
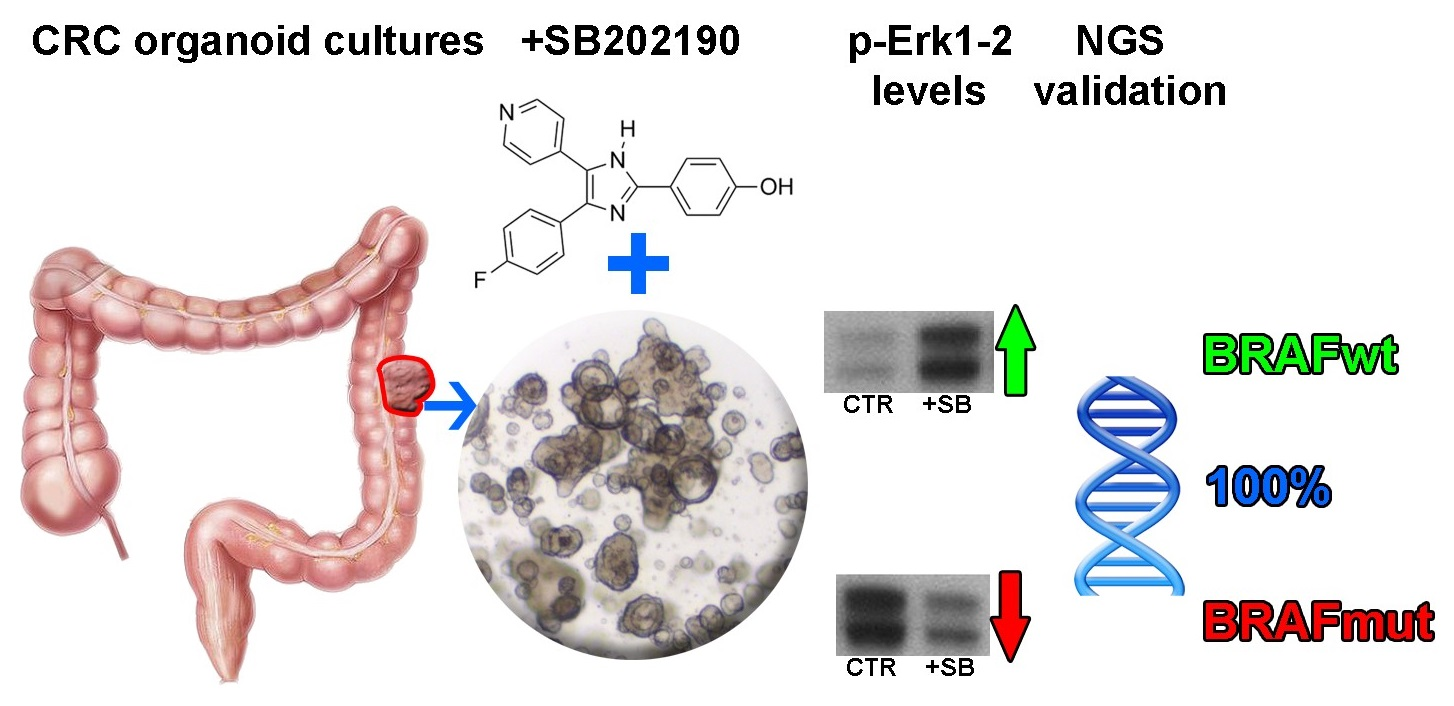
SB202190 Predicts BRAF-Activating Mutations in Primary Colorectal Cancer Organoids via Erk1-2 Modulation

 , , ,
, , ,  and
and 
Abstract
:
1. Introduction
2. Materials and Methods
3. Results
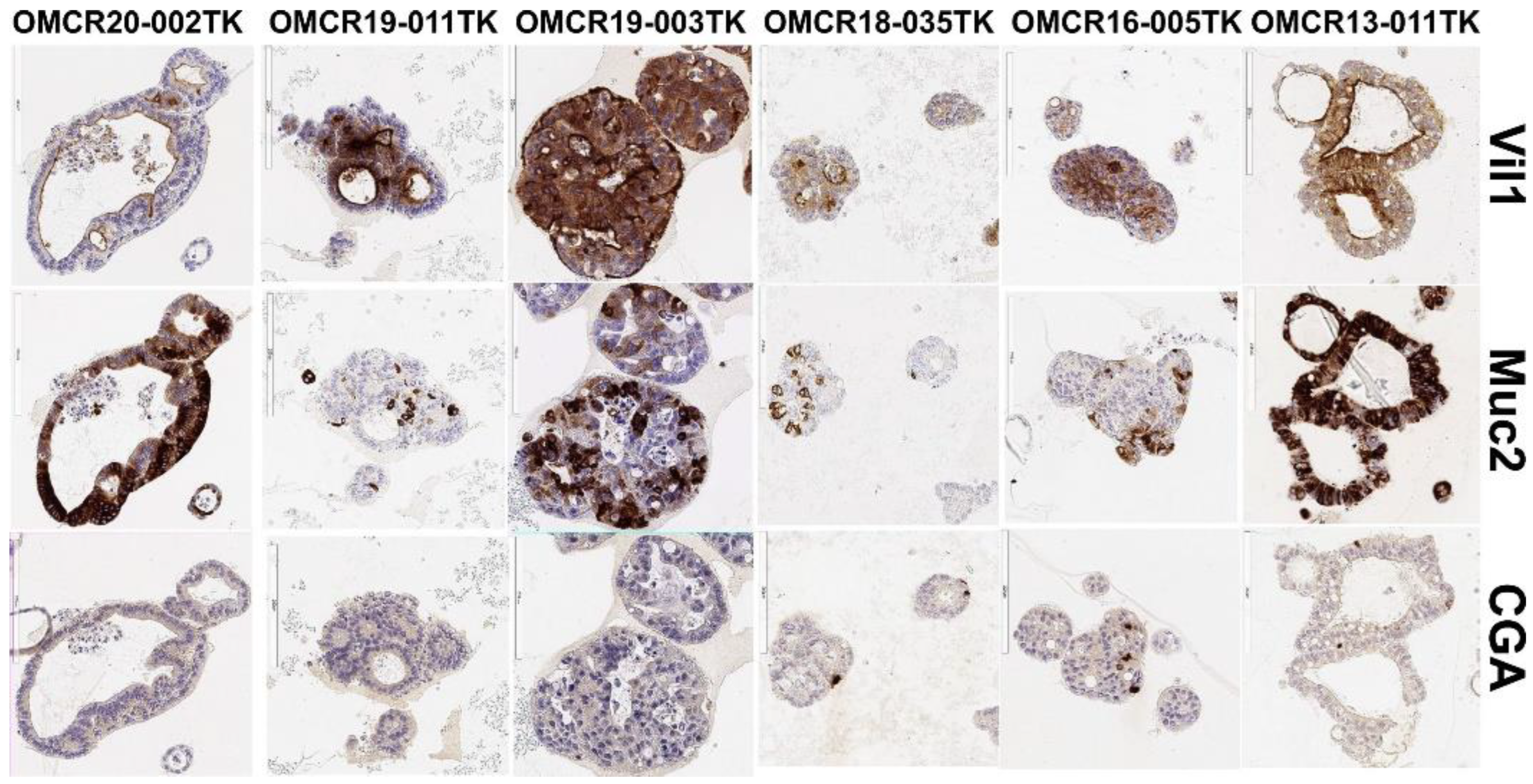
3.1. Organoid Primary Culture Establishment
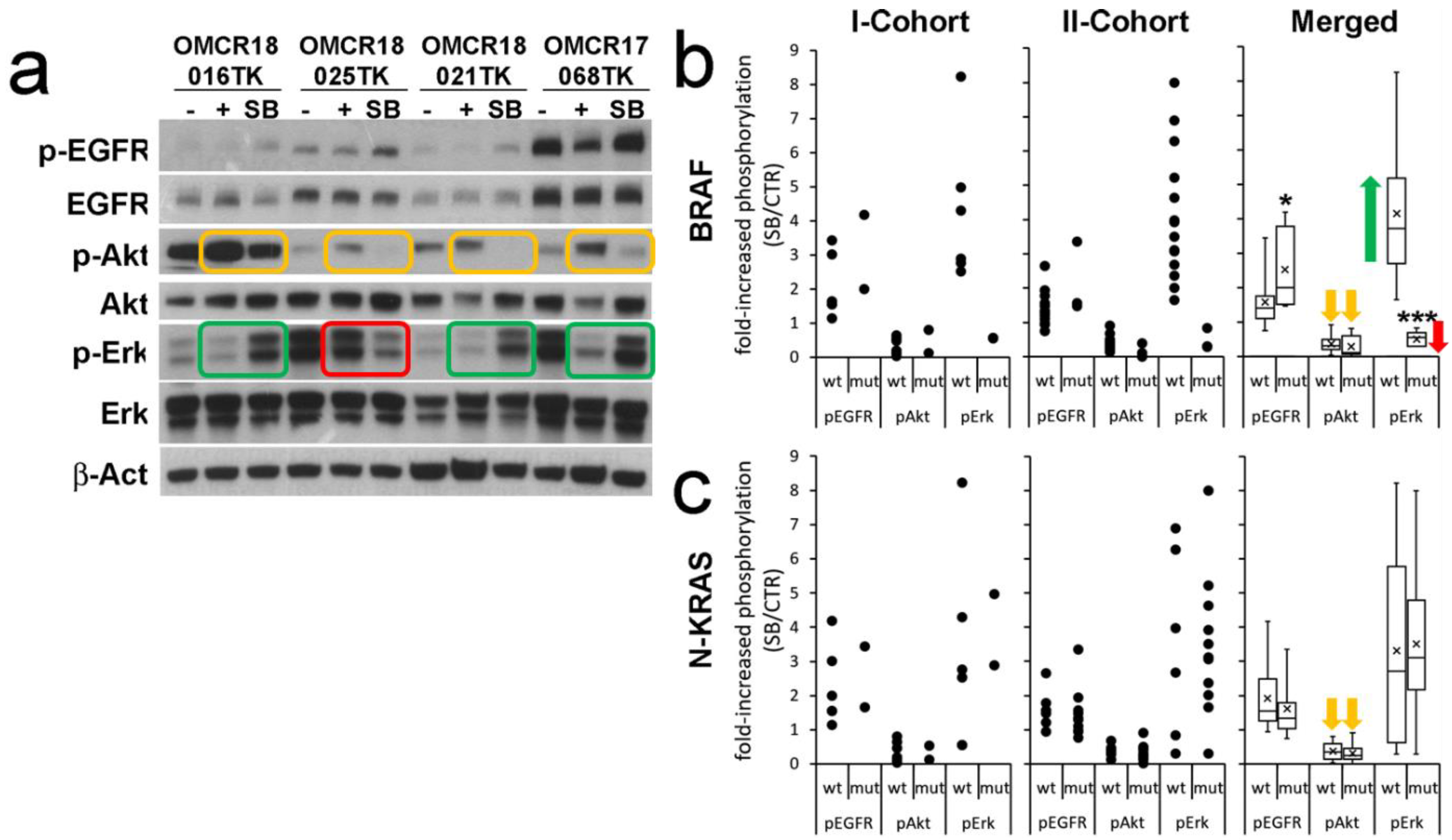
3.2. Influence of SB202190 on the EGFR Signaling Pathway and Its Relation to the Mutational Status of CRC Organoids
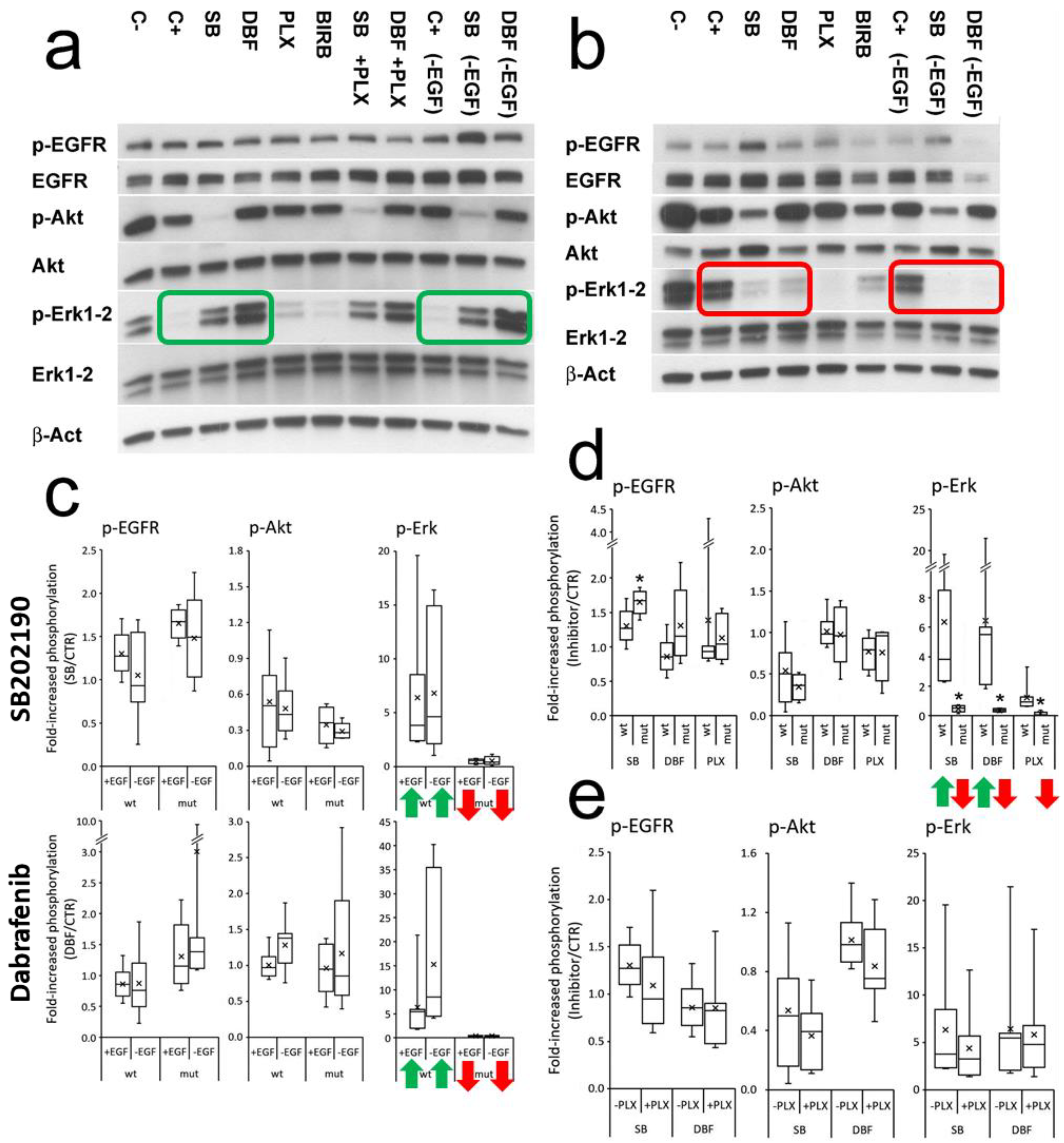
3.3. SB202190 Mimics the Activity of the BRAF V600E Inhibitor Dabrafenib on Erk1-2 Phosphorylation Status
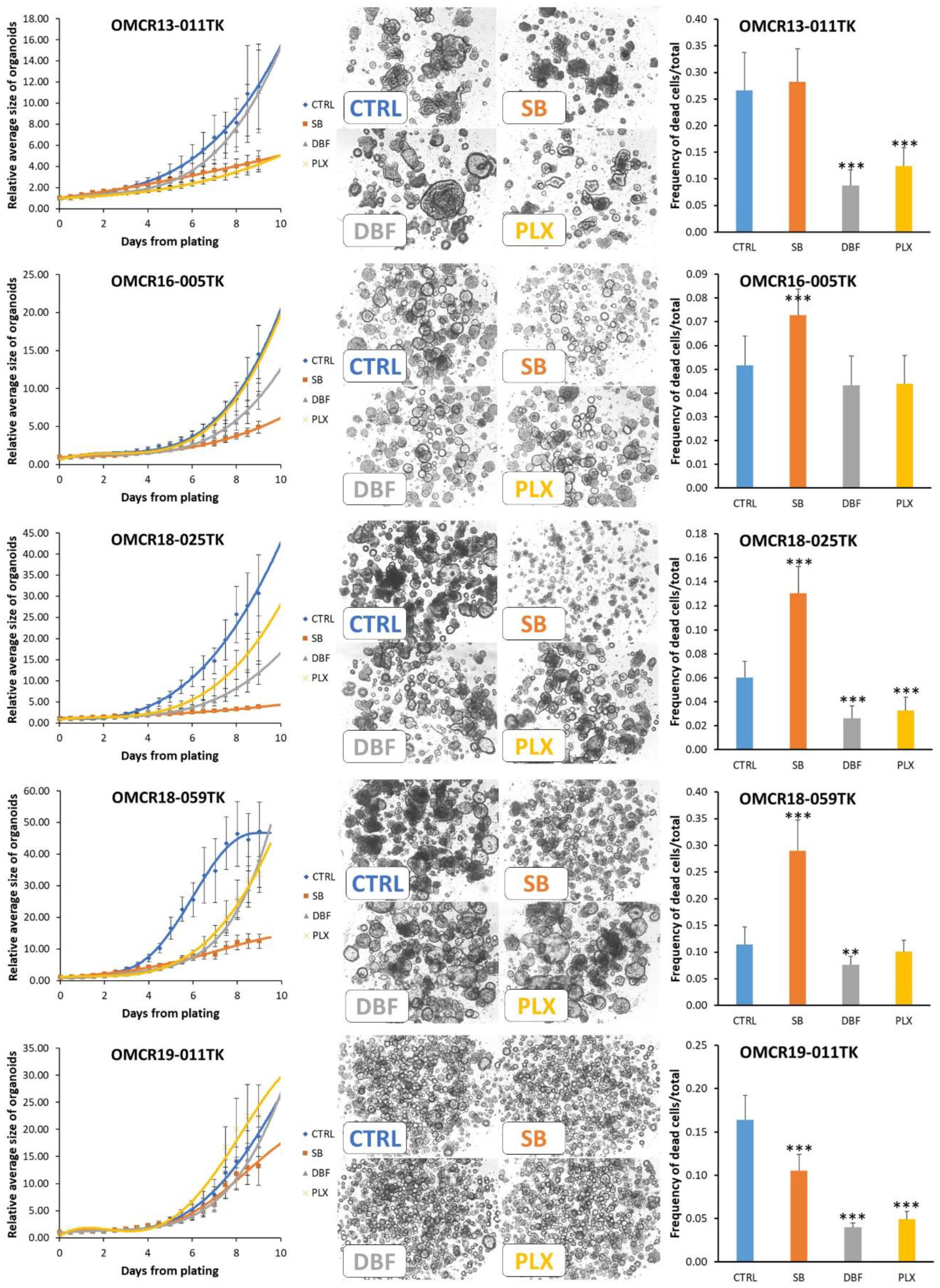
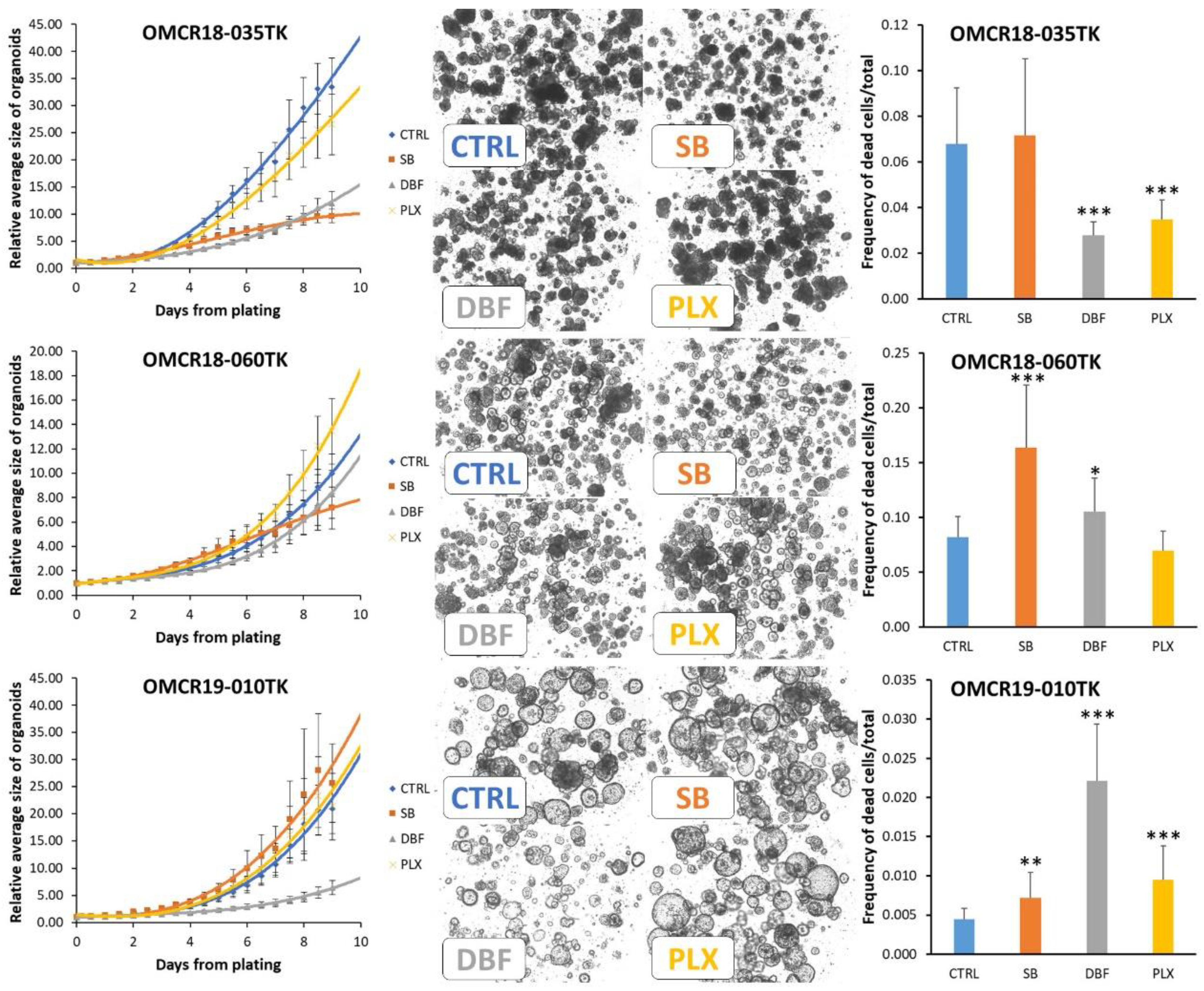
3.4. Effects of SB202190, Dabrafenib and PLX8394 on CRC Organoid Growth
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Uribe, M.L.; Marrocco, I.; Yarden, Y. EGFR in Cancer: Signaling Mechanisms, Drugs, and Acquired Resistance. Cancers 2021, 13, 2748. [Google Scholar] [CrossRef]
