
A Wrong Fate Decision in Adipose Stem Cells upon Obesity

 , ,
, , 
Abstract
:1. Introduction
2. Immunophenotyping and Cellular Heterogeneity
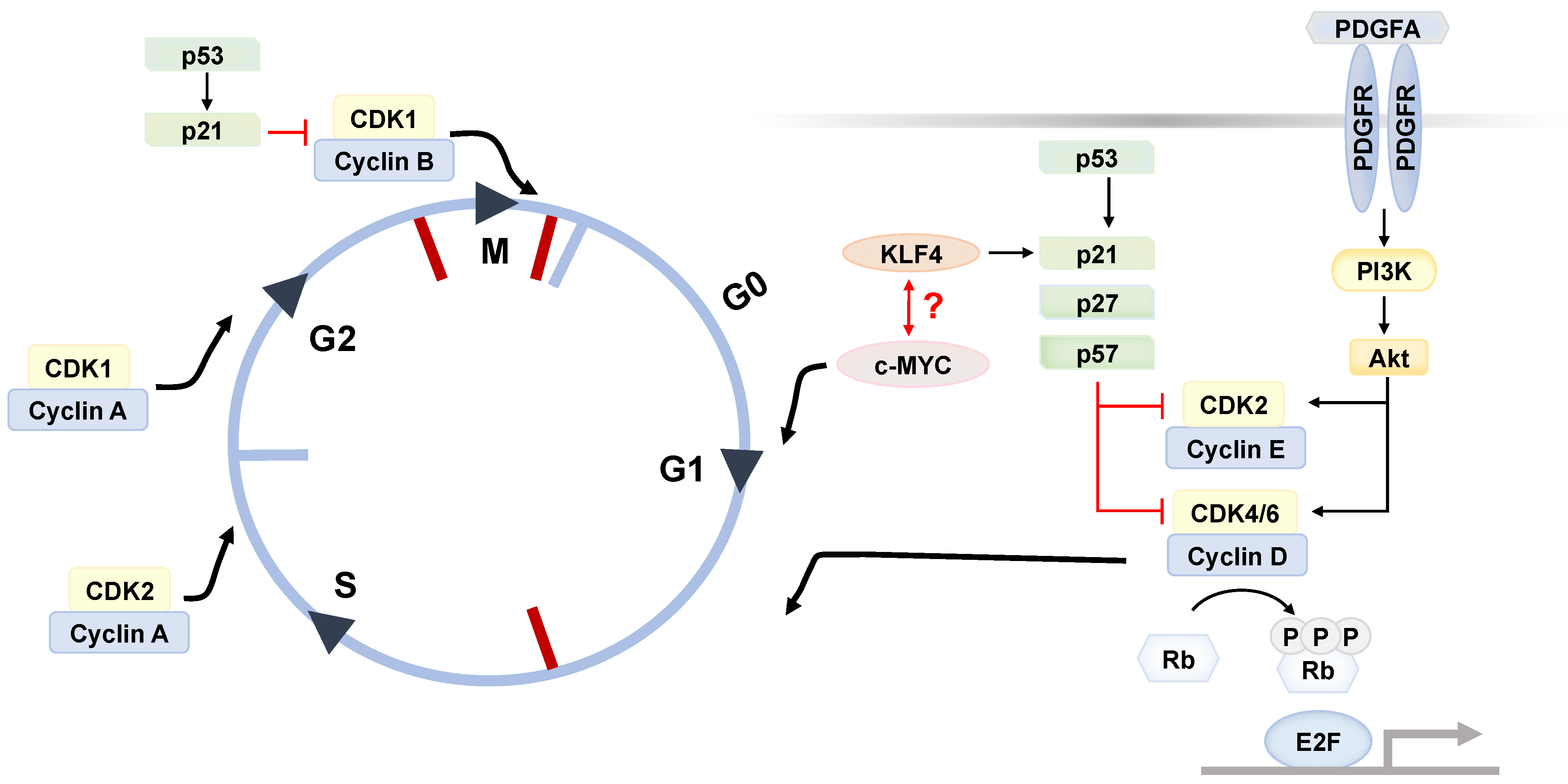
3. ASC Renewal
4. Aberrant Fate Determination as ASC Aging
5. Pathological Alternations in Extracellular Signaling Pathways
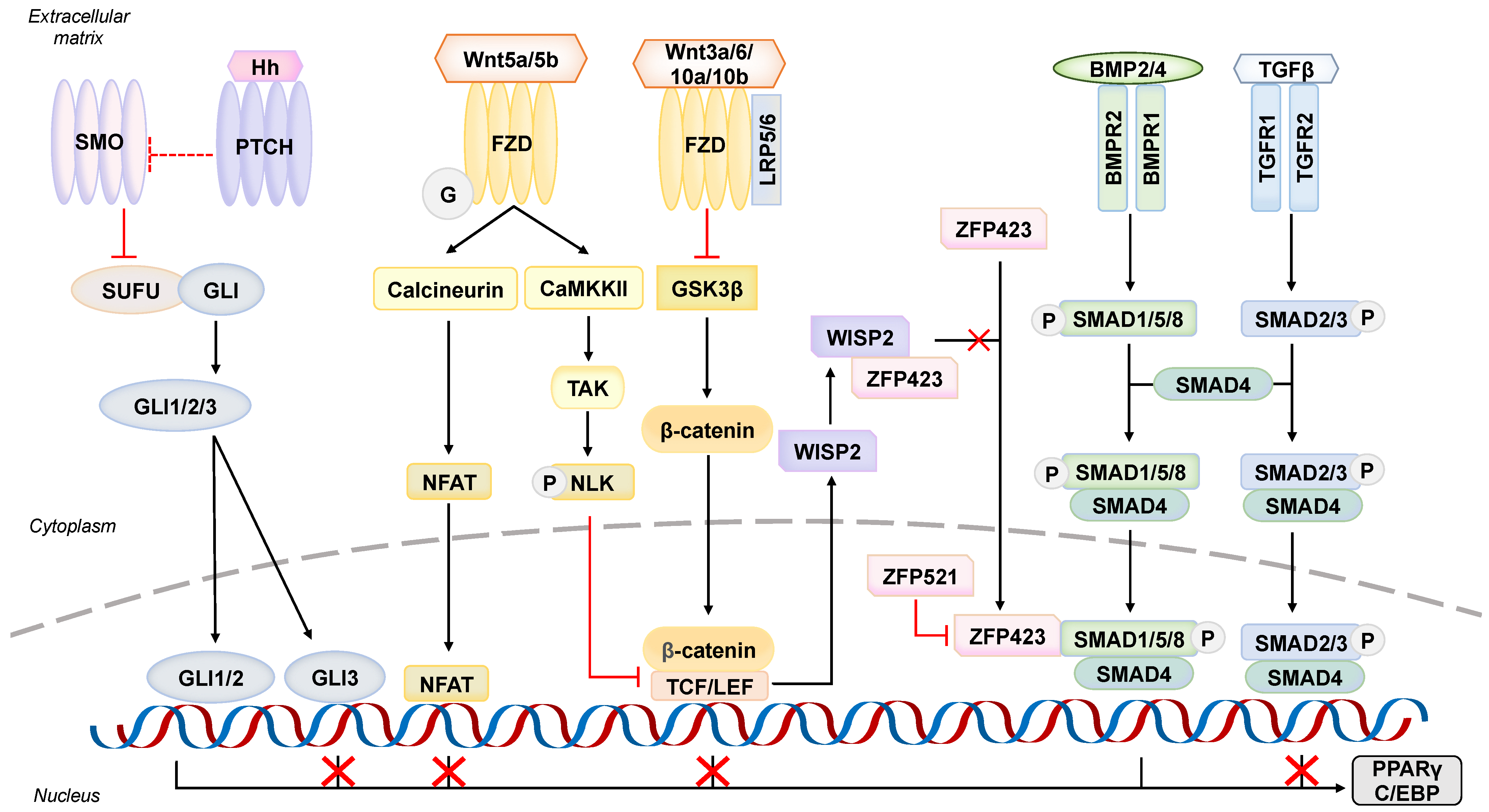
5.1. An Adipogenesis Initiator–Bone Morphogenic Protein Signaling Pathway
5.2. A Regulator towards Wrong Fate–Wingless (Wnt) Signaling Pathway
5.3. An Anti-Adipogenic Player at Primary Cilium–Hedgehog (Hh) Signaling Pathway
5.4. A Contact-Dependent Inhibition–Notch Signaling Pathway
5.5. A Stimulator who Declines over Time—Insulin-like Growth Factor Signaling Pathway
6. Epigenetic Drift

{kind=link}
{kind=link}
| Types | Proteins/RNA | Functions in ASC | Effect on Adipogenesis | Reference |
|---|---|---|---|---|
| DNA modification | ALKBH1 | Demethylates 6mA of Gys1 and Hif1a | + | [115] |
| TET3 | Erases DNA methylation at CEBP binding motif | + | [116] | |
| Histone modification | PRMT1 | Catalyzes H4R3me2a at gene Pparg | + | [117] |
| SIRT1 | Deacetylates gene Sfrp for activating Wnt signaling | - | [118] | |
| SIRT2 | Deacetylates gene Foxo1 for inhibiting Pparg expression | - | [118] | |
| RNA modification | IMP2/IGF2BP2 | Promotes degradation of Fzd8 mRNA | + | [114] |
| miRNA regulation | miR-27a-3p | Targets Pparg mRNA | - | [119] |
| miR-138 | Target the mRNA for lipoprotein lipase | - | [120] | |
| miR-424(322)/503 cluster | Targets γ-synuclein that regulates lipid metabolism | - | [121] | |
| miR-503-3p | Targets Wnt2 and Wnt7b | + | [122] | |
| lncRNA regulation | HOTAIR | Facilitates the actin reorganization and lipogenesis | + | [123] |
| LYPLAL1-AS1 | Modulates the stability of desmoplakin Inhibits Wnt/β-catenin pathway | + | [124] | |
| CircRNA regulation | CircFOXP1 | Indirectly regulates Wnt5a expression | + | [125] |
| circRNA_23525 | Targets miR-30a-3p | - | [126] |
7. Mechanical Stretching
8. Metabolic Reprogramming
9. Emerging Therapeutics
10. Perspectives
11. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| AFF | AF4/FMR2 family member | MCP | Monocyte chemoattractant protein |
| AKT | Protein kinase B | mTOR | Mammalian target of rapamycin |
| AMPK | AMP-activated protein kinase | mTORC1 | Mammalian target of rapamycin complex 1 |
| BDH-1 | D-beta-hydroxybutyrate dehydrogenase | NAD | Nicotinamide adenine dinucleotide |
| BMAL | Basic helix-loop-helix ARNT like | NOT | Negative on TATA-less |
| BMP | Bone morphogen protein | OCT | Octamer-binding transcription factor |
| BMPR | Bone morphogen protein receptor | PD1 | Programmed cell death protein 1 |
| C/EBP | CCAAT/enhancer binding protein | PDL1 | Programmed death ligand 1 |
| CaMKKII | Calcium/calmodulin-dependent protein kinase II | PDGFR | Platelet-derived growth factor receptor |
| CCR | C-C motif chemokine receptor | PER | Period |
| CD | Cluster of differentiation | PI3K | Phosphoinositide 3-kinase |
| CDK | Cyclin-dependent kinase | PKC | Protein kinase C |
| c-MYC | Cellular Myelocytomatosis | PPAR | Peroxisome proliferator-activated receptor |
| CSL | An acronym of CBF1/RBP-J, Su(H), Lag-1 | PRDM16 | PR-domain containing 16 |
| CXCL | (C-X-C) motif ligand | Rb | Retinoblastoma |
| DLK | Delta like non-canonical Notch ligand | ROCK | Rho-associated protein kinase |
| DLL | Delta like canonical Notch ligand | ROR | Receptor tyrosine kinase-like orphan receptor |
| DPP | Dipeptidyl-peptidase | ROS | Reactive oxygen species |
| E2F | E2 factor | RSPO | R-spondin |
| FAD | Factor for adipocyte differentiation | RUNX | Runt-related transcription factor |
| FOXO | Forkhead box O | RYK | Receptor tyrosine kinase-related tyrosine kinase |
| Fzd | Frizzled | SDC | Syndecan |
| GLI | Glioma-associated oncogene | SIRT | Sirtuin |
| GLP-1R | Glucagon-like peptide-1 receptor | SMAD | Suppressor of mothers against decapentaplegic |
| GSK | Glycogen synthase kinase | SNCG | Synuclein |
| HES | Hairy and enhancer of split | SNP | Single nucleotide polymorphism |
| Hh | Hedgehog | SOX | SRY-related HMG-box |
| HIF | Hypoxia inducible factor | TAZ | WW domain-containing transcription regulator 1 |
| ICAM | Intercellular adhesion molecule | TCF/LEF | T-cell factor/lymphoid enhancer factor |
| IL | Interleukin | TGF | Transforming growth factor |
| ILC2s | Type 2 innate lymphoid cells | TNF | Tumor necrosis factor |
| IMP | IGF-2 mRna-binding protein | UTR | Untranslated region |
| JAG | Jagged canonical Notch ligand | VAP | VAMP-associated protein |
| KLF | Krüppel-like factors | WISP | Wnt1-inducible-signaling pathway protein |
| LGR | Leucine-rich repeat-containing G-protein coupled receptor | Wnt | Wingless |
| LRP | LDL receptor-related protein | YAP | Yes-associated protein |
| LY6C | Lymphocyte antigen 6 complex | ZFP | Zinc-finger protein |
| MAPK | Mitogen-activated protein kinase | ZMAT | Zinc finger matrin-type protein |
References
- Cypess, A.M. Reassessing Human Adipose Tissue. N. Engl. J. Med. 2022, 386, 768–779. [Google Scholar] [CrossRef] [PubMed]
- Corvera, S. Cellular Heterogeneity in Adipose Tissues. Annu. Rev. Physiol. 2021, 83, 257–278. [Google Scholar] [CrossRef] [PubMed]
- Hudak, C.S.; Gulyaeva, O.; Wang, Y.; Park, S.M.; Lee, L.; Kang, C.; Sul, H.S. Pref-1 marks very early mesenchymal precursors required for adipose tissue development and expansion. Cell Rep. 2014, 8, 678–687. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vishvanath, L.; MacPherson, K.A.; Hepler, C.; Wang, Q.A.; Shao, M.; Spurgin, S.B.; Wang, M.Y.; Kusminski, C.M.; Morley, T.S.; Gupta, R.K. Pdgfrβ+ Mural Preadipocytes Contribute to Adipocyte Hyperplasia Induced by High-Fat-Diet Feeding and Prolonged Cold Exposure in Adult Mice. Cell. Metab. 2016, 23, 350–359. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Merrick, D.; Sakers, A.; Irgebay, Z.; Okada, C.; Calvert, C.; Morley, M.P.; Percec, I.; Seale, P. Identification of a mesenchymal progenitor cell hierarchy in adipose tissue. Science 2019, 364, eaav2501. [Google Scholar] [CrossRef]
- Ramalho-Santos, M.; Willenbring, H. On the origin of the term "stem cell". Cell Stem Cell 2007, 1, 35–38. [Google Scholar] [CrossRef] [Green Version]
