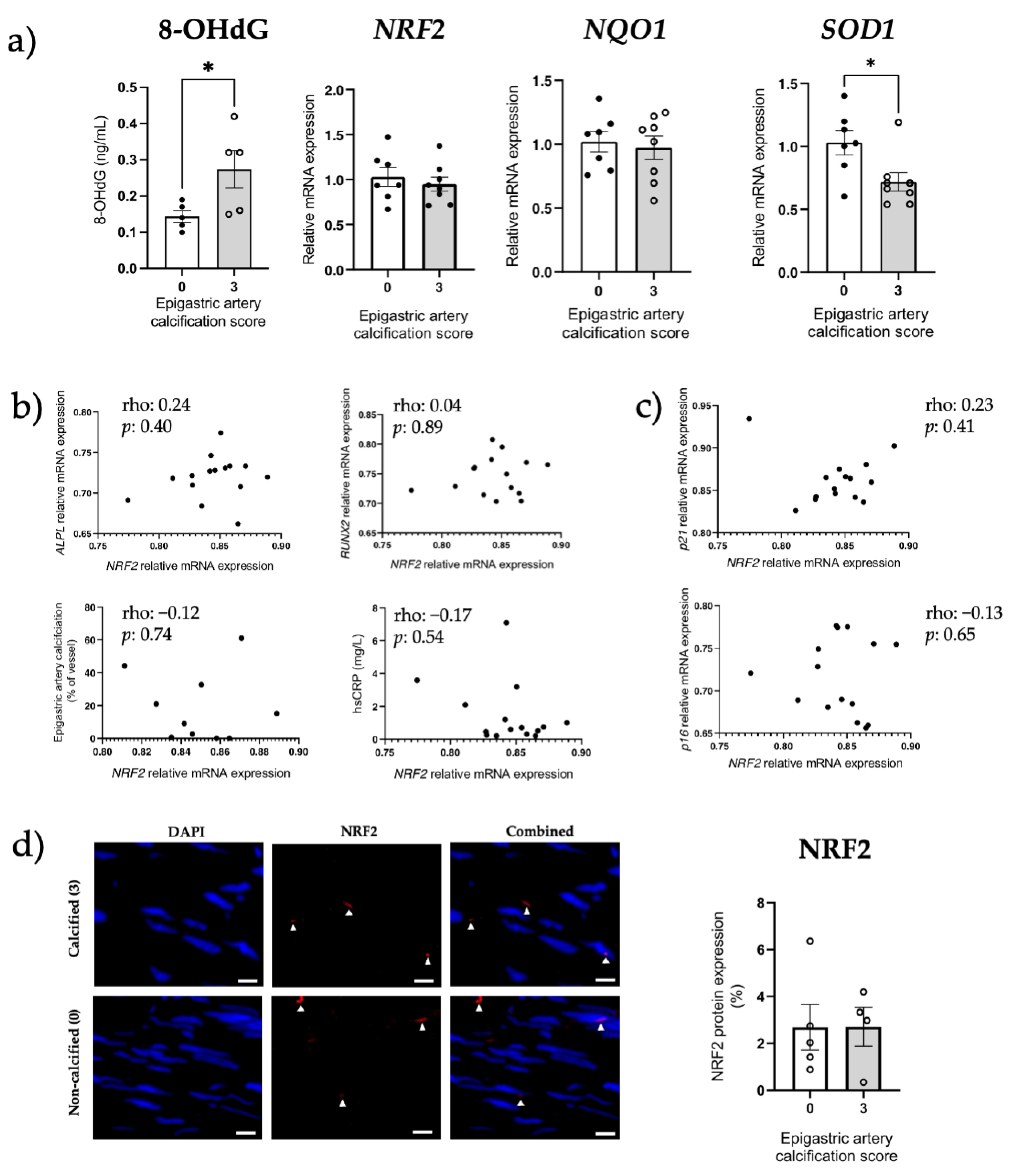
3.5. Increased Senescent Cell Burden, but Not NRF2 in Human Calcified Arteries
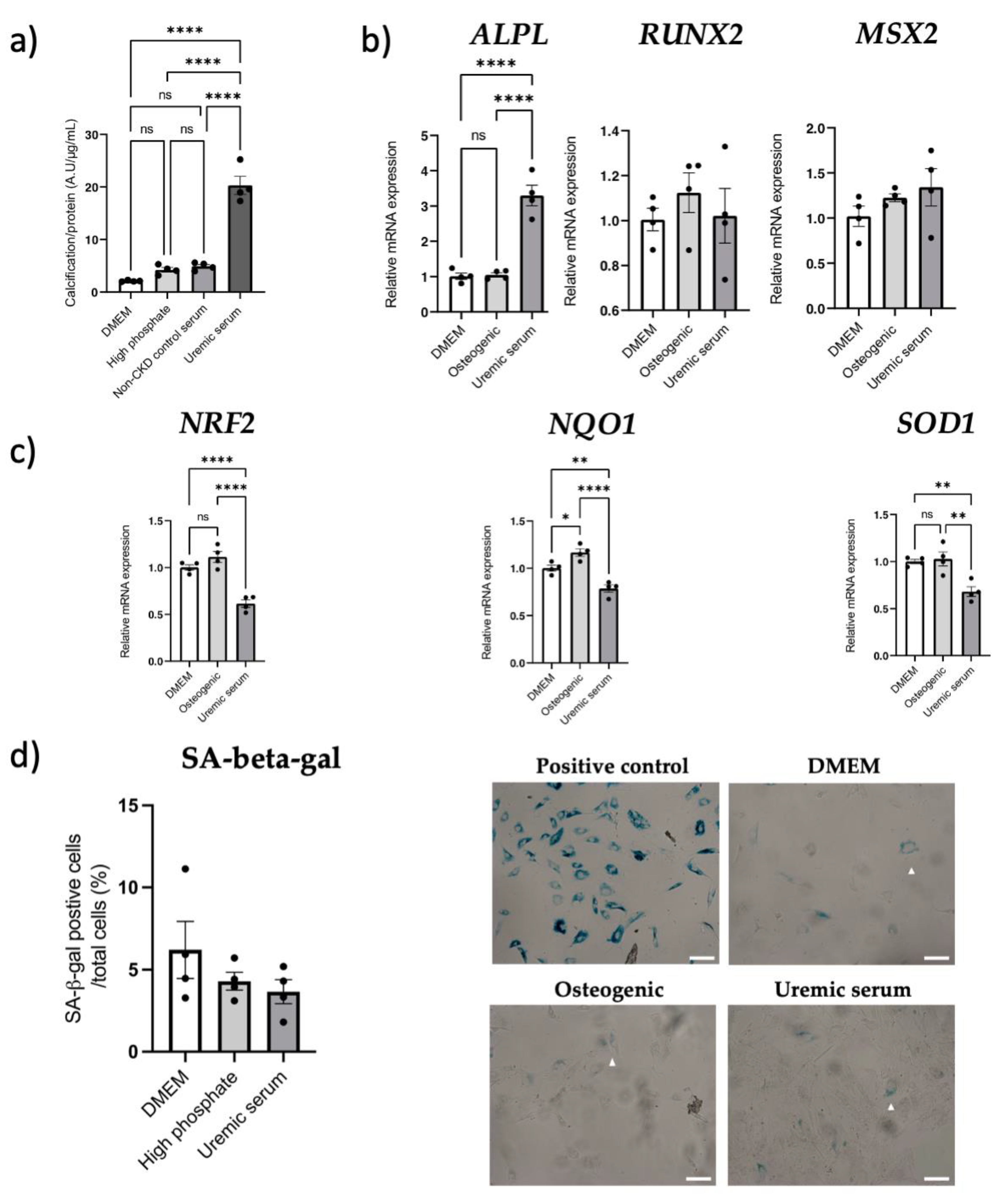
3.6. NRF2 Dysregulation In Vitro
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Cockwell, P.; Fisher, L.A. The global burden of chronic kidney disease. Lancet 2020, 395, 662–664. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kovesdy, C.P. Epidemiology of chronic kidney disease: An update 2022. Kidney Int. Suppl. 2022, 12, 7–11. [Google Scholar] [CrossRef] [PubMed]
- 3. Bikbov, B.; Purcell, C.A.; Levey, A.S.; Smith, M.; Abdoli, A.; Abebe, M.; Adebayo, O.M.; Afarideh, M.; Agarwal, S.K.; Agudelo-Botero, M.; et al. Global, regional, and national burden of chronic kidney disease, 1990–2017: A systematic analysis for the Global Burden of Disease Study 2017. Lancet 2020, 395, 709–733. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jankowski, J.; Floege, J.; Fliser, D.; Böhm, M.; Marx, N. Cardiovascular Disease in Chronic Kidney Disease. Circulation 2021, 143, 1157–1172. [Google Scholar] [CrossRef]
- Chen, J.; Budoff, M.J.; Reilly, M.; Yang, W.; Rosas, S.E.; Rahman, M.; Zhang, X.; Roy, J.A.; Lustigova, E.; Nessel, L.; et al. Coronary Artery Calcification and Risk of Cardiovascular Disease and Death Among Patients With Chronic Kidney Disease. JAMA Cardiol. 2017, 2, 635–643. [Google Scholar] [CrossRef] [Green Version]
- Matsushita, K.; Sang, Y.; Ballew, S.; Shlipak, M.; Katz, R.; Rosas, S.; Peralta, C.; Woodward, M.; Kramer, H.; Jacobs, D.; et al. Subclinical Atherosclerosis Measures for Cardiovascular Prediction in, CKD. J. Am. Soc. Nephrol. 2015, 26, 439–447. [Google Scholar] [CrossRef] [Green Version]
- Wyatt, C.M.; Drueke, T.B. Vascular calcification in chronic kidney disease: Here to stay? Kidney Int. 2017, 92, 276–278. [Google Scholar] [CrossRef]
- Himmelsbach, A.; Ciliox, C.; Goettsch, C. Cardiovascular Calcification in Chronic Kidney Disease—Therapeutic Opportunities. Toxins 2020, 12, 181. [Google Scholar] [CrossRef] [Green Version]
- Shioi, A.; Morioka, T.; Shoji, T.; Emoto, M. The Inhibitory Roles of Vitamin K in Progression of Vascular Calcification. Nutrients 2020, 12, 583. [Google Scholar] [CrossRef] [Green Version]
- Schantl, A.E.; Verhulst, A.; Neven, E.; Behets, G.J.; D’Haese, P.C.; Maillard, M.; Mordasini, D.; Phan, O.; Burnier, M.; Spaggiari, D.; et al. Inhibition of vascular calcification by inositol phosphates derivatized with ethylene glycol oligomers. Nat. Commun. 2020, 11, 721. [Google Scholar] [CrossRef] [Green Version]
- Orth-Alampour, S.; Gayrard, N.; Salem, S.; Bhargava, S.; Jankowski, V.; Jover, B.; Notarnicola, C.; Noels, H.; van der Vorst, E.P.C.; Kuppe, C.; et al. Prevention of vascular calcification by the endogenous chromogranin A-derived mediator that inhibits osteogenic transdifferentiation. Basic Res. Cardiol. 2021, 116, 57. [Google Scholar] [CrossRef] [PubMed]