- Miyamoto, Y.; Suyama, K.; Baba, H. Recent Advances in Targeting the EGFR Signaling Pathway for the Treatment of Metastatic Colorectal Cancer. Int. J. Mol. Sci. 2017, 18, 752. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Shen, J.; Huang, C.; Cao, M.; Shen, L. Clinicopathological Significance of BRAFV600E Mutation in Colorectal Cancer: An Updated Meta-Analysis. J. Cancer 2019, 10, 2332–2341. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dankner, M.; Rose, A.A.N.; Rajkumar, S.; Siegel, P.M.; Watson, I.R. Classifying BRAF Alterations in Cancer: New Rational Therapeutic Strategies for Actionable Mutations. Oncogene 2018, 37, 3183–3199. [Google Scholar] [CrossRef]
- Clarke, C.N.; Kopetz, E.S. BRAF Mutant Colorectal Cancer as a Distinct Subset of Colorectal Cancer: Clinical Characteristics, Clinical Behavior, and Response to Targeted Therapies. J. Gastrointest. Oncol. 2015, 6, 660–667. [Google Scholar] [PubMed]
- Karapetis, C.S.; Jonker, D.; Daneshmand, M.; Hanson, J.E.; O’Callaghan, C.J.; Marginean, C.; Zalcberg, J.R.; Simes, J.; Moore, M.J.; Tebbutt, N.C.; et al. PIK3CA, BRAF, and PTEN Status and Benefit from Cetuximab in the Treatment of Advanced Colorectal Cancer—Results from NCIC CTG/AGITG CO.17. Clin. Cancer Res. 2014, 20, 744–753. [Google Scholar] [CrossRef] [Green Version]
- Drost, J.; Clevers, H. Organoids in Cancer Research. Nat. Rev. Cancer 2018, 18, 407–418. [Google Scholar] [CrossRef] [PubMed]
- Barbáchano, A.; Fernández-Barral, A.; Bustamante-Madrid, P.; Prieto, I.; Rodríguez-Salas, N.; Larriba, M.J.; Muñoz, A. Organoids and Colorectal Cancer. Cancers 2021, 13, 2657. [Google Scholar] [CrossRef]