- Caplan, A.I. Mesenchymal stem cells. J. Orthop. Res. 1991, 9, 641–650. [Google Scholar] [CrossRef]
- Reynolds, B.A.; Weiss, S. Generation of neurons and astrocytes from isolated cells of the adult mammalian central nervous system. Science 1992, 255, 1707–1710. [Google Scholar] [CrossRef] [Green Version]
- Zuk, P.A.; Zhu, M.; Mizuno, H.; Huang, J.; Futrell, J.W.; Katz, A.J.; Benhaim, P.; Lorenz, H.P.; Hedrick, M.H. Multilineage cells from human adipose tissue: Implications for cell-based therapies. Tissue Eng. 2001, 7, 211–228. [Google Scholar] [CrossRef] [Green Version]
- Zuk, P.A.; Zhu, M.; Ashjian, P.; De Ugarte, D.A.; Huang, J.I.; Mizuno, H.; Alfonso, Z.C.; Fraser, J.K.; Benhaim, P.; Hedrick, M.H. Human adipose tissue is a source of multipotent stem cells. Mol. Biol. Cell. 2002, 13, 4279–4295. [Google Scholar] [CrossRef]
- Halvorsen, Y.D.; Franklin, D.; Bond, A.L.; Hitt, D.C.; Auchter, C.; Boskey, A.L.; Paschalis, E.P.; Wilkison, W.O.; Gimble, J.M. Extracellular matrix mineralization and osteoblast gene expression by human adipose tissue-derived stromal cells. Tissue Eng. 2001, 7, 729–741. [Google Scholar] [CrossRef]
- Cawthorn, W.P.; Scheller, E.L.; MacDougald, O.A. Adipose tissue stem cells meet preadipocyte commitment: Going back to the future. J. Lipid. Res. 2012, 53, 227–246. [Google Scholar] [CrossRef] [Green Version]
- Spalding, K.L.; Arner, E.; Westermark, P.O.; Bernard, S.; Buchholz, B.A.; Bergmann, O.; Blomqvist, L.; Hoffstedt, J.; Naslund, E.; Britton, T.; et al. Dynamics of fat cell turnover in humans. Nature 2008, 453, 783–787. [Google Scholar] [CrossRef]
- Salvestrini, V.; Sell, C.; Lorenzini, A. Obesity May Accelerate the Aging Process. Front. Endocrinol. (Lausanne) 2019, 10, 266. [Google Scholar] [CrossRef] [Green Version]
- Gustafson, B.; Nerstedt, A.; Smith, U. Reduced subcutaneous adipogenesis in human hypertrophic obesity is linked to senescent precursor cells. Nat. Commun. 2019, 10, 2757. [Google Scholar] [CrossRef] [Green Version]
- Brunet, A.; Goodell, M.A.; Rando, T.A. Ageing and rejuvenation of tissue stem cells and their niches. Nat. Rev. Mol. Cell Biol. 2023, 24, 45–62. [Google Scholar] [CrossRef]
- Hammarstedt, A.; Gogg, S.; Hedjazifar, S.; Nerstedt, A.; Smith, U. Impaired Adipogenesis and Dysfunctional Adipose Tissue in Human Hypertrophic Obesity. Physiol. Rev. 2018, 98, 1911–1941. [Google Scholar] [CrossRef] [Green Version]
- Bourin, P.; Bunnell, B.A.; Casteilla, L.; Dominici, M.; Katz, A.J.; March, K.L.; Redl, H.; Rubin, J.P.; Yoshimura, K.; Gimble, J.M. Stromal cells from the adipose tissue-derived stromal vascular fraction and culture expanded adipose tissue-derived stromal/stem cells: A joint statement of the International Federation for Adipose Therapeutics and Science (IFATS) and the International Society for Cellular Therapy (ISCT). Cytotherapy 2013, 15, 641–648. [Google Scholar] [CrossRef] [Green Version]
- Sebo, Z.L.; Rodeheffer, M.S. Assembling the adipose organ: Adipocyte lineage segregation and adipogenesis in vivo. Development 2019, 146, dev172098. [Google Scholar] [CrossRef] [Green Version]
- Rosen, E.D.; Spiegelman, B.M. What we talk about when we talk about fat. Cell 2014, 156, 20–44. [Google Scholar] [CrossRef] [Green Version]
- Ikeda, K.; Maretich, P.; Kajimura, S. The Common and Distinct Features of Brown and Beige Adipocytes. Trends Endocrinol. Metab 2018, 29, 191–200. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ussar, S.; Lee, K.Y.; Dankel, S.N.; Boucher, J.; Haering, M.F.; Kleinridders, A.; Thomou, T.; Xue, R.; Macotela, Y.; Cypess, A.M.; et al. ASC-1, PAT2, and P2RX5 are cell surface markers for white, beige, and brown adipocytes. Sci. Transl. Med. 2014, 6, 247ra103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ong, W.K.; Tan, C.S.; Chan, K.L.; Goesantoso, G.G.; Chan, X.H.; Chan, E.; Yin, J.; Yeo, C.R.; Khoo, C.M.; So, J.B.; et al. Identification of specific cell-surface markers of adipose-derived stem cells from subcutaneous and visceral fat depots. Stem Cell Rep. 2014, 2, 171–179. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suga, H.; Matsumoto, D.; Eto, H.; Inoue, K.; Aoi, N.; Kato, H.; Araki, J.; Yoshimura, K. Functional implications of CD34 expression in human adipose-derived stem/progenitor cells. Stem Cells Dev. 2009, 18, 1201–1210. [Google Scholar] [CrossRef] [Green Version]
- Hatzmann, F.M.; Ejaz, A.; Wiegers, G.J.; Mandl, M.; Brucker, C.; Lechner, S.; Rauchenwald, T.; Zwierzina, M.; Baumgarten, S.; Wagner, S.; et al. Quiescence, Stemness and Adipogenic Differentiation Capacity in Human DLK1−/CD34+/CD24+ Adipose Stem/Progenitor Cells. Cells 2021, 10, 214. [Google Scholar] [CrossRef]
- Berry, R.; Rodeheffer, M.S. Characterization of the adipocyte cellular lineage in vivo. Nat. Cell Biol. 2013, 15, 302–308. [Google Scholar] [CrossRef]
- Rodeheffer, M.S.; Birsoy, K.; Friedman, J.M. Identification of white adipocyte progenitor cells in vivo. Cell 2008, 135, 240–249. [Google Scholar] [CrossRef] [Green Version]
- Ferrero, R.; Rainer, P.; Deplancke, B. Toward a Consensus View of Mammalian Adipocyte Stem and Progenitor Cell Heterogeneity. Trends Cell Biol 2020, 30, 937–950. [Google Scholar] [CrossRef]
- Nahmgoong, H.; Jeon, Y.G.; Park, E.S.; Choi, Y.H.; Han, S.M.; Park, J.; Ji, Y.; Sohn, J.H.; Han, J.S.; Kim, Y.Y.; et al. Distinct properties of adipose stem cell subpopulations determine fat depot-specific characteristics. Cell Metab. 2022, 34, 458–472 e456. [Google Scholar] [CrossRef]
- Marcelin, G.; Ferreira, A.; Liu, Y.; Atlan, M.; Aron-Wisnewsky, J.; Pelloux, V.; Botbol, Y.; Ambrosini, M.; Fradet, M.; Rouault, C.; et al. A PDGFRα-Mediated Switch toward CD9high Adipocyte Progenitors Controls Obesity-Induced Adipose Tissue Fibrosis. Cell Metab. 2017, 25, 673–685. [Google Scholar] [CrossRef] [Green Version]
- Joffin, N.; Paschoal, V.A.; Gliniak, C.M.; Crewe, C.; Elnwasany, A.; Szweda, L.I.; Zhang, Q.; Hepler, C.; Kusminski, C.M.; Gordillo, R.; et al. Mitochondrial metabolism is a key regulator of the fibro-inflammatory and adipogenic stromal subpopulations in white adipose tissue. Cell Stem Cell 2021, 28, 702–717 e708. [Google Scholar] [CrossRef]
- Zhang, Q.; Shan, B.; Guo, L.; Shao, M.; Vishvanath, L.; Elmquist, G.; Xu, L.; Gupta, R.K. Distinct functional properties of murine perinatal and adult adipose progenitor subpopulations. Nat. Metab. 2022, 4, 1055–1070. [Google Scholar] [CrossRef]