- Hobson, S.; Arefin, S.; Kublickiene, K.; Shiels, P.; Stenvinkel, P. Senescent Cells in Early Vascular Ageing and Bone Disease of Chronic Kidney Disease—A Novel Target for Treatment. Toxins 2019, 11, 82. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dai, L.; Schurgers, L.J.; Shiels, P.G.; Stenvinkel, P. Early vascular ageing in chronic kidney disease: Impact of inflammation, vitamin K, senescence and genomic damage. Nephrol. Dial. Transplant. 2020, 35 (Suppl. 2), ii31–ii37. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kirkland, J.L.; Tchkonia, T. Cellular Senescence: A Translational Perspective. EBioMedicine 2017, 21, 21–28. [Google Scholar] [CrossRef] [Green Version]
- Demaria, M.; Ohtani, N.; Youssef, S.; Rodier, F.; Toussaint, W.; Mitchell, J.; Laberge, R.-M.; Vijg, J.; Van Steeg, H.; Dollé, M.; et al. An Essential Role for Senescent Cells in Optimal Wound Healing through Secretion of, PDGF-AA. Dev. Cell 2014, 31, 722–733. [Google Scholar] [CrossRef] [Green Version]
- Storer, M.; Mas, A.; Robert-Moreno, À.; Pecoraro, M.; Ortells, M.C.; Di Giacomo, V.; Yosef, R.; Pilpel, N.; Krizhanovsky, V.; Sharpe, J.; et al. Senescence Is a Developmental Mechanism that Contributes to Embryonic Growth and Patterning. Cell 2013, 155, 1119–1130. [Google Scholar] [CrossRef] [Green Version]
- López-Otín, C.; Blasco, M.A.; Partridge, L.; Serrano, M.; Kroemer, G. The Hallmarks of Aging. Cell 2013, 153, 1194–1217. [Google Scholar] [CrossRef] [Green Version]
- Baker, D.J.; Wijshake, T.; Tchkonia, T.; Lebrasseur, N.K.; Childs, B.G.; Van De Sluis, B.; Kirkland, J.L.; Van Deursen, J.M. Clearance of p16Ink4a-positive senescent cells delays ageing-associated disorders. Nature 2011, 479, 232–236. [Google Scholar] [CrossRef] [Green Version]
- Gorgoulis, V.; Adams, P.D.; Alimonti, A.; Bennett, D.C.; Bischof, O.; Bishop, C.; Campisi, J.; Collado, M.; Evangelou, K.; Ferbeyre, G.; et al. Cellular Senescence: Defining a Path Forward. Cell 2019, 179, 813–827. [Google Scholar] [CrossRef]
- Gasek, N.S.; Kuchel, G.A.; Kirkland, J.L.; Xu, M. Strategies for Targeting Senescent Cells in Human Disease. Nat. Aging 2021, 1, 870–879. [Google Scholar] [CrossRef]
- Vomund, S.; Schäfer, A.; Parnham, M.; Brüne, B.; von Knethen, A. Nrf2, the Master Regulator of Anti-Oxidative Responses. Int. J. Mol. Sci. 2017, 18, 2772. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kobayashi, A.; Kang, M.-I.; Okawa, H.; Ohtsuji, M.; Zenke, Y.; Chiba, T.; Igarashi, K.; Yamamoto, M. Oxidative Stress Sensor Keap1 Functions as an Adaptor for Cul3-Based E3 Ligase To Regulate Proteasomal Degradation of Nrf2. Mol. Cell. Biol. 2004, 24, 7130–7139. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suzuki, T.; Motohashi, H.; Yamamoto, M. Toward clinical application of the Keap1-Nrf2 pathway. Trends Pharmacol. Sci. 2013, 34, 340–346. [Google Scholar] [CrossRef]