- Vlachogiannis, G.; Hedayat, S.; Vatsiou, A.; Jamin, Y.; Fernández-Mateos, J.; Khan, K.; Lampis, A.; Eason, K.; Huntingford, I.; Burke, R.; et al. Patient-Derived Organoids Model Treatment Response of Metastatic Gastrointestinal Cancers. Science 2018, 359, 920–926. [Google Scholar] [CrossRef] [Green Version]
- Sato, T.; Stange, D.E.; Ferrante, M.; Vries, R.G.J.; Van Es, J.H.; Van den Brink, S.; Van Houdt, W.J.; Pronk, A.; Van Gorp, J.; Siersema, P.D.; et al. Long-Term Expansion of Epithelial Organoids from Human Colon, Adenoma, Adenocarcinoma, and Barrett’s Epithelium. Gastroenterology 2011, 141, 1762–1772. [Google Scholar] [CrossRef]
- Fujii, M.; Shimokawa, M.; Date, S.; Takano, A.; Matano, M.; Nanki, K.; Ohta, Y.; Toshimitsu, K.; Nakazato, Y.; Kawasaki, K.; et al. A Colorectal Tumor Organoid Library Demonstrates Progressive Loss of Niche Factor Requirements during Tumorigenesis. Cell Stem Cell 2016, 18, 827–838. [Google Scholar] [CrossRef] [Green Version]