- Hepler, C.; Shan, B.; Zhang, Q.; Henry, G.H.; Shao, M.; Vishvanath, L.; Ghaben, A.L.; Mobley, A.B.; Strand, D.; Hon, G.C.; et al. Identification of functionally distinct fibro-inflammatory and adipogenic stromal subpopulations in visceral adipose tissue of adult mice. Elife 2018, 7, e39636. [Google Scholar] [CrossRef]
- Urban, N.; Cheung, T.H. Stem cell quiescence: The challenging path to activation. Development 2021, 148, dev165084. [Google Scholar] [CrossRef]
- Liu, L.; Michowski, W.; Kolodziejczyk, A.; Sicinski, P. The cell cycle in stem cell proliferation, pluripotency and differentiation. Nat. Cell. Biol. 2019, 21, 1060–1067. [Google Scholar] [CrossRef]
- Wang, L.; Jin, S.; Dai, P.; Zhang, T.; Shi, Y.; Ai, G.; Shao, X.; Xie, Y.; Xu, J.; Chen, Z.; et al. p57Kip2 is a master regulator of human adipose derived stem cell quiescence and senescence. Stem Cell Res. 2020, 44, 101759. [Google Scholar] [CrossRef]
- Rivera-Gonzalez, G.C.; Shook, B.A.; Andrae, J.; Holtrup, B.; Bollag, K.; Betsholtz, C.; Rodeheffer, M.S.; Horsley, V. Skin Adipocyte Stem Cell Self-Renewal Is Regulated by a PDGFA/AKT-Signaling Axis. Cell Stem Cell 2016, 19, 738–751. [Google Scholar] [CrossRef] [Green Version]
- Gong, A.H.; Wei, P.; Zhang, S.; Yao, J.; Yuan, Y.; Zhou, A.D.; Lang, F.F.; Heimberger, A.B.; Rao, G.; Huang, S. FoxM1 Drives a Feed-Forward STAT3-Activation Signaling Loop That Promotes the Self-Renewal and Tumorigenicity of Glioblastoma Stem-like Cells. Cancer Res. 2015, 75, 2337–2348. [Google Scholar] [CrossRef] [Green Version]
- Conley, S.M.; Hickson, L.J.; Kellogg, T.A.; McKenzie, T.; Heimbach, J.K.; Taner, T.; Tang, H.; Jordan, K.L.; Saadiq, I.M.; Woollard, J.R.; et al. Human Obesity Induces Dysfunction and Early Senescence in Adipose Tissue-Derived Mesenchymal Stromal/Stem Cells. Front. Cell Dev. Biol. 2020, 8, 197. [Google Scholar] [CrossRef] [Green Version]
- Zhu, X.Y.; Klomjit, N.; Conley, S.M.; Ostlie, M.M.; Jordan, K.L.; Lerman, A.; Lerman, L.O. Impaired immunomodulatory capacity in adipose tissue-derived mesenchymal stem/stromal cells isolated from obese patients. J. Cell. Mol. Med. 2021, 25, 9051–9059. [Google Scholar] [CrossRef]
- Ma, H.; Li, Y.N.; Song, L.; Liu, R.; Li, X.; Shang, Q.; Wang, Y.; Shao, C.; Shi, Y. Macrophages inhibit adipogenic differentiation of adipose tissue derived mesenchymal stem/stromal cells by producing pro-inflammatory cytokines. Cell Biosci. 2020, 10, 88. [Google Scholar] [CrossRef] [PubMed]
- Oestreich, A.K.; Collins, K.H.; Harasymowicz, N.S.; Wu, C.L.; Guilak, F. Is Obesity a Disease of Stem Cells? Cell Stem Cell 2020, 27, 15–18. [Google Scholar] [CrossRef] [PubMed]
- Rosen, E.D.; Sarraf, P.; Troy, A.E.; Bradwin, G.; Moore, K.; Milstone, D.S.; Spiegelman, B.M.; Mortensen, R.M. PPARγ is required for the differentiation of adipose tissue in vivo and in vitro. Mol. Cell. 1999, 4, 611–617. [Google Scholar] [CrossRef] [PubMed]
- Christy, R.J.; Yang, V.W.; Ntambi, J.M.; Geiman, D.E.; Landschulz, W.H.; Friedman, A.D.; Nakabeppu, Y.; Kelly, T.J.; Lane, M.D. Differentiation-induced gene expression in 3T3-L1 preadipocytes: CCAAT/enhancer binding protein interacts with and activates the promoters of two adipocyte-specific genes. Genes Dev. 1989, 3, 1323–1335. [Google Scholar] [CrossRef] [Green Version]
- Ghaben, A.L.; Scherer, P.E. Adipogenesis and metabolic health. Nat. Rev. Mol. Cell. Biol. 2019, 20, 242–258. [Google Scholar] [CrossRef]
- Huang, H.; Song, T.J.; Li, X.; Hu, L.; He, Q.; Liu, M.; Lane, M.D.; Tang, Q.Q. BMP signaling pathway is required for commitment of C3H10T1/2 pluripotent stem cells to the adipocyte lineage. Proc. Natl. Acad. Sci. USA 2009, 106, 12670–12675. [Google Scholar] [CrossRef] [Green Version]
- Qian, S.W.; Tang, Y.; Li, X.; Liu, Y.; Zhang, Y.Y.; Huang, H.Y.; Xue, R.D.; Yu, H.Y.; Guo, L.; Gao, H.D.; et al. BMP4-mediated brown fat-like changes in white adipose tissue alter glucose and energy homeostasis. Proc. Natl. Acad. Sci. USA 2013, 110, E798–E807. [Google Scholar] [CrossRef] [Green Version]
- Derynck, R.; Zhang, Y.E. Smad-dependent and Smad-independent pathways in TGF-β family signalling. Nature 2003, 425, 577–584. [Google Scholar] [CrossRef]
- Gupta, R.K.; Arany, Z.; Seale, P.; Mepani, R.J.; Ye, L.; Conroe, H.M.; Roby, Y.A.; Kulaga, H.; Reed, R.R.; Spiegelman, B.M. Transcriptional control of preadipocyte determination by Zfp423. Nature 2010, 464, 619–623. [Google Scholar] [CrossRef] [Green Version]
- Huang, H.Y.; Hu, L.L.; Song, T.J.; Li, X.; He, Q.; Sun, X.; Li, Y.M.; Lu, H.J.; Yang, P.Y.; Tang, Q.Q. Involvement of cytoskeleton-associated proteins in the commitment of C3H10T1/2 pluripotent stem cells to adipocyte lineage induced by BMP2/4. Mol. Cell. Proteom. 2011, 10, M110.002691. [Google Scholar] [CrossRef] [Green Version]
- Blazquez-Medela, A.M.; Jumabay, M.; Rajbhandari, P.; Sallam, T.; Guo, Y.; Yao, J.; Vergnes, L.; Reue, K.; Zhang, L.; Yao, Y.; et al. Noggin depletion in adipocytes promotes obesity in mice. Mol. Metab. 2019, 25, 50–63. [Google Scholar] [CrossRef]
- Gustafson, B.; Hammarstedt, A.; Hedjazifar, S.; Hoffmann, J.M.; Svensson, P.A.; Grimsby, J.; Rondinone, C.; Smith, U. BMP4 and BMP Antagonists Regulate Human White and Beige Adipogenesis. Diabetes 2015, 64, 1670–1681. [Google Scholar] [CrossRef] [Green Version]
- Bowers, R.R.; Kim, J.W.; Otto, T.C.; Lane, M.D. Stable stem cell commitment to the adipocyte lineage by inhibition of DNA methylation: Role of the BMP-4 gene. Proc. Natl. Acad. Sci. USA 2006, 103, 13022–13027. [Google Scholar] [CrossRef] [Green Version]
- Kawagishi-Hotta, M.; Hasegawa, S.; Igarashi, T.; Date, Y.; Ishii, Y.; Inoue, Y.; Hasebe, Y.; Yamada, T.; Arima, M.; Iwata, Y.; et al. Increase of gremlin 2 with age in human adipose-derived stromal/stem cells and its inhibitory effect on adipogenesis. Regen. Ther. 2019, 11, 324–330. [Google Scholar] [CrossRef]
- Hoffmann, J.M.; Grunberg, J.R.; Church, C.; Elias, I.; Palsdottir, V.; Jansson, J.O.; Bosch, F.; Hammarstedt, A.; Hedjazifar, S.; Smith, U. BMP4 Gene Therapy in Mature Mice Reduces BAT Activation but Protects from Obesity by Browning Subcutaneous Adipose Tissue. Cell Rep. 2017, 20, 1038–1049. [Google Scholar] [CrossRef] [Green Version]