- Finkel, T.; Holbrook, N.J. Oxidants, oxidative stress and the biology of ageing. Nature 2000, 408, 239–247. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.-Y.; Liu, L.-H.; Liu, H.; Wu, K.-F.; An, J.; Wang, Q.; Liu, Y.; Bai, L.-J.; Qi, B.-M.; Qi, B.-L.; et al. Nrf2 protects against diabetic dysfunction of endothelial progenitor cells via regulating cell senescence. Int. J. Mol. Med. 2018, 42, 1327–1340. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hiebert, P.; Wietecha, M.S.; Cangkrama, M.; Haertel, E.; Mavrogonatou, E.; Stumpe, M.; Steenbock, H.; Grossi, S.; Beer, H.-D.; Angel, P.; et al. Nrf2-Mediated Fibroblast Reprogramming Drives Cellular Senescence by Targeting the Matrisome. Dev. Cell 2018, 46, 145–161.e10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kapeta, S.; Chondrogianni, N.; Gonos, E.S. Nuclear Erythroid Factor 2-mediated Proteasome Activation Delays Senescence in Human Fibroblasts. J. Biol. Chem. 2010, 285, 8171–8184. [Google Scholar] [CrossRef] [Green Version]
- Fujihara, C.; Kowala, M.C.; Breyer, M.D.; Sena, C.; Rodrigues, M.; Arias, S.; Fanelli, C.; Malheiros, D.; Jadhav, P.K.; Montrose-Rafizadeh, C.; et al. A Novel Aldosterone Antagonist Limits Renal Injury in 5/6 Nephrectomy. Sci. Rep. 2017, 7, 7899. [Google Scholar] [CrossRef] [Green Version]
- Stevens, P.E. Evaluation and Management of Chronic Kidney Disease: Synopsis of the Kidney Disease: Improving Global Outcomes 2012 Clinical Practice Guideline. Ann. Intern. Med. 2013, 158, 825. [Google Scholar] [CrossRef] [Green Version]
- Cheung, A.K.; Chang, T.I.; Cushman, W.C.; Furth, S.L.; Hou, F.F.; Ix, J.H.; Knoll, G.A.; Muntner, P.; Pecoits-Filho, R.; Sarnak, M.J.; et al. KDIGO 2021 Clinical Practice Guideline for the Management of Blood Pressure in Chronic Kidney Disease. Kidney Int. 2021, 99, S1–S87. [Google Scholar] [CrossRef]
- Kim, B.S.; Chan, N.; Hsu, G.; Makaryus, A.N.; Chopra, M.; Cohen, S.L.; Makaryus, J.N. Sex Differences in Coronary Arterial Calcification in Symptomatic Patients. Am. J. Cardiol. 2021, 149, 16–20. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Zhao, Q.; Chen, Z.; Geng, Y.-J.; Zhang, W.; Zhou, Q.; Yang, W.; Liu, Q.; Liu, H. Estrogen inhibits vascular calcification in rats via hypoxia-induced factor-1α signaling. Vascular 2020, 28, 465–474. [Google Scholar] [CrossRef] [PubMed]
- Stenvinkel, P.; Luttropp, K.; McGuinness, D.; Witasp, A.; Qureshi, A.R.; Wernerson, A.; Nordfors, L.; Schalling, M.; Ripsweden, J.; Wennberg, L.; et al. CDKN2A/p16INK4a expression is associated with vascular progeria in chronic kidney disease. Aging 2017, 9, 494–507. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qureshi, A.R.; Olauson, H.; Witasp, A.; Haarhaus, M.L.; Brandenburg, V.; Wernerson, A.; Lindholm, B.; Söderberg, M.; Wennberg, L.; Nordfors, L.; et al. Increased circulating sclerostin levels in end-stage renal disease predict biopsy-verified vascular medial calcification and coronary artery calcification. Kidney Int. 2015, 88, 1356–1364. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dai, L.; Mukai, H.; Lindholm, B.; Heimbürger, O.; Barany, P.; Stenvinkel, P.; Qureshi, A.R. Clinical global assessment of nutritional status as predictor of mortality in chronic kidney disease patients. PLoS ONE 2017, 12, e0186659. [Google Scholar] [CrossRef] [Green Version]
