
Epigenetics in LMNA-Related Cardiomyopathy

Abstract
:1. Introduction
2. LMNA-Related Dilated Cardiomyopathy
3. Arrhythmogenic Right Ventricular Cardiomyopathy
4. Left Ventricle Noncompaction Cardiomyopathy
5. Restrictive Cardiomyopathy
6. Molecular Mechanisms Resulting in LMNA-Related Cardiomyopathy Pathogenesis
7. Epigenetics in LMNA-Related CARDIOMYOPATHIES
- Lamina-associated domain reorganization and changes in chromatin architecture in LMNA-related cardiomyopathy.
- The role of Polycomb Group Proteins in LMNA-related cardiomyopathy.
8. Advances in Therapeutic Strategies for LMNA-Related Cardiomyopathy
9. Conclusions and Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Schreiber, K.H.; Kennedy, B.K. When lamins go bad: Nuclear structure and disease. Cell 2013, 152, 1365–1375. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Osmanagic-Myers, S.; Foisner, R. The structural and gene expression hypotheses in laminopathic diseases-not so different after all. Mol. Biol. Cell 2019, 30, 1786–1790. [Google Scholar] [CrossRef] [PubMed]
- McKeon, F.D.; Kirschner, M.W.; Caput, D. Homologies in both primary and secondary structure between nuclear envelope and intermediate filament proteins. Nature 1986, 319, 463–468. [Google Scholar] [CrossRef] [PubMed]
- Fisher, D.Z.; Chaudhary, N.; Blobel, G. cDNA sequencing of nuclear lamins A and C reveals primary and secondary structural homology to intermediate filament proteins. Proc. Natl. Acad. Sci. USA 1986, 83, 6450–6454. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gruenbaum, Y.; Foisner, R. Lamins: Nuclear intermediate filament proteins with fundamental functions in nuclear mechanics and genome regulation. Annu. Rev. Biochem. 2015, 84, 131–164. [Google Scholar] [CrossRef] [PubMed]
- Lin, F.; Worman, H.J. Structural organization of the human gene (LMNB1) encoding nuclear lamin B1. Genomics 1995, 27, 230–236. [Google Scholar] [CrossRef]
- Vorburger, K.; Lehner, C.; Kitten, G.; Eppenberger, H.; Nigg, E. A second higher vertebrate B-type lamin: cDNA sequence determination and in vitro processing of chicken lamin B2. J. Mol. Biol. 1989, 208, 405–415. [Google Scholar] [CrossRef]
- Peter, M.; Kitten, G.; Lehner, C.; Vorburger, K.; Bailer, S.; Maridor, G.; Nigg, E. Cloning and sequencing of cDNA clones encoding chicken lamins A and B1 and comparison of the primary structures of vertebrate A-and B-type lamins. J. Mol. Biol. 1989, 208, 393–404. [Google Scholar] [CrossRef]
- Höger, T.H.; Zatloukal, K.; Waizenegger, I.; Krohne, G. Characterization of a second highly conserved B-type lamin present in cells previously thought to contain only a single B-type lamin. Chromosoma 1990, 99, 379–390. [Google Scholar] [CrossRef]
- Turgay, Y.; Eibauer, M.; Goldman, A.E.; Shimi, T.; Khayat, M.; Ben-Harush, K.; Dubrovsky-Gaupp, A.; Sapra, K.T.; Goldman, R.D.; Medalia, O. The molecular architecture of lamins in somatic cells. Nature 2017, 543, 261–264. [Google Scholar] [CrossRef] [Green Version]
- Burke, B.; Stewart, C.L. The nuclear lamins: Flexibility in function. Nat. Rev. Mol. Cell Biol. 2013, 14, 13–24. [Google Scholar] [CrossRef] [PubMed]
- Walling, B.L.; Murphy, P.M. Protean regulation of leukocyte function by nuclear lamins. Trends Immunol. 2021, 42, 323–335. [Google Scholar] [CrossRef]
- Shimi, T.; Kittisopikul, M.; Tran, J.; Goldman, A.E.; Adam, S.A.; Zheng, Y.; Jaqaman, K.; Goldman, R.D. Structural organization of nuclear lamins A, C, B1, and B2 revealed by superresolution microscopy. Mol. Biol. Cell 2015, 26, 4075–4086. [Google Scholar] [CrossRef]
- Nmezi, B.; Xu, J.; Fu, R.; Armiger, T.J.; Rodriguez-Bey, G.; Powell, J.S.; Ma, H.; Sullivan, M.; Tu, Y.; Chen, N.Y. Concentric organization of A-and B-type lamins predicts their distinct roles in the spatial organization and stability of the nuclear lamina. Proc. Natl. Acad. Sci. USA 2019, 116, 4307–4315. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van Steensel, B.; Belmont, A.S. Lamina-Associated Domains: Links with Chromosome Architecture, Heterochromatin, and Gene Repression. Cell 2017, 169, 780–791. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reddy, K.L.; Zullo, J.M.; Bertolino, E.; Singh, H. Transcriptional repression mediated by repositioning of genes to the nuclear lamina. Nature 2008, 452, 243–247. [Google Scholar] [CrossRef] [PubMed]