- Modica, S.; Straub, L.G.; Balaz, M.; Sun, W.; Varga, L.; Stefanicka, P.; Profant, M.; Simon, E.; Neubauer, H.; Ukropcova, B.; et al. Bmp4 Promotes a Brown to White-like Adipocyte Shift. Cell Rep. 2016, 16, 2243–2258. [Google Scholar] [CrossRef] [Green Version]
- Erickson, G.R.; Gimble, J.M.; Franklin, D.M.; Rice, H.E.; Awad, H.; Guilak, F. Chondrogenic potential of adipose tissue-derived stromal cells in vitro and in vivo. Biochem. Biophys. Res. Commun. 2002, 290, 763–769. [Google Scholar] [CrossRef]
- Choy, L.; Derynck, R. Transforming growth factor-beta inhibits adipocyte differentiation by Smad3 interacting with CCAAT/enhancer-binding protein (C/EBP) and repressing C/EBP transactivation function. J. Biol. Chem. 2003, 278, 9609–9619. [Google Scholar] [CrossRef] [Green Version]
- Yadav, H.; Quijano, C.; Kamaraju, A.K.; Gavrilova, O.; Malek, R.; Chen, W.; Zerfas, P.; Zhigang, D.; Wright, E.C.; Stuelten, C.; et al. Protection from obesity and diabetes by blockade of TGF-β/Smad3 signaling. Cell. Metab. 2011, 14, 67–79. [Google Scholar] [CrossRef] [Green Version]
- MacDonald, B.T.; Tamai, K.; He, X. Wnt/beta-catenin signaling: Components, mechanisms, and diseases. Dev. Cell. 2009, 17, 9–26. [Google Scholar] [CrossRef] [Green Version]
- Ross, S.E.; Hemati, N.; Longo, K.A.; Bennett, C.N.; Lucas, P.C.; Erickson, R.L.; MacDougald, O.A. Inhibition of adipogenesis by Wnt signaling. Science 2000, 289, 950–953. [Google Scholar] [CrossRef] [PubMed]
- Bagchi, D.P.; MacDougald, O.A. Wnt Signaling: From Mesenchymal Cell Fate to Lipogenesis and Other Mature Adipocyte Functions. Diabetes 2021, 70, 1419–1430. [Google Scholar] [CrossRef] [PubMed]
- Gustafson, B.; Smith, U. The WNT inhibitor Dickkopf 1 and bone morphogenetic protein 4 rescue adipogenesis in hypertrophic obesity in humans. Diabetes 2012, 61, 1217–1224. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Niehrs, C. The complex world of WNT receptor signalling. Nat. Rev. Mol. Cell. Biol. 2012, 13, 767–779. [Google Scholar] [CrossRef] [PubMed]
- Bowers, R.R.; Lane, M.D. Wnt signaling and adipocyte lineage commitment. Cell Cycle 2008, 7, 1191–1196. [Google Scholar] [CrossRef] [Green Version]
- de Lau, W.; Peng, W.C.; Gros, P.; Clevers, H. The R-spondin/Lgr5/Rnf43 module: Regulator of Wnt signal strength. Genes Dev 2014, 28, 305–316. [Google Scholar] [CrossRef] [Green Version]
- Kang, S.; Bennett, C.N.; Gerin, I.; Rapp, L.A.; Hankenson, K.D.; Macdougald, O.A. Wnt signaling stimulates osteoblastogenesis of mesenchymal precursors by suppressing CCAAT/enhancer-binding protein alpha and peroxisome proliferator-activated receptor gamma. J. Biol. Chem. 2007, 282, 14515–14524. [Google Scholar] [CrossRef]
- Cawthorn, W.P.; Bree, A.J.; Yao, Y.; Du, B.; Hemati, N.; Martinez-Santibanez, G.; MacDougald, O.A. Wnt6, Wnt10a and Wnt10b inhibit adipogenesis and stimulate osteoblastogenesis through a β-catenin-dependent mechanism. Bone 2012, 50, 477–489. [Google Scholar] [CrossRef] [Green Version]
- Hammarstedt, A.; Hedjazifar, S.; Jenndahl, L.; Gogg, S.; Grunberg, J.; Gustafson, B.; Klimcakova, E.; Stich, V.; Langin, D.; Laakso, M.; et al. WISP2 regulates preadipocyte commitment and PPARγ activation by BMP4. Proc. Natl. Acad. Sci. USA 2013, 110, 2563–2568. [Google Scholar] [CrossRef] [Green Version]
- Li, H.X.; Luo, X.; Liu, R.X.; Yang, Y.J.; Yang, G.S. Roles of Wnt/β-catenin signaling in adipogenic differentiation potential of adipose-derived mesenchymal stem cells. Mol. Cell. Endocrinol. 2008, 291, 116–124. [Google Scholar] [CrossRef]
- Loh, N.Y.; Neville, M.J.; Marinou, K.; Hardcastle, S.A.; Fielding, B.A.; Duncan, E.L.; McCarthy, M.I.; Tobias, J.H.; Gregson, C.L.; Karpe, F.; et al. LRP5 regulates human body fat distribution by modulating adipose progenitor biology in a dose- and depot-specific fashion. Cell. Metab. 2015, 21, 262–273. [Google Scholar] [CrossRef] [Green Version]
- Dong, H.; Sun, W.; Shen, Y.; Balaz, M.; Balazova, L.; Ding, L.; Loffler, M.; Hamilton, B.; Kloting, N.; Bluher, M.; et al. Identification of a regulatory pathway inhibiting adipogenesis via RSPO2. Nat. Metab. 2022, 4, 90–105. [Google Scholar] [CrossRef]
- Loh, N.Y.; Minchin, J.E.N.; Pinnick, K.E.; Verma, M.; Todorcevic, M.; Denton, N.; Moustafa, J.E.; Kemp, J.P.; Gregson, C.L.; Evans, D.M.; et al. RSPO3 impacts body fat distribution and regulates adipose cell biology in vitro. Nat. Commun. 2020, 11, 2797. [Google Scholar] [CrossRef]
- Lee, G.J.; Kim, Y.J.; Park, B.; Yim, S.; Park, C.; Roh, H.; Moon, Y.; Seong, J.K.; Park, H. YAP-dependent Wnt5a induction in hypertrophic adipocytes restrains adiposity. Cell Death Dis. 2022, 13, 407. [Google Scholar] [CrossRef]
- Santos, A.; Bakker, A.D.; de Blieck-Hogervorst, J.M.; Klein-Nulend, J. WNT5A induces osteogenic differentiation of human adipose stem cells via rho-associated kinase ROCK. Cytotherapy 2010, 12, 924–932. [Google Scholar] [CrossRef]
- Catalan, V.; Gomez-Ambrosi, J.; Rodriguez, A.; Perez-Hernandez, A.I.; Gurbindo, J.; Ramirez, B.; Mendez-Gimenez, L.; Rotellar, F.; Valenti, V.; Moncada, R.; et al. Activation of noncanonical Wnt signaling through WNT5A in visceral adipose tissue of obese subjects is related to inflammation. J. Clin. Endocrinol. Metab. 2014, 99, E1407–E1417. [Google Scholar] [CrossRef] [Green Version]
- Hilgendorf, K.I.; Johnson, C.T.; Jackson, P.K. The primary cilium as a cellular receiver: Organizing ciliary GPCR signaling. Curr. Opin. Cell Biol. 2016, 39, 84–92. [Google Scholar] [CrossRef] [Green Version]
- Suh, J.M.; Gao, X.; McKay, J.; McKay, R.; Salo, Z.; Graff, J.M. Hedgehog signaling plays a conserved role in inhibiting fat formation. Cell Metab. 2006, 3, 25–34. [Google Scholar] [CrossRef] [Green Version]
- Briscoe, J.; Therond, P.P. The mechanisms of Hedgehog signalling and its roles in development and disease. Nat. Rev. Mol. Cell Biol. 2013, 14, 416–429. [Google Scholar] [CrossRef]
- James, A.W.; Leucht, P.; Levi, B.; Carre, A.L.; Xu, Y.; Helms, J.A.; Longaker, M.T. Sonic Hedgehog influences the balance of osteogenesis and adipogenesis in mouse adipose-derived stromal cells. Tissue Eng. Part. A 2010, 16, 2605–2616. [Google Scholar] [CrossRef] [Green Version]