- Sert, N.P.; Ahluwalia, A.; Avey, M.T.; Baker, M.; Browne, W.J.; Clark, A.; Cuthill, I.C.; Dirnagl, U.; Emerson, M.; Garner, P.; et al. Reporting animal research: Explanation and elaboration for the ARRIVE guidelines 2.0. PLoS Biol. 2020, 18, e3000411. [Google Scholar]
- Georgiadis, G.S.; Argyriou, C.; Antoniou, G.A.; Kantartzi, K.; Kriki, P.; Theodoridis, M.; Thodis, E.; Lazarides, M.K. Upper limb vascular calcification score as a predictor of mortality in diabetic hemodialysis patients. J. Vasc. Surg. 2015, 61, 1529–1537. [Google Scholar] [CrossRef] [Green Version]
- Jung, Y.S.; Qian, Y.; Chen, X. Examination of the expanding pathways for the regulation of p21 expression and activity. Cell Signal. 2010, 22, 1003–1012. [Google Scholar] [CrossRef] [Green Version]
- Ha, C.-M.; Park, S.; Choi, Y.-K.; Jeong, J.-Y.; Oh, C.J.; Bae, K.-H.; Lee, S.J.; Kim, J.-H.; Park, K.-G.; Jun, D.Y.; et al. Activation of Nrf2 by dimethyl fumarate improves vascular calcification. Vasc. Pharmacol. 2014, 63, 29–36. [Google Scholar] [CrossRef]
- Wei, R.; Enaka, M.; Muragaki, Y. Activation of KEAP1/NRF2/P62 signaling alleviates high phosphate-induced calcification of vascular smooth muscle cells by suppressing reactive oxygen species production. Sci. Rep. 2019, 9, 10366. [Google Scholar] [CrossRef] [Green Version]
- Shobeiri, N.; Adams, M.A.; Holden, R.M. Vascular calcification in animal models of CKD: A review. Am. J. Nephrol. 2010, 31, 471–481. [Google Scholar] [CrossRef] [PubMed]
- Hortells, L.; Sosa, C.; Guillén, N.; Lucea, S.; Millán, Á.; Sorribas, V. Identifying early pathogenic events during vascular calcification in uremic rats. Kidney Int. 2017, 92, 1384–1394. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shanahan, C.M.; Crouthamel, M.H.; Kapustin, A.; Giachelli, C.M. Arterial Calcification in Chronic Kidney Disease: Key Roles for Calcium and Phosphate. Circ. Res. 2011, 109, 697–711. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Steitz, S.A.; Speer, M.Y.; Curinga, G.; Yang, H.Y.; Haynes, P.; Aebersold, R.; Schinke, T.; Karsenty, G.; Giachelli, C.M. Smooth muscle cell phenotypic transition associated with calcification: Upregulation of Cbfa1 and downregulation of smooth muscle lineage markers. Circ. Res. 2001, 89, 1147–1154. [Google Scholar] [CrossRef] [PubMed]
- Yamada, S.; Leaf, E.M.; Chia, J.J.; Cox, T.C.; Speer, M.Y.; Giachelli, C.M. PiT-2, a type III sodium-dependent phosphate transporter, protects against vascular calcification in mice with chronic kidney disease fed a high-phosphate diet. Kidney Int. 2018, 94, 716–727. [Google Scholar] [CrossRef] [PubMed]
- Savinov, A.Y.; Salehi, M.; Yadav, M.C.; Radichev, I.; Millán, J.L.; Savinova, O.V. Transgenic Overexpression of Tissue-Nonspecific Alkaline Phosphatase (TNAP) in Vascular Endothelium Results in Generalized Arterial Calcification. J. Am. Hear. Assoc. 2015, 4, 2499. [Google Scholar] [CrossRef] [Green Version]
- Sheen, C.R.; Kuss, P.; Narisawa, S.; Yadav, M.C.; Nigro, J.; Wang, W.; Chhea, T.N.; Sergienko, E.A.; Kapoor, K.; Jackson, M.R.; et al. Pathophysiological Role of Vascular Smooth Muscle Alkaline Phosphatase in Medial Artery Calcification. J. Bone Miner. Res. 2014, 30, 824–836. [Google Scholar] [CrossRef] [Green Version]