- van de Wetering, M.; Francies, H.E.; Francis, J.M.; Bounova, G.; Iorio, F.; Pronk, A.; van Houdt, W.; van Gorp, J.; Taylor-Weiner, A.; Kester, L.; et al. Prospective Derivation of a Living Organoid Biobank of Colorectal Cancer Patients. Cell 2015, 161, 933–945. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Benelli, R.; Costa, D.; Salvini, L.; Tardito, S.; Tosetti, F.; Villa, F.; Zocchi, M.R.; Poggi, A. Targeting of Colorectal Cancer Organoids with Zoledronic Acid Conjugated to the Anti-EGFR Antibody Cetuximab. J. Immunother. Cancer 2022, 10, e005660. [Google Scholar] [CrossRef] [PubMed]
- Vanni, I.; Coco, S.; Truini, A.; Rusmini, M.; Dal Bello, M.G.; Alama, A.; Banelli, B.; Mora, M.; Rijavec, E.; Barletta, G.; et al. Next-Generation Sequencing Workflow for NSCLC Critical Samples Using a Targeted Sequencing Approach by Ion Torrent PGMTM Platform. Int. J. Mol. Sci. 2015, 16, 28765–28782. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tate, J.G.; Bamford, S.; Jubb, H.C.; Sondka, Z.; Beare, D.M.; Bindal, N.; Boutselakis, H.; Cole, C.G.; Creatore, C.; Dawson, E.; et al. COSMIC: The Catalogue of Somatic Mutations in Cancer. Nucleic Acids Res. 2019, 47, D941–D947. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sequence Read Archive (SRA). Available online: https://www.ncbi.nlm.nih.gov/sra (accessed on 2 September 2021).
- Coco, S.; De Mariano, M.; Valdora, F.; Servidei, T.; Ridola, V.; Andolfo, I.; Oberthuer, A.; Tonini, G.P.; Longo, L. Identification of ALK Germline Mutation (3605delG) in Pediatric Anaplastic Medulloblastoma. J. Hum. Genet. 2012, 57, 682–684. [Google Scholar] [CrossRef] [Green Version]
- Primer3. Available online: https://bioinfo.ut.ee/primer3/ (accessed on 2 September 2021).
- Primer-BLAST. Available online: https://www.ncbi.nlm.nih.gov/tools/primer-blast/ (accessed on 2 September 2021).