- Dwyer, J.R.; Sever, N.; Carlson, M.; Nelson, S.F.; Beachy, P.A.; Parhami, F. Oxysterols are novel activators of the hedgehog signaling pathway in pluripotent mesenchymal cells. J. Biol. Chem. 2007, 282, 8959–8968. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fontaine, C.; Cousin, W.; Plaisant, M.; Dani, C.; Peraldi, P. Hedgehog signaling alters adipocyte maturation of human mesenchymal stem cells. Stem Cells 2008, 26, 1037–1046. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Long, F. Hedgehog signaling via Gli2 prevents obesity induced by high-fat diet in adult mice. Elife 2017, 6, e31649. [Google Scholar] [CrossRef] [PubMed]
- Yao, Q.; Liu, J.; Xiao, L.; Wang, N. Sonic hedgehog signaling instigates high-fat diet-induced insulin resistance by targeting PPARγ stability. J. Biol. Chem. 2019, 294, 3284–3293. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vaisse, C.; Reiter, J.F.; Berbari, N.F. Cilia and Obesity. Cold Spring Harb. Perspect. Biol. 2017, 9, a028217. [Google Scholar] [CrossRef] [Green Version]
- Dalbay, M.T.; Thorpe, S.D.; Connelly, J.T.; Chapple, J.P.; Knight, M.M. Adipogenic Differentiation of hMSCs is Mediated by Recruitment of IGF-1r Onto the Primary Cilium Associated With Cilia Elongation. Stem. Cells 2015, 33, 1952–1961. [Google Scholar] [CrossRef]
- Ritter, A.; Friemel, A.; Kreis, N.N.; Hoock, S.C.; Roth, S.; Kielland-Kaisen, U.; Bruggmann, D.; Solbach, C.; Louwen, F.; Yuan, J. Primary Cilia Are Dysfunctional in Obese Adipose-Derived Mesenchymal Stem Cells. Stem. Cell Rep. 2018, 10, 583–599. [Google Scholar] [CrossRef] [Green Version]
- Meshorer, E.; Gruenbaum, Y. Gone with the Wnt/Notch: Stem cells in laminopathies, progeria, and aging. J. Cell Biol. 2008, 181, 9–13. [Google Scholar] [CrossRef] [Green Version]
- Bi, P.; Kuang, S. Notch signaling as a novel regulator of metabolism. Trends Endocrinol. Metab. 2015, 26, 248–255. [Google Scholar] [CrossRef] [Green Version]
- Zhou, B.; Lin, W.; Long, Y.; Yang, Y.; Zhang, H.; Wu, K.; Chu, Q. Notch signaling pathway: Architecture, disease, and therapeutics. Signal. Transduct. Target Ther. 2022, 7, 95. [Google Scholar] [CrossRef]
- Ba, K.; Yang, X.; Wu, L.; Wei, X.; Fu, N.; Fu, Y.; Cai, X.; Yao, Y.; Ge, Y.; Lin, Y. Jagged-1-mediated activation of notch signalling induces adipogenesis of adipose-derived stem cells. Cell Prolif. 2012, 45, 538–544. [Google Scholar] [CrossRef]
- Osathanon, T.; Subbalekha, K.; Sastravaha, P.; Pavasant, P. Notch signalling inhibits the adipogenic differentiation of single-cell-derived mesenchymal stem cell clones isolated from human adipose tissue. Cell Biol. Int. 2012, 36, 1161–1170. [Google Scholar] [CrossRef]
- Ugarte, F.; Ryser, M.; Thieme, S.; Fierro, F.A.; Navratiel, K.; Bornhauser, M.; Brenner, S. Notch signaling enhances osteogenic differentiation while inhibiting adipogenesis in primary human bone marrow stromal cells. Exp. Hematol. 2009, 37, 867–875 e861. [Google Scholar] [CrossRef]
- Sandel, D.A.; Liu, M.; Ogbonnaya, N.; Newman, J.J. Notch3 is involved in adipogenesis of human adipose-derived stromal/stem cells. Biochimie 2018, 150, 31–36. [Google Scholar] [CrossRef]
- Gulyaeva, O.; Nguyen, H.; Sambeat, A.; Heydari, K.; Sul, H.S. Sox9-Meis1 Inactivation Is Required for Adipogenesis, Advancing Pref-1+ to PDGFRα+ Cells. Cell Rep. 2018, 25, 1002–1017 e1004. [Google Scholar] [CrossRef] [Green Version]
- Lei, T.; Bi, Y.; Gao, M.J.; Gao, S.M.; Zhou, L.L.; Zheng, H.L.; Chen, X.D. HES1 inhibits adipogenesis of porcine mesenchymal stem cells via transcriptional repression of FAD24. Domest. Anim. Endocrinol. 2013, 45, 28–32. [Google Scholar] [CrossRef]
- Boucher, J.M.; Ryzhova, L.; Harrington, A.; Davis-Knowlton, J.; Turner, J.E.; Cooper, E.; Maridas, D.; Ryzhov, S.; Rosen, C.J.; Vary, C.P.H.; et al. Pathological Conversion of Mouse Perivascular Adipose Tissue by Notch Activation. Arterioscler. Thromb. Vasc. Biol. 2020, 40, 2227–2243. [Google Scholar] [CrossRef]
- Boulet, N.; Briot, A.; Jargaud, V.; Esteve, D.; Remaury, A.; Belles, C.; Viana, P.; Fontaine, J.; Murphy, L.; Deon, C.; et al. Notch activation shifts the fate decision of senescent progenitors toward myofibrogenesis in human adipose tissue. Aging Cell 2023, e13776. [Google Scholar] [CrossRef]
- Yamaguchi, K.; Hayashi, M.; Uchida, Y.; Cheng, X.W.; Nakayama, T.; Matsushita, T.; Murohara, T.; Takeshita, K. Notch1 haploinsufficiency in mice accelerates adipogenesis. Sci. Rep. 2021, 11, 16761. [Google Scholar] [CrossRef]
- Wang, Y.; Sul, H.S. Pref-1 regulates mesenchymal cell commitment and differentiation through Sox9. Cell Metab. 2009, 9, 287–302. [Google Scholar] [CrossRef] [Green Version]
- Jensen, C.H.; Kosmina, R.; Ryden, M.; Baun, C.; Hvidsten, S.; Andersen, M.S.; Christensen, L.L.; Gastaldelli, A.; Marraccini, P.; Arner, P.; et al. The imprinted gene Delta like non-canonical notch ligand 1 (Dlk1) associates with obesity and triggers insulin resistance through inhibition of skeletal muscle glucose uptake. EBioMedicine 2019, 46, 368–380. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, L.; Yang, G.; Hagg, D.; Sun, G.; Ahn, J.M.; Jiang, N.; Ricupero, C.L.; Wu, J.; Rodhe, C.H.; Ascherman, J.A.; et al. IGF1 Promotes Adipogenesis by a Lineage Bias of Endogenous Adipose Stem/Progenitor Cells. Stem Cells 2015, 33, 2483–2495. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boucher, J.; Mori, M.A.; Lee, K.Y.; Smyth, G.; Liew, C.W.; Macotela, Y.; Rourk, M.; Bluher, M.; Russell, S.J.; Kahn, C.R. Impaired thermogenesis and adipose tissue development in mice with fat-specific disruption of insulin and IGF-1 signalling. Nat. Commun. 2012, 3, 902. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakae, J.; Kitamura, T.; Kitamura, Y.; Biggs, W.H., 3rd; Arden, K.C.; Accili, D. The forkhead transcription factor Foxo1 regulates adipocyte differentiation. Dev. Cell 2003, 4, 119–129. [Google Scholar] [CrossRef] [Green Version]
- Xu, J.; Liao, K. Protein kinase B/AKT 1 plays a pivotal role in insulin-like growth factor-1 receptor signaling induced 3T3-L1 adipocyte differentiation. J. Biol. Chem. 2004, 279, 35914–35922. [Google Scholar] [CrossRef] [Green Version]
- Wang, C.; Li, X.; Dang, H.; Liu, P.; Zhang, B.O.; Xu, F. Insulin-like growth factor 2 regulates the proliferation and differentiation of rat adipose-derived stromal cells via IGF-1R and IR. Cytotherapy 2019, 21, 619–630. [Google Scholar] [CrossRef] [PubMed]