- Jin, D.; Lin, L.; Xie, Y.; Jia, M.; Qiu, H.; Xun, K. NRF2-suppressed vascular calcification by regulating the antioxidant pathway in chronic kidney disease. FASEB J. 2021, 36, e22098. [Google Scholar] [CrossRef]
- Lastres-Becker, I.; Innamorato, N.; Jaworski, T.; Rábano, A.; Kügler, S.; Van Leuven, F.; Cuadrado, A. Fractalkine activates NRF2/NFE2L2 and heme oxygenase 1 to restrain tauopathy-induced microgliosis. Brain 2014, 137, 78–91. [Google Scholar] [CrossRef]
- Pajares, M.; Jiménez-Moreno, N.; García-Yagüe, Á.; Escoll, M.; de Ceballos, M.; Van Leuven, F.; Rábano, A.; Yamamoto, M.; Rojo, A.; Cuadrado, A. Transcription factor NFE2L2/NRF2 is a regulator of macroautophagy genes. Autophagy 2016, 12, 1902–1916. [Google Scholar] [CrossRef] [Green Version]
- Nelson, A.J.; Raggi, P.; Wolf, M.; Gold, A.M.; Chertow, G.M.; Roe, M.T. Targeting Vascular Calcification in Chronic Kidney Disease. JACC Basic Transl. Sci. 2020, 5, 398–412. [Google Scholar] [CrossRef] [PubMed]
- Stockler-Pinto, M.; Soulage, C.; Borges, N.; Cardozo, L.; Dolenga, C.; Nakao, L.; Pecoits-Filho, R.; Fouque, D.; Mafra, D. From bench to the hemodialysis clinic: Protein-bound uremic toxins modulate NF-κB/Nrf2 expression. Int. Urol. Nephrol. 2018, 50, 347–354. [Google Scholar] [CrossRef] [PubMed]
- Pedruzzi, L.M.; Cardozo, L.; Daleprane, J.B.; Stockler-Pinto, M.B.; Monteiro, E.B.; Leite, M., Jr.; Vaziri, N.D.; Mafra, D. Systemic inflammation and oxidative stress in hemodialysis patients are associated with down-regulation of Nrf2. J. Nephrol. 2015, 28, 495–501. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmidlin, C.J.; Dodson, M.B.; Madhavan, L.; Zhang, D.D. Redox regulation by NRF2 in aging and disease. Free Radic. Biol. Med. 2019, 134, 702–707. [Google Scholar] [CrossRef]
- Barnes, P.J. Oxidative Stress in Chronic Obstructive Pulmonary Disease. Antioxidants 2022, 11, 965. [Google Scholar] [CrossRef]
- Dodson, M.; de la Vega, M.R.; Cholanians, A.B.; Schmidlin, C.J.; Chapman, E.; Zhang, D.D. Modulating NRF2 in Disease: Timing Is Everything. Annu. Rev. Pharmacol. Toxicol. 2019, 59, 555–575. [Google Scholar] [CrossRef]
- Wu, S.; Lu, H.; Bai, Y. Nrf2 in cancers: A double-edged sword. Cancer Med. 2019, 8, 2252–2267. [Google Scholar] [CrossRef]
- Aranda-Rivera, A.K.; Cruz-Gregorio, A.; Pedraza-Chaverri, J.; Scholze, A. Nrf2 Activation in Chronic Kidney Disease: Promises and Pitfalls. Antioxidants 2022, 11, 1112. [Google Scholar] [CrossRef]
- Neilson, L.E.; Quinn, J.F.; Gray, N.E. Peripheral Blood NRF2 Expression as a Biomarker in Human Health and Disease. Antioxidants 2020, 10, 28. [Google Scholar] [CrossRef]
- Nakano-Kurimoto, R.; Ikeda, K.; Uraoka, M.; Nakagawa, Y.; Yutaka, K.; Koide, M.; Takahashi, T.; Matoba, S.; Yamada, H.; Okigaki, M.; et al. Replicative senescence of vascular smooth muscle cells enhances the calcification through initiating the osteoblastic transition. Am. J. Physiol.-Heart Circ. Physiol. 2009, 297, H1673–H1684. [Google Scholar] [CrossRef]