- Wen, B.; Wu, H.; Shinkai, Y.; Irizarry, R.A.; Feinberg, A.P. Large histone H3 lysine 9 dimethylated chromatin blocks distinguish differentiated from embryonic stem cells. Nat. Genet. 2009, 41, 246–250. [Google Scholar] [CrossRef] [Green Version]
- Gesson, K.; Rescheneder, P.; Skoruppa, M.P.; von Haeseler, A.; Dechat, T.; Foisner, R. A-type lamins bind both hetero- and euchromatin, the latter being regulated by lamina-associated polypeptide 2 alpha. Genome Res. 2016, 26, 462–473. [Google Scholar] [CrossRef] [Green Version]
- Jahed, Z.; Mofrad, M.R. The nucleus feels the force, LINCed in or not! Curr. Opin. Cell Biol. 2019, 58, 114–119. [Google Scholar] [CrossRef]
- Stroud, M.J.; Banerjee, I.; Veevers, J.; Chen, J. Linker of nucleoskeleton and cytoskeleton complex proteins in cardiac structure, function, and disease. Circ. Res. 2014, 114, 538–548. [Google Scholar] [CrossRef] [Green Version]
- Lityagina, O.; Dobreva, G. The LINC Between Mechanical Forces and Chromatin. Front. Physiol. 2021, 12, 710809. [Google Scholar] [CrossRef]
- Gerull, B.; Klaassen, S.; Brodehl, A. The Genetic Landscape of Cardiomyopathies. In Genetic Causes of Cardiac Disease; Springer Nature: Berlin/Heidelberg, Germany, 2019. [Google Scholar]
- McNally, E.M.; Mestroni, L. Dilated Cardiomyopathy: Genetic Determinants and Mechanisms. Circ. Res. 2017, 121, 731–748. [Google Scholar] [CrossRef] [PubMed]
- Quarta, G.; Syrris, P.; Ashworth, M.; Jenkins, S.; Zuborne Alapi, K.; Morgan, J.; Muir, A.; Pantazis, A.; McKenna, W.J.; Elliott, P.M. Mutations in the Lamin A/C gene mimic arrhythmogenic right ventricular cardiomyopathy. Eur. Heart J. 2012, 33, 1128–1136. [Google Scholar] [CrossRef] [Green Version]
- Van Rijsingen, I.A.; Arbustini, E.; Elliott, P.M.; Mogensen, J.; Hermans-van Ast, J.F.; Van Der Kooi, A.J.; Van Tintelen, J.P.; Van Den Berg, M.P.; Pilotto, A.; Pasotti, M. Risk factors for malignant ventricular arrhythmias in lamin A/C mutation carriers: A European cohort study. J. Am. Coll. Cardiol. 2012, 59, 493–500. [Google Scholar] [CrossRef] [PubMed]
- Fatkin, D.; MacRae, C.; Sasaki, T.; Wolff, M.R.; Porcu, M.; Frenneaux, M.; Atherton, J.; Vidaillet Jr, H.J.; Spudich, S.; De Girolami, U. Missense mutations in the rod domain of the lamin A/C gene as causes of dilated cardiomyopathy and conduction-system disease. N. Engl. J. Med. 1999, 341, 1715–1724. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, S.; Baldinger, S.H.; Gandjbakhch, E.; Maury, P.; Sellal, J.-M.; Androulakis, A.F.; Waintraub, X.; Charron, P.; Rollin, A.; Richard, P. Long-term arrhythmic and nonarrhythmic outcomes of lamin A/C mutation carriers. J. Am. Coll. Cardiol. 2016, 68, 2299–2307. [Google Scholar] [CrossRef]
- Nishiuchi, S.; Makiyama, T.; Aiba, T.; Nakajima, K.; Hirose, S.; Kohjitani, H.; Yamamoto, Y.; Harita, T.; Hayano, M.; Wuriyanghai, Y. Gene-based risk stratification for cardiac disorders in LMNA mutation carriers. Circ. Cardiovasc. Genet. 2017, 10, e001603. [Google Scholar] [CrossRef] [Green Version]
- van Engelen, B.G.; Muchir, A.; Hutchison, C.J.; van der Kooi, A.J.; Bonne, G.; Lammens, M. The lethal phenotype of a homozygous nonsense mutation in the lamin A/C gene. Neurology 2005, 64, 374–376. [Google Scholar] [CrossRef]
- Roncarati, R.; Viviani Anselmi, C.; Krawitz, P.; Lattanzi, G.; von Kodolitsch, Y.; Perrot, A.; di Pasquale, E.; Papa, L.; Portararo, P.; Columbaro, M.; et al. Doubly heterozygous LMNA and TTN mutations revealed by exome sequencing in a severe form of dilated cardiomyopathy. Eur. J. Hum. Genet. 2013, 21, 1105–1111. [Google Scholar] [CrossRef] [Green Version]