- Boucher, J.; Tseng, Y.H.; Kahn, C.R. Insulin and insulin-like growth factor-1 receptors act as ligand-specific amplitude modulators of a common pathway regulating gene transcription. J. Biol. Chem. 2010, 285, 17235–17245. [Google Scholar] [CrossRef] [Green Version]
- Junnila, R.K.; List, E.O.; Berryman, D.E.; Murrey, J.W.; Kopchick, J.J. The GH/IGF-1 axis in ageing and longevity. Nat. Rev. Endocrinol. 2013, 9, 366–376. [Google Scholar] [CrossRef] [Green Version]
- Juiz-Valina, P.; Pena-Bello, L.; Cordido, M.; Outeirino-Blanco, E.; Pertega, S.; Varela-Rodriguez, B.; Garcia-Brao, M.J.; Mena, E.; Sangiao-Alvarellos, S.; Cordido, F. Altered GH-IGF-1 Axis in Severe Obese Subjects is Reversed after Bariatric Surgery-Induced Weight Loss and Related with Low-Grade Chronic Inflammation. J. Clin. Med. 2020, 9, 2614. [Google Scholar] [CrossRef]
- Kerr, A.G.; Wang, Z.; Wang, N.; Kwok, K.H.M.; Jalkanen, J.; Ludzki, A.; Lecoutre, S.; Langin, D.; Bergo, M.O.; Dahlman, I.; et al. The long noncoding RNA ADIPINT regulates human adipocyte metabolism via pyruvate carboxylase. Nat. Commun. 2022, 13, 2958. [Google Scholar] [CrossRef]
- Spinelli, R.; Florese, P.; Parrillo, L.; Zatterale, F.; Longo, M.; D’Esposito, V.; Desiderio, A.; Nerstedt, A.; Gustafson, B.; Formisano, P.; et al. ZMAT3 hypomethylation contributes to early senescence of preadipocytes from healthy first-degree relatives of type 2 diabetics. Aging Cell 2022, 21, e13557. [Google Scholar] [CrossRef] [PubMed]
- Dai, N. The Diverse Functions of IMP2/IGF2BP2 in Metabolism. Trends Endocrinol. Metab 2020, 31, 670–679. [Google Scholar] [CrossRef] [PubMed]
- Zeggini, E.; Weedon, M.N.; Lindgren, C.M.; Frayling, T.M.; Elliott, K.S.; Lango, H.; Timpson, N.J.; Perry, J.R.; Rayner, N.W.; Freathy, R.M.; et al. Replication of genome-wide association signals in UK samples reveals risk loci for type 2 diabetes. Science 2007, 316, 1336–1341. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Regue, L.; Wang, W.; Ji, F.; Avruch, J.; Wang, H.; Dai, N. Human T2D-Associated Gene IMP2/IGF2BP2 Promotes the Commitment of Mesenchymal Stem Cells Into Adipogenic Lineage. Diabetes 2023, 72, 33–44. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Chen, Y.; Wang, Y.; Jiang, S.; Lin, W.; Wu, Y.; Li, Q.; Guo, Y.; Liu, W.; Yuan, Q. DNA demethylase ALKBH1 promotes adipogenic differentiation via regulation of HIF-1 signaling. J. Biol. Chem. 2022, 298, 101499. [Google Scholar] [CrossRef]
- Park, J.; Lee, D.H.; Ham, S.; Oh, J.; Noh, J.R.; Lee, Y.K.; Park, Y.J.; Lee, G.; Han, S.M.; Han, J.S.; et al. Targeted erasure of DNA methylation by TET3 drives adipogenic reprogramming and differentiation. Nat. Metab. 2022, 4, 918–931. [Google Scholar] [CrossRef]
- Zhu, Q.; Wang, D.; Liang, F.; Tong, X.; Liang, Z.; Wang, X.; Chen, Y.; Mo, D. Protein arginine methyltransferase PRMT1 promotes adipogenesis by modulating transcription factors C/EBPβ and PPARγ. J. Biol. Chem. 2022, 298, 102309. [Google Scholar] [CrossRef]
- Perrini, S.; Porro, S.; Nigro, P.; Cignarelli, A.; Caccioppoli, C.; Genchi, V.A.; Martines, G.; De Fazio, M.; Capuano, P.; Natalicchio, A.; et al. Reduced SIRT1 and SIRT2 expression promotes adipogenesis of human visceral adipose stem cells and associates with accumulation of visceral fat in human obesity. Int. J. Obes. (Lond.) 2020, 44, 307–319. [Google Scholar] [CrossRef]
- Wu, H.; Pula, T.; Tews, D.; Amri, E.Z.; Debatin, K.M.; Wabitsch, M.; Fischer-Posovszky, P.; Roos, J. microRNA-27a-3p but Not -5p Is a Crucial Mediator of Human Adipogenesis. Cells 2021, 10, 3205. [Google Scholar] [CrossRef]
- Wang, Y.; Lin, L.; Huang, Y.; Sun, J.; Wang, X.; Wang, P. MicroRNA-138 Suppresses Adipogenic Differentiation in Human Adipose Tissue-Derived Mesenchymal Stem Cells by Targeting Lipoprotein Lipase. Yonsei Med. J. 2019, 60, 1187–1194. [Google Scholar] [CrossRef]
- Rodriguez-Barrueco, R.; Latorre, J.; Devis-Jauregui, L.; Lluch, A.; Bonifaci, N.; Llobet, F.J.; Olivan, M.; Coll-Iglesias, L.; Gassner, K.; Davis, M.L.; et al. A microRNA Cluster Controls Fat Cell Differentiation and Adipose Tissue Expansion By Regulating SNCG. Adv. Sci. (Weinh) 2022, 9, e2104759. [Google Scholar] [CrossRef]
- Luo, Y.; Ding, X.; Ji, H.; Li, M.; Song, H.; Li, S.; Wang, C.; Wu, H.; Du, H. MicroRNA-503-3p affects osteogenic differentiation of human adipose-derived stem cells by regulation of Wnt2 and Wnt7b under cyclic strain. Stem Cell Res. Ther. 2020, 11, 318. [Google Scholar] [CrossRef]
- Kuo, F.C.; Huang, Y.C.; Yen, M.R.; Lee, C.H.; Hsu, K.F.; Yang, H.Y.; Wu, L.W.; Lu, C.H.; Hsu, Y.J.; Chen, P.Y. Aberrant overexpression of HOTAIR inhibits abdominal adipogenesis through remodelling of genome-wide DNA methylation and transcription. Mol. Metab. 2022, 60, 101473. [Google Scholar] [CrossRef]
- Yang, Y.; Fan, J.; Xu, H.; Fan, L.; Deng, L.; Li, J.; Li, D.; Li, H.; Zhang, F.; Zhao, R.C. Long noncoding RNA LYPLAL1-AS1 regulates adipogenic differentiation of human mesenchymal stem cells by targeting desmoplakin and inhibiting the Wnt/beta-catenin pathway. Cell Death Discov. 2021, 7, 105. [Google Scholar] [CrossRef]
- Cherubini, A.; Barilani, M.; Rossi, R.L.; Jalal, M.M.K.; Rusconi, F.; Buono, G.; Ragni, E.; Cantarella, G.; Simpson, H.; Peault, B.; et al. FOXP1 circular RNA sustains mesenchymal stem cell identity via microRNA inhibition. Nucleic Acids Res. 2019, 47, 5325–5340. [Google Scholar] [CrossRef] [Green Version]
- Guo, Z.; Zhao, L.; Ji, S.; Long, T.; Huang, Y.; Ju, R.; Tang, W.; Tian, W.; Long, J. CircRNA-23525 regulates osteogenic differentiation of adipose-derived mesenchymal stem cells via miR-30a-3p. Cell Tissue Res. 2021, 383, 795–807. [Google Scholar] [CrossRef]
- Bannister, A.J.; Kouzarides, T. Regulation of chromatin by histone modifications. Cell Res. 2011, 21, 381–395. [Google Scholar] [CrossRef]
- Morris, K.V.; Mattick, J.S. The rise of regulatory RNA. Nat. Rev. Genet. 2014, 15, 423–437. [Google Scholar] [CrossRef] [Green Version]
- Pope, B.D.; Warren, C.R.; Parker, K.K.; Cowan, C.A. Microenvironmental Control of Adipocyte Fate and Function. Trends Cell Biol. 2016, 26, 745–755. [Google Scholar] [CrossRef]