- Liu, Y.; Drozdov, I.; Shroff, R.; Beltran, L.E.; Shanahan, C.M. Prelamin A accelerates vascular calcification via activation of the DNA damage response and senescence-associated secretory phenotype in vascular smooth muscle cells. Circ. Res. 2013, 112, e99–e109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamada, S.; Tatsumoto, N.; Tokumoto, M.; Noguchi, H.; Ooboshi, H.; Kitazono, T.; Tsuruya, K. Phosphate Binders Prevent Phosphate-Induced Cellular Senescence of Vascular Smooth Muscle Cells and Vascular Calcification in a Modified, Adenine-Based Uremic Rat Model. Calcif. Tissue Int. 2014, 96, 347–358. [Google Scholar] [CrossRef] [PubMed]
- Takemura, A.; Iijima, K.; Ota, H.; Son, B.-K.; Ito, Y.; Ogawa, S.; Eto, M.; Akishita, M.; Ouchi, Y. Sirtuin 1 Retards Hyperphosphatemia-Induced Calcification of Vascular Smooth Muscle Cells. Arter. Thromb. Vasc. Biol. 2011, 31, 2054–2062. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, H.C.; Zuo, Y.; Fogo, A.B. Models of chronic kidney disease. Drug Discov. Today Dis. Models 2010, 7, 13–19. [Google Scholar] [CrossRef] [Green Version]
- Agharazii, M.; St-Louis, R.; Gautier-Bastien, A.; Ung, R.-V.; Mokas, S.; Larivière, R.; Richard, D.E. Inflammatory Cytokines and Reactive Oxygen Species as Mediators of Chronic Kidney Disease-Related Vascular Calcification. Am. J. Hypertens. 2014, 28, 746–755. [Google Scholar] [CrossRef] [Green Version]
- Pazár, B.; Ea, H.-K.; Narayan, S.; Kolly, L.; Bagnoud, N.; Chobaz, V.; Roger, T.; Lioté, F.; So, A.; Busso, N. Basic Calcium Phosphate Crystals Induce Monocyte/Macrophage IL-1β Secretion through the NLRP3 Inflammasome In Vitro. J. Immunol. 2011, 186, 2495–2502. [Google Scholar] [CrossRef] [Green Version]
- Nadra, I.; Mason, J.; Philippidis, P.; Florey, O.; Smythe, C.; McCarthy, G.; Landis, R.; Haskard, D. Proinflammatory activation of macrophages by basic calcium phosphate crystals via protein kinase C and MAP kinase pathways: A vicious cycle of inflammation and arterial calcification? Circ. Res. 2005, 96, 1248–1256. [Google Scholar] [CrossRef] [Green Version]
- Wu, C.M.; Zheng, L.; Wang, Q.; Hu, Y.W. The emerging role of cell senescence in atherosclerosis. Clin. Chem. Lab. Med. 2021, 59, 27–38. [Google Scholar] [CrossRef]
- Sharpless, N.E.; Sherr, C.J. Forging a signature of in vivo senescence. Nat. Rev. Cancer 2015, 15, 397–408. [Google Scholar] [CrossRef]
- Stein, G.H.; Drullinger, L.F.; Soulard, A.; Dulić, V. Differential Roles for Cyclin-Dependent Kinase Inhibitors p21 and p16 in the Mechanisms of Senescence and Differentiation in Human Fibroblasts. Mol. Cell Biol. 1999, 19, 2109–2117. [Google Scholar] [CrossRef] [Green Version]
- Sanchis, P.; Ho, C.Y.; Liu, Y.; Beltran, L.; Ahmad, S.; Jacob, A.; Furmanik, M.; Laycock, J.; Long, D.; Shroff, R.; et al. Arterial “inflammaging” drives vascular calcification in children on dialysis. Kidney Int. 2019, 95, 958–972. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tanikawa, T.; Okada, Y.; Tanikawa, R.; Tanaka, Y. Advanced Glycation End Products Induce Calcification of Vascular Smooth Muscle Cells through RAGE/p38 MAPK. J. Vasc. Res. 2009, 46, 572–580. [Google Scholar] [CrossRef]