- Galata, Z.; Kloukina, I.; Kostavasili, I.; Varela, A.; Davos, C.H.; Makridakis, M.; Bonne, G.; Capetanaki, Y. Amelioration of desmin network defects by αB-crystallin overexpression confers cardioprotection in a mouse model of dilated cardiomyopathy caused by LMNA gene mutation. J. Mol. Cell. Cardiol. 2018, 125, 73–86. [Google Scholar] [CrossRef]
- Maggi, L.; Mavroidis, M.; Psarras, S.; Capetanaki, Y.; Lattanzi, G. Skeletal and cardiac muscle disorders caused by mutations in genes encoding intermediate filament proteins. Int. J. Mol. Sci. 2021, 22, 4256. [Google Scholar] [CrossRef] [PubMed]
- Vincenzo, M.; Michelangelo, M.; Costanza, S.; Francesca, T.; Lucia, C.; Greta, A.; Anna, R.; Fulvia, B.; Adelaide, C.M.; Giovanna, L. A Single mtDNA Deletion in Association with a LMNA Gene New Frameshift Variant: A Case Report. J. Neuromuscul. Dis. 2022, 9, 457–462. [Google Scholar]
- Meinke, P.; Mattioli, E.; Haque, F.; Antoku, S.; Columbaro, M.; Straatman, K.R.; Worman, H.J.; Gundersen, G.G.; Lattanzi, G.; Wehnert, M. Muscular dystrophy-associated SUN1 and SUN2 variants disrupt nuclear-cytoskeletal connections and myonuclear organization. PLoS Genet. 2014, 10, e1004605. [Google Scholar] [CrossRef] [Green Version]
- Mounkes, L.C.; Kozlov, S.V.; Rottman, J.N.; Stewart, C.L. Expression of an LMNA-N195K variant of A-type lamins results in cardiac conduction defects and death in mice. Hum. Mol. Genet. 2005, 14, 2167–2180. [Google Scholar] [CrossRef] [PubMed]
- Cai, Z.-J.; Lee, Y.-K.; Lau, Y.-M.; Ho, J.C.-Y.; Lai, W.-H.; Wong, N.L.-Y.; Huang, D.; Hai, J.-j.; Ng, K.-M.; Tse, H.-F. Expression of Lmna-R225X nonsense mutation results in dilated cardiomyopathy and conduction disorders (DCM-CD) in mice: Impact of exercise training. Int. J. Cardiol. 2020, 298, 85–92. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, Y.K.; Lau, Y.M.; Cai, Z.J.; Lai, W.H.; Wong, L.Y.; Tse, H.F.; Ng, K.M.; Siu, C.W. Modeling treatment response for lamin A/C related dilated cardiomyopathy in human induced pluripotent stem cells. J. Am. Heart Assoc. 2017, 6, e005677. [Google Scholar] [CrossRef]
- Lee, J.; Termglinchan, V.; Diecke, S.; Itzhaki, I.; Lam, C.K.; Garg, P.; Lau, E.; Greenhaw, M.; Seeger, T.; Wu, H. Activation of PDGF pathway links LMNA mutation to dilated cardiomyopathy. Nature 2019, 572, 335–340. [Google Scholar] [CrossRef]
- MacLeod, H.M.; Culley, M.R.; Huber, J.M.; McNally, E.M. Lamin A/C truncation in dilated cardiomyopathy with conduction disease. BMC Med. Genet. 2003, 4, 4. [Google Scholar] [CrossRef] [Green Version]
- Sebillon, P.; Bouchier, C.; Bidot, L.; Bonne, G.; Ahamed, K.; Charron, P.; Drouin-Garraud, V.; Millaire, A.; Desrumeaux, G.; Benaiche, A. Expanding the phenotype of LMNA mutations in dilated cardiomyopathy and functional consequences of these mutations. J. Med. Genet. 2003, 40, 560–567. [Google Scholar] [CrossRef]
- Shah, P.P.; Lv, W.; Rhoades, J.H.; Poleshko, A.; Abbey, D.; Caporizzo, M.A.; Linares-Saldana, R.; Heffler, J.G.; Sayed, N.; Thomas, D. Pathogenic LMNA variants disrupt cardiac lamina-chromatin interactions and de-repress alternative fate genes. Cell Stem Cell 2021, 28, 938–954.e939. [Google Scholar] [CrossRef]
- Hussain, I.; Patni, N.; Ueda, M.; Sorkina, E.; Valerio, C.M.; Cochran, E.; Brown, R.J.; Peeden, J.; Tikhonovich, Y.; Tiulpakov, A. A novel generalized lipodystrophy-associated progeroid syndrome due to recurrent heterozygous LMNA p. T10I mutation. J. Clin. Endocrinol. Metab. 2018, 103, 1005–1014. [Google Scholar] [CrossRef] [PubMed]
- Sahinoz, M.; Khairi, S.; Cuttitta, A.; Brady, G.F.; Rupani, A.; Meral, R.; Tayeh, M.K.; Thomas, P.; Riebschleger, M.; Camelo-Piragua, S. Potential association of LMNA-associated generalized lipodystrophy with juvenile dermatomyositis. Clin. Diabetes Endocrinol. 2018, 4, 6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mory, P.B.; Crispim, F.; Kasamatsu, T.; Gabbay, M.A.; Dib, S.A.; Moisés, R.S. Atypical generalized lipoatrophy and severe insulin resistance due to a heterozygous LMNA p. T10I mutation. Arq. Bras. Endocrinol. Metabol. 2008, 52, 1252–1256. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, J.; Argenziano, M.A.; Burgos Angulo, M.; Bertalovitz, A.; Beidokhti, M.N.; McDonald, T.V. Phenotypic Variability in iPSC-Induced Cardiomyocytes and Cardiac Fibroblasts Carrying Diverse LMNA Mutations. Front. Physiol. 2021, 12, 2162. [Google Scholar] [CrossRef] [PubMed]