- Kechagia, J.Z.; Ivaska, J.; Roca-Cusachs, P. Integrins as biomechanical sensors of the microenvironment. Nat. Rev. Mol. Cell Biol. 2019, 20, 457–473. [Google Scholar] [CrossRef]
- McBeath, R.; Pirone, D.M.; Nelson, C.M.; Bhadriraju, K.; Chen, C.S. Cell shape, cytoskeletal tension, and RhoA regulate stem cell lineage commitment. Dev. Cell 2004, 6, 483–495. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hyvari, L.; Ojansivu, M.; Juntunen, M.; Kartasalo, K.; Miettinen, S.; Vanhatupa, S. Focal Adhesion Kinase and ROCK Signaling Are Switch-Like Regulators of Human Adipose Stem Cell Differentiation towards Osteogenic and Adipogenic Lineages. Stem Cells Int. 2018, 2018, 2190657. [Google Scholar] [CrossRef] [PubMed]
- Panciera, T.; Azzolin, L.; Cordenonsi, M.; Piccolo, S. Mechanobiology of YAP and TAZ in physiology and disease. Nat. Rev. Mol. Cell Biol 2017, 18, 758–770. [Google Scholar] [CrossRef] [PubMed]
- Hong, J.H.; Hwang, E.S.; McManus, M.T.; Amsterdam, A.; Tian, Y.; Kalmukova, R.; Mueller, E.; Benjamin, T.; Spiegelman, B.M.; Sharp, P.A.; et al. TAZ, a transcriptional modulator of mesenchymal stem cell differentiation. Science 2005, 309, 1074–1078. [Google Scholar] [CrossRef] [Green Version]
- Yanes, O.; Clark, J.; Wong, D.M.; Patti, G.J.; Sanchez-Ruiz, A.; Benton, H.P.; Trauger, S.A.; Desponts, C.; Ding, S.; Siuzdak, G. Metabolic oxidation regulates embryonic stem cell differentiation. Nat. Chem. Biol. 2010, 6, 411–417. [Google Scholar] [CrossRef] [Green Version]
- Tatapudy, S.; Aloisio, F.; Barber, D.; Nystul, T. Cell fate decisions: Emerging roles for metabolic signals and cell morphology. EMBO Rep. 2017, 18, 2105–2118. [Google Scholar] [CrossRef]
- Tormos, K.V.; Anso, E.; Hamanaka, R.B.; Eisenbart, J.; Joseph, J.; Kalyanaraman, B.; Chandel, N.S. Mitochondrial complex III ROS regulate adipocyte differentiation. Cell Metab. 2011, 14, 537–544. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.H.; Kim, S.H.; Song, S.Y.; Kim, W.S.; Song, S.U.; Yi, T.; Jeon, M.S.; Chung, H.M.; Xia, Y.; Sung, J.H. Hypoxia induces adipocyte differentiation of adipose-derived stem cells by triggering reactive oxygen species generation. Cell Biol. Int. 2014, 38, 32–40. [Google Scholar] [CrossRef]
- Park, H.S.; Kim, J.H.; Sun, B.K.; Song, S.U.; Suh, W.; Sung, J.H. Hypoxia induces glucose uptake and metabolism of adipose-derived stem cells. Mol. Med. Rep. 2016, 14, 4706–4714. [Google Scholar] [CrossRef] [Green Version]
- Sanchez-Ramirez, E.; Ung, T.P.L.; Alarcon Del Carmen, A.; del Toro-Rios, X.; Fajardo-Orduna, G.R.; Noriega, L.G.; Cortes-Morales, V.A.; Tovar, A.R.; Montesinos, J.J.; Orozco-Solis, R.; et al. Coordinated metabolic transitions and gene expression by NAD+ during adipogenesis. J. Cell Biol. 2022, 221, e202111137. [Google Scholar] [CrossRef]
- Tian, Q.; Zhao, J.; Yang, Q.; Wang, B.; Deavila, J.M.; Zhu, M.J.; Du, M. Dietary alpha-ketoglutarate promotes beige adipogenesis and prevents obesity in middle-aged mice. Aging Cell 2020, 19, e13059. [Google Scholar] [CrossRef]
- Wang, W.; Ishibashi, J.; Trefely, S.; Shao, M.; Cowan, A.J.; Sakers, A.; Lim, H.W.; O’Connor, S.; Doan, M.T.; Cohen, P.; et al. A PRDM16-Driven Metabolic Signal from Adipocytes Regulates Precursor Cell Fate. Cell Metab. 2019, 30, 174–189 e175. [Google Scholar] [CrossRef]
- Muller, T.D.; Bluher, M.; Tschop, M.H.; DiMarchi, R.D. Anti-obesity drug discovery: Advances and challenges. Nat. Rev. Drug. Discov. 2022, 21, 201–223. [Google Scholar] [CrossRef]
- Cheang, W.S.; Tian, X.Y.; Wong, W.T.; Lau, C.W.; Lee, S.S.; Chen, Z.Y.; Yao, X.; Wang, N.; Huang, Y. Metformin protects endothelial function in diet-induced obese mice by inhibition of endoplasmic reticulum stress through 5′ adenosine monophosphate-activated protein kinase-peroxisome proliferator-activated receptor delta pathway. Arterioscler. Thromb. Vasc. Biol. 2014, 34, 830–836. [Google Scholar] [CrossRef] [Green Version]
- Chinnapaka, S.; Yang, K.S.; Flowers, Q.; Faisal, M.; Nerone, W.V.; Rubin, J.P.; Ejaz, A. Metformin Improves Stemness of Human Adipose-Derived Stem Cells by Downmodulation of Mechanistic Target of Rapamycin (mTOR) and Extracellular Signal-Regulated Kinase (ERK) Signaling. Biomedicines 2021, 9, 1782. [Google Scholar] [CrossRef]
- Tang, W.; Zeve, D.; Seo, J.; Jo, A.Y.; Graff, J.M. Thiazolidinediones regulate adipose lineage dynamics. Cell Metab. 2011, 14, 116–122. [Google Scholar] [CrossRef] [Green Version]
- White, U.; Fitch, M.D.; Beyl, R.A.; Hellerstein, M.K.; Ravussin, E. Adipose depot-specific effects of 16 weeks of pioglitazone on in vivo adipogenesis in women with obesity: A randomised controlled trial. Diabetologia 2021, 64, 159–167. [Google Scholar] [CrossRef]
- Hoffmann, J.M.; Grunberg, J.R.; Hammarstedt, A.; Kroon, T.; Greiner, T.U.; Maurer, S.; Elias, I.; Palsdottir, V.; Bosch, F.; Boucher, J.; et al. BMP4 gene therapy enhances insulin sensitivity but not adipose tissue browning in obese mice. Mol. Metab. 2020, 32, 15–26. [Google Scholar] [CrossRef]
- Casana, E.; Jimenez, V.; Sacristan, V.; Munoz, S.; Jambrina, C.; Rodo, J.; Garcia, M.; Mallol, C.; Leon, X.; Franckhauser, S.; et al. BMP7 overexpression in adipose tissue induces white adipogenesis and improves insulin sensitivity in ob/ob mice. Int. J. Obes. (Lond.) 2021, 45, 449–460. [Google Scholar] [CrossRef]
- Kim, S.; Choe, S.; Lee, D.K. BMP-9 enhances fibroblast growth factor 21 expression and suppresses obesity. Biochim. Biophys. Acta 2016, 1862, 1237–1246. [Google Scholar] [CrossRef]
- Gutierrez-Cuevas, J.; Sandoval-Rodriguez, A.; Monroy-Ramirez, H.C.; Vazquez-Del Mercado, M.; Santos-Garcia, A.; Armendariz-Borunda, J. Prolonged-release pirfenidone prevents obesity-induced cardiac steatosis and fibrosis in a mouse NASH model. Cardiovasc. Drugs Ther. 2021, 35, 927–938. [Google Scholar] [CrossRef] [PubMed]
- Ouchi, N.; Higuchi, A.; Ohashi, K.; Oshima, Y.; Gokce, N.; Shibata, R.; Akasaki, Y.; Shimono, A.; Walsh, K. Sfrp5 is an anti-inflammatory adipokine that modulates metabolic dysfunction in obesity. Science 2010, 329, 454–457. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bi, P.; Shan, T.; Liu, W.; Yue, F.; Yang, X.; Liang, X.R.; Wang, J.; Li, J.; Carlesso, N.; Liu, X.; et al. Inhibition of Notch signaling promotes browning of white adipose tissue and ameliorates obesity. Nat. Med. 2014, 20, 911–918. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Noda, K.; Nakajima, S.; Godo, S.; Saito, H.; Ikeda, S.; Shimizu, T.; Enkhjargal, B.; Fukumoto, Y.; Tsukita, S.; Yamada, T.; et al. Rho-kinase inhibition ameliorates metabolic disorders through activation of AMPK pathway in mice. PLoS ONE 2014, 9, e110446. [Google Scholar] [CrossRef]