- Gayrard, N.; Muyor, K.; Notarnicola, C.; Duranton, F.; Jover, B.; Argilés, À. Optimisation of cell and ex vivo culture conditions to study vascular calcification. PLoS ONE 2020, 15, e0230201. [Google Scholar] [CrossRef] [PubMed]
- O'Rourke, C.; Shelton, G.; Hutcheson, J.; Burke, M.; Martyn, T.; Thayer, T.; Shakartzi, H.; Buswell, M.; Tainsh, R.; Yu, B.; et al. Calcification of Vascular Smooth Muscle Cells and Imaging of Aortic Calcification and Inflammation. J. Vis. Exp. 2016, 31, 54017. [Google Scholar]
- Kirkland, J.L.; Tchkonia, T. Senolytic drugs: From discovery to translation. J. Intern. Med. 2020, 288, 518–536. [Google Scholar] [CrossRef] [PubMed]
- Bover, J.; Ureña-Torres, P.; Górriz, J.L.; Lloret, M.J.; da Silva, I.; Ruiz-García, C.; Chang, P.; Rodríguez, M.; Ballarín, J. Cardiovascular calcifications in chronic kidney disease: Potential therapeutic implications. Nefrología 2016, 36, 597–608. [Google Scholar] [CrossRef] [PubMed] [Green Version]
Disclaimer/Publisher’s Note: The statements, opinions, and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions, or products referred to in the content. |








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Non-Calcified Epigastric Artery Group (n = 8) | Severely Calcified Epigastric Artery Group (n = 8) | p-Value | |
|---|---|---|---|
| Age (years) | 47 (15) | 54 (13) | 0.695 |
| Sex (males/females) | 8/0 | 8/0 | - |
| BMI (kg/m2) | 23.9 (4.8) | 24.1 (4.7) | 0.785 |
| Systolic blood pressure (mmHg) | 157 (31) | 156 (44) | 0.242 |
| Diastolic blood pressure (mmHg) | 94 (14) | 80.0 (27) | 0.113 |
| eGFR (mL/min/1.73 m2) | 8 (2) | 10 (4) | 0.136 |
| hsCRP (mg/L) | 2.1 (2.7) | 5.0 (7.9) | 0.878 |
| Calcium (mmol/L) | 2.3 (0.1) | 2.3 (0.4) | 0.422 |
| Phosphate (mmol/L) | 1.5 (0.4) | 1.4 (0.6) | 0.798 |
| Cardiovascular disease (yes/no) | 0/8 | 0/8 | - |
| Statin user (yes/no) | 6/2 | 2/6 | 0.132 |
| Diabetes (yes/no) | 1/7 | 2/6 | 1.000 |
| CAC score (AU) | 0 (0) | 463 (1165) | 0.018 |
| Control (n = 6) | SNX (n= 5) | SNX 0.1 (n = 6) | SNX 0.4 (n = 6) | p-Value | |
|---|---|---|---|---|---|
| Final Weight (g) | 491 ± 24 | 530 ± 39 | 538 ± 21 | 474 ± 25 | 0.3026 |
| Arterial Pressure | |||||
| Systolic Arterial Pressure (mmHg) | 155 ± 8 | 190 ± 18 | 161 ± 16 | 184 ± 13 | 0.2502 |
| Diastolic Arterial Pressure (mmHg) | 118 ± 8 | 133 ± 11 | 105 ± 12 | 97 ± 9 | 0.0983 |
| Pulse Pressure (mmHg) | 38 ± 1 | 57 ± 7 #t | 56 ± 9 # | 87 ± 10 *** | 0.0017 |
| Hematocrit (%) | 45 ± 1 | 38 ± 2 | 39 ± 3 | 36 ± 3 | 0.0533 |
| Plasma | |||||
| Creatinine in Plasma (µM) | 26 ± 1 | 91 ± 20 | 94 ± 18 | 104 ± 34 | 0.0675 |
| Urea in Plasma (µM) | 5.6 ± 0.2 | 14.4 ± 2.9 * | 14.4 ± 3.0 * | 11.7 ± 1.8 | 0.0253 |
| Calcium in Plasma (mM) | 2.51 ± 0.02 | 2.72 ± 0.04 | 2.77 ± 0.06 | 2.85 ± 0.12 * | 0.0284 |
| Phosphate in Plasma (mM) | 1.79 ± 0.03 | 2.11 ± 0.11 | 2.54 ± 0.32 | 2.88 ± 0.43 | 0.0559 |
| Product [Ca × P] in Plasma (mM) | 4.50 ± 0.12 | 5.73 ± 0.32 | 7.11 ± 1.02 | 8.07 ± 0.97 * | 0.0264 |
| Urine | |||||