- Muchir, A.; Bonne, G.; van der Kooi, A.J.; van Meegen, M.; Baas, F.; Bolhuis, P.A.; de Visser, M.; Schwartz, K. Identification of mutations in the gene encoding lamins A/C in autosomal dominant limb girdle muscular dystrophy with atrioventricular conduction disturbances (LGMD1B). Hum. Mol. Genet. 2000, 9, 1453–1459. [Google Scholar] [CrossRef]
- Charniot, J.C.; Pascal, C.; Bouchier, C.; Sébillon, P.; Salama, J.; Duboscq-Bidot, L.; Peuchmaurd, M.; Desnos, M.; Artigou, J.Y.; Komajda, M. Functional consequences of an LMNA mutation associated with a new cardiac and non-cardiac phenotype. Hum. Mutat. 2003, 21, 473–481. [Google Scholar] [CrossRef]
- van Tintelen, J.P.; Hofstra, R.M.; Katerberg, H.; Rossenbacker, T.; Wiesfeld, A.C.; du Marchie Sarvaas, G.J.; Wilde, A.A.; van Langen, I.M.; Nannenberg, E.A.; van der Kooi, A.J.; et al. High yield of LMNA mutations in patients with dilated cardiomyopathy and/or conduction disease referred to cardiogenetics outpatient clinics. Am. Heart J. 2007, 154, 1130–1139. [Google Scholar] [CrossRef] [PubMed]
- West, G.; Gullmets, J.; Virtanen, L.; Li, S.-P.; Keinänen, A.; Shimi, T.; Mauermann, M.; Heliö, T.; Kaartinen, M.; Ollila, L. Deleterious assembly of the lamin A/C mutant p. S143P causes ER stress in familial dilated cardiomyopathy. J. Cell Sci. 2016, 129, 2732–2743. [Google Scholar]
- West, G.; Turunen, M.; Aalto, A.; Virtanen, L.; Li, S.-P.; Heliö, T.; Meinander, A.; Taimen, P. A heterozygous p. S143P mutation in LMNA associates with proteasome dysfunction and enhanced autophagy-mediated degradation of mutant lamins A and C. Front. Cell Dev. Biol. 2022, 10, 932983. [Google Scholar] [CrossRef]
- Bonne, G.; Mercuri, E.; Muchir, A.; Urtizberea, A.; Bécane, H.M.; Recan, D.; Merlini, L.; Wehnert, M.; Boor, R.; Reuner, U.; et al. Clinical and molecular genetic spectrum of autosomal dominant Emery-Dreifuss muscular dystrophy due to mutations of the lamin A/C gene. Ann. Neurol. 2000, 48, 170–180. [Google Scholar] [CrossRef]
- Perrot, A.; Hussein, S.; Ruppert, V.; Schmidt, H.H.; Wehnert, M.S.; Duong, N.T.; Posch, M.G.; Panek, A.; Dietz, R.; Kindermann, I.; et al. Identification of mutational hot spots in LMNA encoding lamin A/C in patients with familial dilated cardiomyopathy. Basic Res. Cardiol. 2009, 104, 90–99. [Google Scholar] [CrossRef] [PubMed]
- Salvarani, N.; Crasto, S.; Miragoli, M.; Bertero, A.; Paulis, M.; Kunderfranco, P.; Serio, S.; Forni, A.; Lucarelli, C.; Dal Ferro, M.; et al. The K219T-Lamin mutation induces conduction defects through epigenetic inhibition of SCN5A in human cardiac laminopathy. Nat. Commun. 2019, 10, 2267. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mewborn, S.K.; Puckelwartz, M.J.; Abuisneineh, F.; Fahrenbach, J.P.; Zhang, Y.; MacLeod, H.; Dellefave, L.; Pytel, P.; Selig, S.; Labno, C.M. Altered chromosomal positioning, compaction, and gene expression with a lamin A/C gene mutation. PLoS ONE 2010, 5, e14342. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, L.; Sun, J.; Chen, Z.; Liu, L.; Sun, Y.; Lin, J.; Hu, X.; Zhao, M.; Ma, Y.; Lu, D. The LMNA p. R541C mutation causes dilated cardiomyopathy in human and mice. Int. J. Cardiol. 2022, 363, 149–158. [Google Scholar] [CrossRef]
- Hermida-Prieto, M.; Monserrat, L.; Castro-Beiras, A.; Laredo, R.; Soler, R.; Peteiro, J.; Rodríguez, E.; Bouzas, B.; Álvarez, N.; Muñiz, J. Familial dilated cardiomyopathy and isolated left ventricular noncompaction associated with lamin A/C gene mutations. Am. J. Cardiol. 2004, 94, 50–54. [Google Scholar] [CrossRef] [Green Version]
- Chatzifrangkeskou, M.; Yadin, D.; Marais, T.; Chardonnet, S.; Cohen-Tannoudji, M.; Mougenot, N.; Schmitt, A.; Crasto, S.; Di Pasquale, E.; Macquart, C. Cofilin-1 phosphorylation catalyzed by ERK1/2 alters cardiac actin dynamics in dilated cardiomyopathy caused by lamin A/C gene mutation. Hum. Mol. Genet. 2018, 27, 3060–3078. [Google Scholar] [CrossRef] [Green Version]