- Palmer, A.K.; Xu, M.; Zhu, Y.; Pirtskhalava, T.; Weivoda, M.M.; Hachfeld, C.M.; Prata, L.G.; van Dijk, T.H.; Verkade, E.; Casaclang-Verzosa, G.; et al. Targeting senescent cells alleviates obesity-induced metabolic dysfunction. Aging Cell 2019, 18, e12950. [Google Scholar] [CrossRef]
- Zhang, L.; Pitcher, L.E.; Prahalad, V.; Niedernhofer, L.J.; Robbins, P.D. Targeting cellular senescence with senotherapeutics: Senolytics and senomorphics. FEBS J. 2022. [Google Scholar] [CrossRef]
- Aggarwal, A.; Costa, M.J.; Rivero-Gutierrez, B.; Ji, L.; Morgan, S.L.; Feldman, B.J. The Circadian Clock Regulates Adipogenesis by a Per3 Crosstalk Pathway to Klf15. Cell Rep. 2017, 21, 2367–2375. [Google Scholar] [CrossRef] [Green Version]
- Maury, E.; Navez, B.; Brichard, S.M. Circadian clock dysfunction in human omental fat links obesity to metabolic inflammation. Nat. Commun. 2021, 12, 2388. [Google Scholar] [CrossRef]
- Zhang, Y.; Sowers, J.R.; Ren, J. Targeting autophagy in obesity: From pathophysiology to management. Nat. Rev. Endocrinol. 2018, 14, 356–376. [Google Scholar] [CrossRef]
- Chen, Y.; Li, Q.; Liu, Y.; Chen, X.; Jiang, S.; Lin, W.; Zhang, Y.; Liu, R.; Shao, B.; Chen, C.; et al. AFF4 regulates cellular adipogenic differentiation via targeting autophagy. PLoS Genet 2022, 18, e1010425. [Google Scholar] [CrossRef]
- Kaushik, S.; Juste, Y.R.; Lindenau, K.; Dong, S.; Macho-Gonzalez, A.; Santiago-Fernandez, O.; McCabe, M.; Singh, R.; Gavathiotis, E.; Cuervo, A.M. Chaperone-mediated autophagy regulates adipocyte differentiation. Sci. Adv. 2022, 8, eabq2733. [Google Scholar] [CrossRef]
- Mahlakoiv, T.; Flamar, A.L.; Johnston, L.K.; Moriyama, S.; Putzel, G.G.; Bryce, P.J.; Artis, D. Stromal cells maintain immune cell homeostasis in adipose tissue via production of interleukin-33. Sci. Immunol. 2019, 4, aax0416. [Google Scholar] [CrossRef]
- Zhou, K.; Guo, S.; Tong, S.; Sun, Q.; Li, F.; Zhang, X.; Qiao, Y.; Liang, G. Immunosuppression of Human Adipose-Derived Stem Cells on T Cell Subsets via the Reduction of NF-kappaB Activation Mediated by PD-L1/PD-1 and Gal-9/TIM-3 Pathways. Stem Cells Dev. 2018, 27, 1191–1202. [Google Scholar] [CrossRef]
- Johnson, D.B.; Nebhan, C.A.; Moslehi, J.J.; Balko, J.M. Immune-checkpoint inhibitors: Long-term implications of toxicity. Nat. Rev. Clin. Oncol. 2022, 19, 254–267. [Google Scholar] [CrossRef]
- Chan, J.S.K.; Lakhani, I.; Lee, T.T.L.; Chou, O.H.I.; Lee, Y.H.A.; Cheung, Y.M.; Yeung, H.W.; Tang, P.; Ng, K.; Dee, E.C.; et al. Cardiovascular Outcomes and Hospitalizations in Asian Patients Receiving Immune Checkpoint Inhibitors: A Population-based Study. Curr. Probl. Cardiol. 2023, 48, 101380. [Google Scholar] [CrossRef]
- Porsche, C.E.; Delproposto, J.B.; Geletka, L.; O’Rourke, R.; Lumeng, C.N. Obesity results in adipose tissue T cell exhaustion. JCI Insight 2021, 6, e139793. [Google Scholar] [CrossRef]
- Pushpakom, S.; Iorio, F.; Eyers, P.A.; Escott, K.J.; Hopper, S.; Wells, A.; Doig, A.; Guilliams, T.; Latimer, J.; McNamee, C.; et al. Drug repurposing: Progress, challenges and recommendations. Nat. Rev. Drug Discov. 2019, 18, 41–58. [Google Scholar] [CrossRef]


| White Adipocyte | Beige Adipocyte | Brown Adipocyte | ||
|---|---|---|---|---|
| Development origin | Dermomyotome in somite and lateral plate mesoderm (except for craniofacial adipocytes as ectodermal descendant) | |||
| Embryogenic progenitor | PDGFRa + CD29 + CD44 + | (1) MYH11, ACTA2 (2) Mature white adipocyte | EN1+ PAX7+ MYF5+ | |
| Lineage commitment during embryogenesis | FGF10 | (1) Selective expression of BMP7 (2) Unclear | Selective expression of PRDM16 and BMP7 | |
| Predominant depots | Human | Omental (visceral) Gluteofemoral (subcutaneous) | Within subcutaneous WAT | Supraclavicular |
| Rodent | Gonadal (visceral) Inguinal (subcutaneous) | Interscapular | ||
| Phenotypes | Single but large lipid droplet Few mitochondria | Multiple but small lipid droplets Abundant mitochondria | ||
| Molecular patterns | ASC1+ UCP1low HOXC9+ | PRDM16+ UCP1high PGC1α+ | CD40+ CD142+ UCP1high | |
| Major Functions | Energy storage Endocrine function | Thermogenesis Clearance of ectopic lipid | Thermogenesis Anti-inflammation | |
| As a secretory organ | Adiponectin, leptin, adipsin | Prostaglandins, 12,13-diHOME, FGF21, NRG4, IL6 | ||
| Changes upon obesity | Hypertrophy Cellular senescence | Whitening (transdifferentiation) | Inflammation Oxidative damage | |
| Dysfunction in | Treatment | Strategy | Reference |
|---|---|---|---|
| BMP signaling | AAV-BMP4 | Ligand overexpression | [148] |
| AAV-BMP7 | Ligand overexpression | [149] | |
| Recombinant BMP9 | Reinforces stimuli to BMPR | [150] | |
| TGFβ signaling | Anti-TGFβ | Blocks TGFβ activity | [59] |
| Pirfenidone | Inhibits TGFβ production | [151] | |
| Wnt signaling | AAV-SFRP5 | Inhibits the Wnt ligands | [152] |
| Hh signaling | Vismodegib | Inhibits SMO activity | [84] |
| Notch signaling | Dibenzazepine | Inhibits γ-secretase | [153] |
| Anti-DLL4 | Blocks DLL4 activity | [92] | |
| ROCK signaling | Fasudil | Inhibits ROCK | [154] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cheung, Y.-M.; Chook, C.-Y.-B.; Yeung, H.-W.; Leung, F.-P.; Wong, W.-T. A Wrong Fate Decision in Adipose Stem Cells upon Obesity. Cells 2023, 12, 662. https://doi.org/10.3390/cells12040662
Cheung Y-M, Chook C-Y-B, Yeung H-W, Leung F-P, Wong W-T. A Wrong Fate Decision in Adipose Stem Cells upon Obesity. Cells. 2023; 12(4):662. https://doi.org/10.3390/cells12040662
Chicago/Turabian StyleCheung, Yiu-Ming, Chui-Yiu-Bamboo Chook, Hoi-Wa Yeung, Fung-Ping Leung, and Wing-Tak Wong. 2023. "A Wrong Fate Decision in Adipose Stem Cells upon Obesity" Cells 12, no. 4: 662. https://doi.org/10.3390/cells12040662
APA StyleCheung, Y. -M., Chook, C. -Y. -B., Yeung, H. -W., Leung, F. -P., & Wong, W. -T. (2023). A Wrong Fate Decision in Adipose Stem Cells upon Obesity. Cells, 12(4), 662. https://doi.org/10.3390/cells12040662