- Roepstorff, K.; Grandal, M.V.; Henriksen, L.; Knudsen, S.L.J.; Lerdrup, M.; Grøvdal, L.; Willumsen, B.M.; van Deurs, B. Differential Effects of EGFR Ligands on Endocytic Sorting of the Receptor. Traffic 2009, 10, 1115–1127. [Google Scholar] [CrossRef]
- Menon, M.B.; Kotlyarov, A.; Gaestel, M. SB202190-Induced Cell Type-Specific Vacuole Formation and Defective Autophagy Do Not Depend on P38 MAP Kinase Inhibition. PLoS ONE 2011, 6, e23054. [Google Scholar] [CrossRef]
- Jones, J.C.; Renfro, L.A.; Al-Shamsi, H.O.; Schrock, A.B.; Rankin, A.; Zhang, B.Y.; Kasi, P.M.; Voss, J.S.; Leal, A.D.; Sun, J.; et al. Non-V600 BRAF Mutations Define a Clinically Distinct Molecular Subtype of Metastatic Colorectal Cancer. J. Clin. Oncol. 2017, 35, 2624–2630. [Google Scholar] [CrossRef]
- Hatzivassiliou, G.; Song, K.; Yen, I.; Brandhuber, B.J.; Anderson, D.J.; Alvarado, R.; Ludlam, M.J.C.; Stokoe, D.; Gloor, S.L.; Vigers, G.; et al. RAF Inhibitors Prime Wild-Type RAF to Activate the MAPK Pathway and Enhance Growth. Nature 2010, 464, 431–435. [Google Scholar] [CrossRef] [Green Version]
- Bollag, G.; Hirth, P.; Tsai, J.; Zhang, J.; Ibrahim, P.N.; Cho, H.; Spevak, W.; Zhang, C.; Zhang, Y.; Habets, G.; et al. Clinical Efficacy of a RAF Inhibitor Needs Broad Target Blockade in BRAF-Mutant Melanoma. Nature 2010, 467, 596–599. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lavoie, H.; Thevakumaran, N.; Gavory, G.; Li, J.J.; Padeganeh, A.; Guiral, S.; Duchaine, J.; Mao, D.Y.L.; Bouvier, M.; Sicheri, F.; et al. Inhibitors That Stabilize a Closed RAF Kinase Domain Conformation Induce Dimerization. Nat. Chem. Biol. 2013, 9, 428–436. [Google Scholar] [CrossRef] [PubMed]
- Yao, Z.; Gao, Y.; Su, W.; Yaeger, R.; Tao, J.; Na, N.; Zhang, Y.; Zhang, C.; Rymar, A.; Tao, A.; et al. RAF Inhibitor PLX8394 Selectively Disrupts BRAF Dimers and RAS-Independent BRAF-Mutant-Driven Signaling. Nat. Med. 2019, 25, 284–291. [Google Scholar] [CrossRef] [PubMed]
- Bain, J.; Plater, L.; Elliott, M.; Shpiro, N.; Hastie, C.J.; McLauchlan, H.; Klevernic, I.; Arthur, J.S.C.; Alessi, D.R.; Cohen, P. The Selectivity of Protein Kinase Inhibitors: A Further Update. Biochem. J. 2007, 408, 297–315. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cohen, R.; Cervera, P.; Svrcek, M.; Pellat, A.; Dreyer, C.; de Gramont, A.; André, T. BRAF-Mutated Colorectal Cancer: What Is the Optimal Strategy for Treatment? Curr. Treat. Options Oncol. 2017, 18, 9. [Google Scholar] [CrossRef]
- Reynolds, A.; Wharton, N.; Parris, A.; Mitchell, E.; Sobolewski, A.; Kam, C.; Bigwood, L.; El Hadi, A.; Münsterberg, A.; Lewis, M.; et al. Canonical Wnt Signals Combined with Suppressed TGFβ/BMP Pathways Promote Renewal of the Native Human Colonic Epithelium. Gut 2014, 63, 610–621. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sapkota, G.; Alarcón, C.; Spagnoli, F.M.; Brivanlou, A.H.; Massagué, J. Balancing BMP Signaling through Integrated Inputs into the Smad1 Linker. Mol. Cell 2007, 25, 441–454. [Google Scholar] [CrossRef]
- Yang, J.; Davies, R.J.; Southwood, M.; Long, L.; Yang, X.; Sobolewski, A.; Upton, P.D.; Trembath, R.C.; Morrell, N.W. Mutations in Bone Morphogenetic Protein Type II Receptor Cause Dysregulation of Id Gene Expression in Pulmonary Artery Smooth Muscle Cells: Implications for Familial Pulmonary Arterial Hypertension. Circ. Res. 2008, 102, 1212–1221. [Google Scholar] [CrossRef] [Green Version]
- Zhan, T.; Ambrosi, G.; Wandmacher, A.M.; Rauscher, B.; Betge, J.; Rindtorff, N.; Häussler, R.S.; Hinsenkamp, I.; Bamberg, L.; Hessling, B.; et al. MEK Inhibitors Activate Wnt Signalling and Induce Stem Cell Plasticity in Colorectal Cancer. Nat. Commun. 2019, 10, 2197. [Google Scholar] [CrossRef] [Green Version]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| ID | Set | Loc | MS Status | Stage UICC | T | N | M | G | Molecular Diagnostic of Tumor Tissue | Mutation Status by NGS | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| CTNNB1 | FBXW7 | MET | ERBB2 | KRAS | NRAS | BRAF | DDR2 | PIK3CA | PTEN | ALK | SMAD4 | TP53 | ||||||||||