- Liu, Z.; Shan, H.; Huang, J.; Li, N.; Hou, C.; Pu, J. A novel lamin A/C gene missense mutation (445 V > E) in immunoglobulin-like fold associated with left ventricular non-compaction. Europace 2016, 18, 617–622. [Google Scholar] [CrossRef]
- Rankin, J.; Auer-Grumbach, M.; Bagg, W.; Colclough, K.; Duong, N.T.; Fenton-May, J.; Hattersley, A.; Hudson, J.; Jardine, P.; Josifova, D. Extreme phenotypic diversity and nonpenetrance in families with the LMNA gene mutation R644C. Am. J. Med. Genet. Part A 2008, 146, 1530–1542. [Google Scholar] [CrossRef]
- Parent, J.J.; Towbin, J.A.; Jefferies, J.L. Left ventricular noncompaction in a family with lamin A/C gene mutation. Tex. Heart Inst. J. 2015, 42, 73. [Google Scholar] [CrossRef] [Green Version]
- Paller, M.S.; Martin, C.M.; Pierpont, M.E. Restrictive cardiomyopathy: An unusual phenotype of a lamin A variant. ESC Heart Fail. 2018, 5, 724–726. [Google Scholar] [CrossRef]
- Kim, Y.; Zheng, Y. Generation and characterization of a conditional deletion allele for Lmna in mice. Biochem. Biophys. Res. Commun. 2013, 440, 8–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Elsherbiny, A.; Kessler, L.; Cordero, J.; Shi, H.; Serke, H.; Lityagina, O.; Trogisch, F.A.; Mohammadi, M.M.; El-Battrawy, I. Lamin A/C-dependent chromatin architecture safeguards nave pluripotency to prevent aberrant cardiovascular cell fate and function. Nat. Commun. 2022, 13, 6663. [Google Scholar] [CrossRef] [PubMed]
- Wolf, C.M.; Wang, L.; Alcalai, R.; Pizard, A.; Burgon, P.G.; Ahmad, F.; Sherwood, M.; Branco, D.M.; Wakimoto, H.; Fishman, G.I. Lamin A/C haploinsufficiency causes dilated cardiomyopathy and apoptosis-triggered cardiac conduction system disease. J. Mol. Cell. Cardiol. 2008, 44, 293–303. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nikolova, V.; Leimena, C.; Mcmahon, A.C.; Tan, A.C.; Chandar, S.; Jogia, D.; Kesteven, S.H.; Michalicek, J.; Otway, R.; Verheyen, F. Defects in nuclear structure and function promote dilated cardiomyopathy in lamin A/C–deficient mice. J. Clin. Investig. 2004, 113, 357–369. [Google Scholar] [CrossRef] [PubMed]
- Kubben, N.; Voncken, J.W.; Konings, G.; van Weeghel, M.; van den Hoogenhof, M.M.; Gijbels, M.; van Erk, A.; Schoonderwoerd, K.; van den Bosch, B.; Dahlmans, V.; et al. Post-natal myogenic and adipogenic developmental: Defects and metabolic impairment upon loss of A-type lamins. Nucleus 2011, 2, 195–207. [Google Scholar] [CrossRef] [Green Version]
- Auguste, G.; Rouhi, L.; Matkovich, S.J.; Coarfa, C.; Marian, A.J. BET bromodomain inhibition attenuates cardiac phenotype in myocyte-specific Lamin A/C-deficient mice. J. Clin. Investig. 2020, 130, 4740–4758. [Google Scholar] [CrossRef]
- Fong, L.G.; Ng, J.K.; Lammerding, J.; Vickers, T.A.; Young, S.G. Prelamin A and lamin A appear to be dispensable in the nuclear lamina. J. Clin. Investig. 2006, 116, 743–752. [Google Scholar] [CrossRef]
- Coffinier, C.; Jung, H.J.; Li, Z.; Nobumori, C.; Yun, U.J.; Farber, E.A.; Davies, B.S.; Weinstein, M.M.; Yang, S.H.; Lammerding, J. Direct Synthesis of Lamin A, Bypassing Prelamin A Processing, Causes Misshapen Nuclei in Fibroblasts but No Detectable Pathology in Mice. J. Biol. Chem. 2010, 285, 20818–20826. [Google Scholar] [CrossRef] [Green Version]
- Dan, L.; Lian, H.; Zhang, X.; Shao, H.; Lan, H.; Qin, C.; Zhang, L.; Wu, G.S. LMNA E82K Mutation Activates FAS and Mitochondrial Pathways of Apoptosis in Heart Tissue Specific Transgenic Mice. PLoS ONE 2010, 5, e15167. [Google Scholar]
- Bertrand, A.T.; Renou, L.; Papadopoulos, A.; Beuvin, M.; Lacène, E.; Massart, C.; Ottolenghi, C.; Decostre, V.; Maron, S.; Schlossarek, S.; et al. DelK32-lamin A/C has abnormal location and induces incomplete tissue maturation and severe metabolic defects leading to premature death. Hum. Mol. Genet. 2012, 21, 1037–1048. [Google Scholar] [CrossRef] [Green Version]