| Creatinine in Urine (mM) | 5.20 ± 0.62 | 2.33 ± 0.73 *t | 2.26 ± 0.49 * | 3.29 ± 0.94 | 0.029 |
| Urea in Urine (µM) | 329 ± 30 | 136 ± 48 ** | 132 ± 23 ** | 179 ± 35 * | 0.0015 |
| Daily Urine Volume (mL/24 h) | 14 ± 1 | 26 ± 4 * | 29 ± 4 ** | 24 ± 2 | 0.009 |
| Proteins in Urine (mg/24 h) | 6.5 ± 1.1 | 60.0 ± 21.2 *t | 60.6 ± 8.0 *t | 50.8 ± 20.4 | 0.0494 |
| Creatinine Clearance (mL/min) | 1.87 ± 0.19 | 0.47 ± 0.14 ** | 0.58 ± 0.20 ** | 0.83 ± 0.36 * | 0.0026 |
| Kidney Fibrosis (% area stained) | 4.7 ± 0.7 | 8.2 ± 1.2 | 10.3 ± 2.7 | 10.6 ± 2.1 | 0.1724 |
| Left Ventricle weight (g) | 1.00 ± 0.05 | 1.57 ± 0.07 *** # | 1.54 ± 0.05 *** # | 1.33 ± 0.03 ** | <0.0001 |
| Left Ventricle weight (% of body weight) | 0.21 ± 0.01 | 0.30 ± 0.02 ** | 0.29 ± 0.02 ** | 0.28 ± 0.02 * | 0.0019 |
| Heart Fibrosis (% area stained) | 2.7 ± 0.3 | 3.3 ± 0.8 | 2.8 ± 0.5 # | 8.6 ± 2.7 * | 0.0242 |
| r | p-Value | |
|---|---|---|
| VC regulation and phenotypic transition | ||
| Opn | 0.873 | 0.0002 |
| a-SMA | −0.837 | 0.0007 |
| Alpl | −0.820 | 0.0011 |
| Mmp2 | −0.807 | 0.0015 |
| Runx2 | 0.763 | 0.0039 |
| Mgp | 0.737 | 0.0062 |
| Pit-1 | 0.731 | 0.0069 |
| Pit-2 | −0.658 | 0.0198 |
| Enpp1 | 0.637 | 0.0351 |
| Msx2 | 0.600 | 0.0393 |
| Bmp2 | 0.596 | 0.0412 |
| Tgf-β1 | 0.402 | 0.1962 |
| Markers of senescent cells | ||
| p16 Ink4a | 0.669 | 0.0174 |
| p21Cip1 | −0.575 | 0.0506 |
| p53 | −0.041 | 0.8984 |
| Cytokines contributing to the SASP | ||
| Mcp-1 | 0.640 | 0.0250 |
| IL-1b | 0.546 | 0.0751 |
| TNFa | 0.390 | 0.2096 |
| IL-6 | 0.254 | 0.4259 |
| NRF2 pathway | ||
| Nrf2 | 0.663 | 0.0186 |
| Nqo-1 | 0.813 | 0.0013 |
| SOD1 | −0.462 | 0.1307 |
| Catalase | −0.293 | 0.3559 |
| GPx4 | −0.111 | 0.7312 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Laget, J.; Hobson, S.; Muyor, K.; Duranton, F.; Cortijo, I.; Bartochowski, P.; Jover, B.; Lajoix, A.-D.; Söderberg, M.; Ebert, T.; et al. Implications of Senescent Cell Burden and NRF2 Pathway in Uremic Calcification: A Translational Study. Cells 2023, 12, 643. https://doi.org/10.3390/cells12040643
Laget J, Hobson S, Muyor K, Duranton F, Cortijo I, Bartochowski P, Jover B, Lajoix A-D, Söderberg M, Ebert T, et al. Implications of Senescent Cell Burden and NRF2 Pathway in Uremic Calcification: A Translational Study. Cells. 2023; 12(4):643. https://doi.org/10.3390/cells12040643
Chicago/Turabian StyleLaget, Jonas, Sam Hobson, Karen Muyor, Flore Duranton, Irene Cortijo, Piotr Bartochowski, Bernard Jover, Anne-Dominique Lajoix, Magnus Söderberg, Thomas Ebert, and et al. 2023. "Implications of Senescent Cell Burden and NRF2 Pathway in Uremic Calcification: A Translational Study" Cells 12, no. 4: 643. https://doi.org/10.3390/cells12040643
APA StyleLaget, J., Hobson, S., Muyor, K., Duranton, F., Cortijo, I., Bartochowski, P., Jover, B., Lajoix, A. -D., Söderberg, M., Ebert, T., Stenvinkel, P., Argilés, À., Kublickiene, K., & Gayrard, N. (2023). Implications of Senescent Cell Burden and NRF2 Pathway in Uremic Calcification: A Translational Study. Cells, 12(4), 643. https://doi.org/10.3390/cells12040643