| OMCR17-062TK | Exp | T | MSS | III | 3 | 2a | 3 | Q61R | 3236T>C | R248Q | ||||||||||||
| OMCR17-068TK | Exp | L | MSS | III | 3 | 1b | 2 | TACTT->A | R273C | |||||||||||||
| OMCR18-016TK | Exp | T | MSI | II | 3 | 0 | 2 | R465H | L755S | |||||||||||||
| OMCR18-021TK | Exp | S | MSS | IV | 3 | 2a | 1a | 2 | KRAS G12V | G12V | ||||||||||||
| OMCR18-025TK | Exp | R | MSS | I | 2 | 0 | 2 | G34E | R505C | V600E | ||||||||||||
| OMCR18-035TK | Exp | R | MSS | II | 4b | 0 | 2 | S241 fs*22 | ||||||||||||||
| OMCR18-059TK | Exp | RT | MSS | I | 2 | 0 | 2 | G60V | V471dup | N131 del | ||||||||||||
| OMCR18-060TK | Exp | RT | MSS | II | 3 | 0 | 2 | KRAS/BRAF/NRAS WT | ||||||||||||||
| OMCR19-003TK | Val | R | MSS | III | 2 | 1b | 3 | KRAS G12A | N873G | G12A | R282W | |||||||||||
| OMCR19-006TK | Val | RT | MSS | I | 2 | 0 | 2 | 1418+1G>A | A59T | R1212H | R196P | |||||||||||
| OMCR19-009TK | Val | R | MSS | III | 4a | 2a | 3 | KRAS G12R; PIK3CA Q546K | G12R | Q546K | R273C | |||||||||||
| OMCR19-010TK | Val | T | MSS | II | 3 | 0 | 1 | T232N fsTer11; N323M fsTer21 | L1165H | R181H | ||||||||||||
| OMCR19-011TK | Val | R | MSS | III | 3 | 1b | 3 | A146P | G469V | M1043V | R282W | |||||||||||
| OMCR19-015TK | Val | S | MSS | III | 3 | 1b | 2 | KRAS G12A | R988C | G12A | ||||||||||||
| OMCR19-016TK | Val | R | MSI | II | 3 | 0 | 3 | BRAF wt | T41A | E545K | 801+2T>C | P152R fsTer18 | ||||||||||
| OMCR19-017TK | Val | RT | MSS | II | 3 | 0 | 2 | |||||||||||||||
| OMCR19-024TK | Val | T | MSS | II | 3 | 0 | 2 | G12C | M441I | |||||||||||||
| OMCR19-030TK | Val | T | MSS | III | 3 | 2a | 2 | KRAS G12D | G12D | R135 Ter | Q104R fsTer19 | |||||||||||
| OMCR19-034TK | Val | R | MSS | IV | 3 | 2b | 1a | 2 | KRAS G13V | G13V | E542K | R175H | ||||||||||
| OMCR19-040TK | Val | R | MSS | III | 3 | 2a | 2 | G12A | R175H | |||||||||||||
| OMCR19-041TK | Val | R | MSS | III | 3 | 2a | 3 | R505C | G12V | G1049R | S178 Ter | R213 Ter | ||||||||||
| OMCR20-002TK | Val | R | MSS | II | 3 | 0 | 2 | G12D | ||||||||||||||
| OMCR13-011TK | Ad | R | MSS | III | 4a | 2b | 3 | BRAF K601E | K601E | |||||||||||||
| OMCR15-045TK | Ad | R | MSS | II | 3 | 0 | 2 | N375S | G12V | R282W | ||||||||||||
| OMCR16-005TK | Ad | R | MSS | IV | 4a | 2a | 1b | 4 | BRAF V600E | S23N | V600E | G244S | ||||||||||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Costa, D.; Venè, R.; Coco, S.; Longo, L.; Tosetti, F.; Scabini, S.; Mastracci, L.; Grillo, F.; Poggi, A.; Benelli, R. SB202190 Predicts BRAF-Activating Mutations in Primary Colorectal Cancer Organoids via Erk1-2 Modulation. Cells 2023, 12, 664. https://doi.org/10.3390/cells12040664
Costa D, Venè R, Coco S, Longo L, Tosetti F, Scabini S, Mastracci L, Grillo F, Poggi A, Benelli R. SB202190 Predicts BRAF-Activating Mutations in Primary Colorectal Cancer Organoids via Erk1-2 Modulation. Cells. 2023; 12(4):664. https://doi.org/10.3390/cells12040664
Chicago/Turabian StyleCosta, Delfina, Roberta Venè, Simona Coco, Luca Longo, Francesca Tosetti, Stefano Scabini, Luca Mastracci, Federica Grillo, Alessandro Poggi, and Roberto Benelli. 2023. "SB202190 Predicts BRAF-Activating Mutations in Primary Colorectal Cancer Organoids via Erk1-2 Modulation" Cells 12, no. 4: 664. https://doi.org/10.3390/cells12040664
APA StyleCosta, D., Venè, R., Coco, S., Longo, L., Tosetti, F., Scabini, S., Mastracci, L., Grillo, F., Poggi, A., & Benelli, R. (2023). SB202190 Predicts BRAF-Activating Mutations in Primary Colorectal Cancer Organoids via Erk1-2 Modulation. Cells, 12(4), 664. https://doi.org/10.3390/cells12040664