- Cattin, M.E.; Ferry, A.; Vignaud, A.; Mougenot, N.; Jacquet, A.; Wahbi, K.; Bertrand, A.T.; Bonne, G. Mutation in lamin A/C sensitizes the myocardium to exercise-induced mechanical stress but has no effect on skeletal muscles in mouse. Neuromuscul. Disord. NMD 2016, 26, 490–499. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cattin, M.E.; Bertrand, A.T.; Schlossarek, S.; Le Bihan, M.C.; Skov Jensen, S.; Neuber, C.; Crocini, C.; Maron, S.; Lainé, J.; Mougenot, N.; et al. Heterozygous LmnadelK32 mice develop dilated cardiomyopathy through a combined pathomechanism of haploinsufficiency and peptide toxicity. Hum. Mol. Genet. 2013, 22, 3152–3164. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wada, E.; Kato, M.; Yamashita, K.; Kokuba, H.; Hayashi, Y.K. Deficiency of emerin contributes differently to the pathogenesis of skeletal and cardiac muscles in LmnaH222P/H222P mutant mice. PLoS ONE 2019, 14, e0221512. [Google Scholar] [CrossRef] [PubMed]
- Guenantin, A.C.; Jebeniani, I.; Leschik, J.; Watrin, E.; Puceat, M. Targeting the histone demethylase LSD1 prevents cardiomyopathy in a mouse model of laminopathy. J. Clin. Investig. 2021, 131, e136488. [Google Scholar] [CrossRef]
- Arimura, T.; Helbling-Leclerc, A.; Massart, C.; Varnous, S.; Niel, F.; Lacene, E.; Fromes, Y.; Toussaint, M.; Mura, A.-M.; Keller, D.I. Mouse model carrying H222P-Lmna mutation develops muscular dystrophy and dilated cardiomyopathy similar to human striated muscle laminopathies. Hum. Mol. Genet. 2005, 14, 155–169. [Google Scholar] [CrossRef]
- Wang, Y.; Herron, A.J.; Worman, H.J. Pathology and nuclear abnormalities in hearts of transgenic mice expressing M371K lamin A encoded by an LMNA mutation causing Emery-Dreifuss muscular dystrophy. Hum. Mol. Genet. 2006, 15, 2479–2489. [Google Scholar] [CrossRef] [Green Version]
- Chen, S.N.; Lombardi, R.; Karmouch, J.; Tsai, J.Y.; Czernuszewicz, G.; Taylor, M.R.G.; Mestroni, L.; Coarfa, C.; Gurha, P.; Marian, A.J. DNA Damage Response/TP53 Pathway Is Activated and Contributes to the Pathogenesis of Dilated Cardiomyopathy Associated With LMNA (Lamin A/C) Mutations. Circ. Res. 2019, 124, 856–873. [Google Scholar] [CrossRef]
- Mounkes, L.C.; Kozlov, S.; Hernandez, L.; Sullivan, T.; Stewart, C.L. A progeroid syndrome in mice is caused by defects in A-type lamins. Nature 2003, 423, 298–301. [Google Scholar] [CrossRef] [Green Version]
- Osorio, F.G.; Navarro, C.L.; Cadianos, J.; López-Mejía, I.; López-Otín, C. Splicing-Directed Therapy in a New Mouse Model of Human Accelerated Aging. Sci. Transl. Med. 2011, 3, 106ra107. [Google Scholar] [CrossRef]
- Yang, S.H.; Meta, M.; Qiao, X.; Frost, D.; Bauch, J.; Coffinier, C.; Majumdar, S.; Bergo, M.O.; Young, S.G.; Fong, L.G. A farnesyltransferase inhibitor improves disease phenotypes in mice with a Hutchinson-Gilford progeria syndrome mutation. J. Clin. Investig. 2006, 116, 2115–2121. [Google Scholar] [CrossRef] [Green Version]
- Yang, S.H.; Andres, D.A.; Spielmann, H.P.; Young, S.G.; Fong, L.G. Progerin elicits disease phenotypes of progeria in mice whether or not it is farnesylated. J. Clin. Investig. 2008, 118, 3291–3300. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, S.H.; Chang, S.Y.; Ren, S.; Wang, Y.; Andres, D.A.; Spielmann, H.P.; Fong, L.G.; Young, S.G. Absence of progeria-like disease phenotypes in knock-in mice expressing a non-farnesylated version of progerin. Hum. Mol. Genet. 2011, 20, 436–444. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Varga, R.; Eriksson, M.; Erdos, M.R.; Olive, M.; Harten, I.; Kolodgie, F.; Capell, B.C.; Cheng, J.; Faddah, D.; Perkins, S. Progressive vascular smooth muscle cell defects in a mouse model of Hutchinson–Gilford progeria syndrome. Proc. Natl. Acad. Sci. USA 2006, 103, 3250–3255. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sagelius, H.; Rosengardten, Y.; Hanif, M.; Erdos, M.R.; Rozell, B.; Collins, F.S.; Eriksson, M. Targeted transgenic expression of the mutation causing Hutchinson-Gilford progeria syndrome leads to proliferative and degenerative epidermal disease. J. Cell Sci. 2008, 121, 969–978. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Panteleyev, A.A.; Owens, D.M.; Djabali, K.; Stewart, C.L.; Worman, H.J. Epidermal expression of the truncated prelamin A causing Hutchinson-Gilford progeria syndrome: Effects on keratinocytes, hair and skin. Hum. Mol. Genet. 2008, 17, 2357–2369. [Google Scholar] [CrossRef] [Green Version]
- Lee, Y.K.; Jiang, Y.; Ran, X.R.; Lau, Y.M.; Ng, K.M.; Lai, W.H.; Siu, C.W.; Tse, H.F. Recent advances in animal and human pluripotent stem cell modeling of cardiac laminopathy. Stem Cell Res. 2016, 7, 139. [Google Scholar] [CrossRef] [Green Version]
- Gupta, P.; Bilinska, Z.T.; Sylvius, N.; Boudreau, E.; Veinot, J.P.; Labib, S.; Bolongo, P.M.; Hamza, A.; Jackson, T.; Ploski, R. Genetic and ultrastructural studies in dilated cardiomyopathy patients: A large deletion in the lamin A/C gene is associated with cardiomyocyte nuclear envelope disruption. Basic Res. Cardiol. 2010, 105, 365–377. [Google Scholar] [CrossRef] [Green Version]
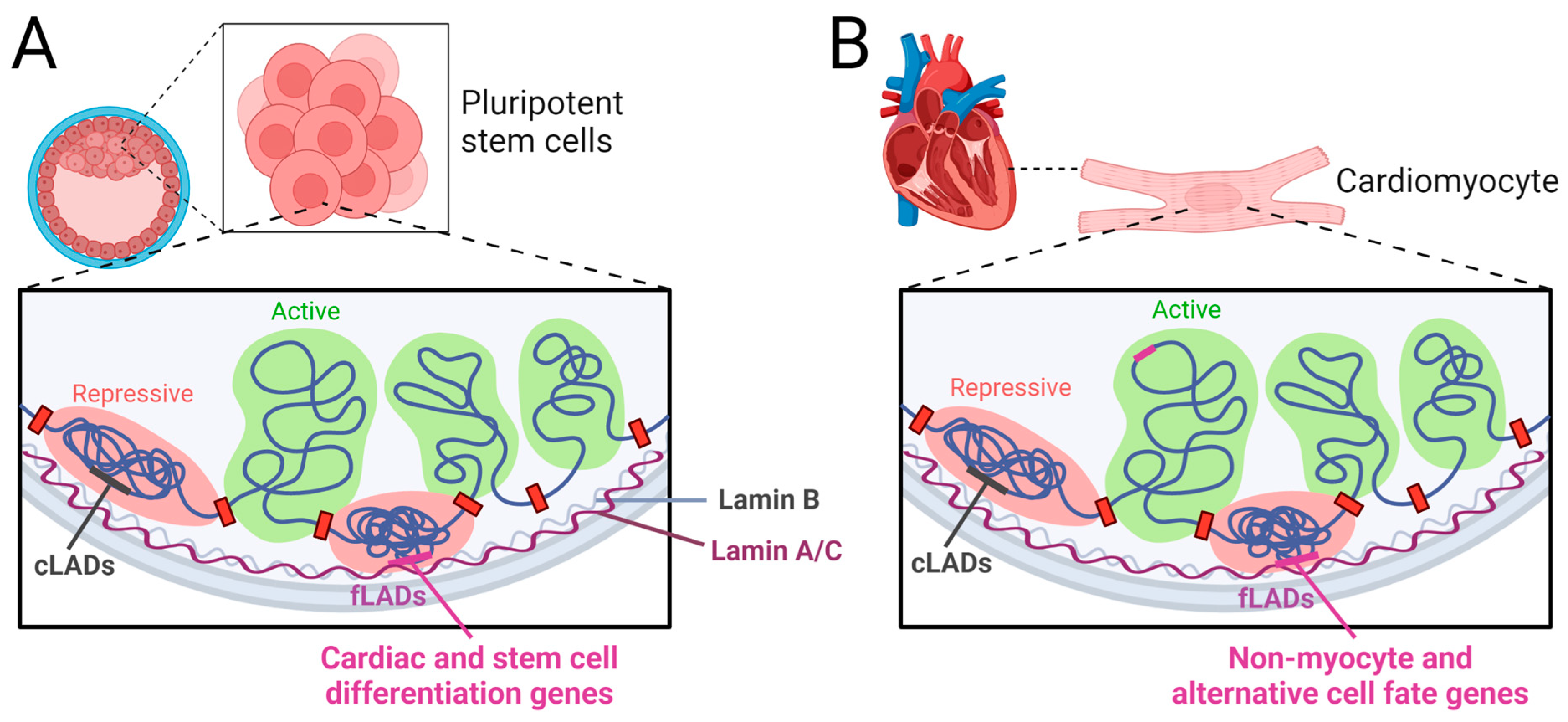
- Cheedipudi, S.M.; Matkovich, S.J.; Coarfa, C.; Hu, X.; Robertson, M.J.; Sweet, M.; Taylor, M.; Mestroni, L.; Cleveland, J.; Willerson, J.T.; et al. Genomic Reorganization of Lamin-Associated Domains in Cardiac Myocytes Is Associated With Differential Gene Expression and DNA Methylation in Human Dilated Cardiomyopathy. Circ. Res. 2019, 124, 1198–1213. [Google Scholar] [CrossRef]
- Lin, E.W.; Brady, G.F.; Kwan, R.; Nesvizhskii, A.I.; Omary, M.B. Genotype-phenotype analysis of LMNA-related diseases predicts phenotype-selective alterations in lamin phosphorylation. FASEB J. 2020, 34, 9051–9073. [Google Scholar] [CrossRef]
- Gerull, B.; Brodehl, A. Insights Into Genetics and Pathophysiology of Arrhythmogenic Cardiomyopathy. Curr. Heart Fail. Rep. 2021, 18, 378–390. [Google Scholar] [CrossRef]
- Marcus, F.I.; McKenna, W.J.; Sherrill, D.; Basso, C.; Bauce, B.; Bluemke, D.A.; Calkins, H.; Corrado, D.; Cox, M.G.; Daubert, J.P. Diagnosis of arrhythmogenic right ventricular cardiomyopathy/dysplasia: Proposed modification of the task force criteria. Circulation 2010, 121, 1533–1541. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Joshi-Mukherjee, R.; Coombs, W.; Musa, H.; Oxford, E.; Taffet, S.; Delmar, M. Characterization of the molecular phenotype of two arrhythmogenic right ventricular cardiomyopathy (ARVC)-related plakophilin-2 (PKP2) mutations. Heart Rhythm. 2008, 5, 1715–1723. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Awad, M.M.; Dalal, D.; Tichnell, C.; James, C.; Tucker, A.; Abraham, T.; Spevak, P.J.; Calkins, H.; Judge, D.P. Recessive arrhythmogenic right ventricular dysplasia due to novel cryptic splice mutation in PKP2. Hum. Mutat. 2006, 27, 1157. [Google Scholar] [CrossRef] [Green Version]
- Yang, Z.; Bowles, N.E.; Scherer, S.E.; Taylor, M.D.; Kearney, D.L.; Ge, S.; Nadvoretskiy, V.V.; DeFreitas, G.; Carabello, B.; Brandon, L.I. Desmosomal dysfunction due to mutations in desmoplakin causes arrhythmogenic right ventricular dysplasia/cardiomyopathy. Circ. Res. 2006, 99, 646–655. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Awad, M.M.; Dalal, D.; Cho, E.; Amat-Alarcon, N.; James, C.; Tichnell, C.; Tucker, A.; Russell, S.D.; Bluemke, D.A.; Dietz, H.C. DSG2 mutations contribute to arrhythmogenic right ventricular dysplasia/cardiomyopathy. Am. J. Hum. Genet. 2006, 79, 136–142. [Google Scholar] [CrossRef] [Green Version]
- Corrado, D.; Thiene, G. Arrhythmogenic right ventricular cardiomyopathy/dysplasia: Clinical impact of molecular genetic studies. Circulation 2006, 113, 1634–1637. [Google Scholar] [CrossRef]
- Brodehl, A.; Weiss, J.; Debus, J.D.; Stanasiuk, C.; Klauke, B.; Deutsch, M.A.; Fox, H.; Bax, J.; Ebbinghaus, H.; Gartner, A.; et al. A homozygous DSC2 deletion associated with arrhythmogenic cardiomyopathy is caused by uniparental isodisomy. J. Mol. Cell. Cardiol. 2020, 141, 17–29. [Google Scholar] [CrossRef]
- Tiso, N.; Stephan, D.A.; Nava, A.; Bagattin, A.; Devaney, J.M.; Stanchi, F.; Larderet, G.; Brahmbhatt, B.; Brown, K.; Bauce, B. Identification of mutations in the cardiac ryanodine receptor gene in families affected with arrhythmogenic right ventricular cardiomyopathy type 2 (ARVD2). Hum. Mol. Genet. 2001, 10, 189–194. [Google Scholar] [CrossRef] [Green Version]
- Zwaag, P.; Rijsingen, I.; Ruite, R. Recurrent and founder mutations in the Netherlands—Phospholamban p.Arg14del mutation causes arrhythmogenic cardiomyopathy. Neth. Heart J. 2013, 21, 286–293. [Google Scholar] [CrossRef] [Green Version]
- Mayosi, B.M.; Fish, M.; Shaboodien, G.; Mastantuono, E.; Kraus, S.; Wieland, T.; Kotta, M.-C.; Chin, A.; Laing, N.; Ntusi, N.B.A.; et al. Identification of Cadherin 2 (CDH2) Mutations in Arrhythmogenic Right Ventricular Cardiomyopathy. Circ. Cardiovasc. Genet. 2017, 10, e001605. [Google Scholar] [CrossRef] [Green Version]
- Brodehl, A.; Rezazadeh, S.; Williams, T.; Munsie, N.M.; Liedtke, D.; Oh, T.; Ferrier, R.; Shen, Y.; Jones, S.J.M.; Stiegler, A.L.; et al. Mutations in ILK, encoding integrin-linked kinase, are associated with arrhythmogenic cardiomyopathy. Transl. Res. J. Lab. Clin. Med. 2019, 208, 15–29. [Google Scholar] [CrossRef] [PubMed]
- Beffagna, G.; Occhi, G.; Nava, A.; Vitiello, L.; Ditadi, A.; Basso, C.; Bauce, B.; Carraro, G.; Thiene, G.; Towbin, J.A. Regulatory mutations in transforming growth factor-β3 gene cause arrhythmogenic right ventricular cardiomyopathy type 1. Cardiovasc. Res. 2005, 65, 366–373. [Google Scholar] [CrossRef] [Green Version]
- Taylor, M.; Graw, S.; Sinagra, G.; Barnes, C.; Slavov, D.; Brun, F.; Pinamonti, B.; Salcedo, E.E.; Sauer, W.; Pyxaras, S. Genetic variation in titin in arrhythmogenic right ventricular cardiomyopathy–overlap syndromes. Circulation 2011, 124, 876–885. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Protonotarios, A.; Brodehl, A.; Asimaki, A.; Jager, J.; Quinn, E.; Stanasiuk, C.; Ratnavadivel, S.; Futema, M.; Akhtar, M.M.; Gossios, T.D.; et al. The Novel Desmin Variant p.Leu115Ile Is Associated With a Unique Form of Biventricular Arrhythmogenic Cardiomyopathy. Can. J. Cardiol. 2021, 37, 857–866. [Google Scholar] [CrossRef] [PubMed]
- Hodgkinson, K.; Connors, S.; Merner, N.; Haywood, A.; Young, T.L.; McKenna, W.; Gallagher, B.; Curtis, F.; Bassett, A.; Parfrey, P. The natural history of a genetic subtype of arrhythmogenic right ventricular cardiomyopathy caused by a p. S358L mutation in TMEM43. Clin. Genet. 2013, 83, 321–331. [Google Scholar] [CrossRef] [PubMed]
- Merner, N.D.; Hodgkinson, K.A.; Haywood, A.F.; Connors, S.; French, V.M.; Drenckhahn, J.-D.; Kupprion, C.; Ramadanova, K.; Thierfelder, L.; McKenna, W. Arrhythmogenic right ventricular cardiomyopathy type 5 is a fully penetrant, lethal arrhythmic disorder caused by a missense mutation in the TMEM43 gene. Am. J. Hum. Genet. 2008, 82, 809–821. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kato, K.; Takahashi, N.; Fujii, Y.; Umehara, A.; Nishiuchi, S.; Makiyama, T.; Ohno, S.; Horie, M. LMNA cardiomyopathy detected in Japanese arrhythmogenic right ventricular cardiomyopathy cohort. J. Cardiol. 2016, 68, 346–351. [Google Scholar] [CrossRef] [Green Version]
- Liang, J.J.; Goodsell, K.; Grogan, M.; Ackerman, M.J. LMNA-mediated arrhythmogenic right ventricular cardiomyopathy and charcot-marie-tooth type 2B1: A patient-discovered unifying diagnosis. J. Cardiovasc. Electrophysiol. 2016, 27, 868–871. [Google Scholar] [CrossRef]
- Lombardi, R.; Dong, J.; Rodriguez, G.; Bell, A.; Leung, T.K.; Schwartz, R.J.; Willerson, J.T.; Brugada, R.; Marian, A.J. Genetic fate mapping identifies second heart field progenitor cells as a source of adipocytes in arrhythmogenic right ventricular cardiomyopathy. Circ. Res. 2009, 104, 1076–1084. [Google Scholar] [CrossRef] [PubMed]
- Lombardi, R.; Chen, S.N.; Ruggiero, A.; Gurha, P.; Czernuszewicz, G.Z.; Willerson, J.T.; Marian, A.J. Cardiac fibro-adipocyte progenitors express desmosome proteins and preferentially differentiate to adipocytes upon deletion of the desmoplakin gene. Circ. Res. 2016, 119, 41–54. [Google Scholar] [CrossRef]
- Matthes, S.A.; Taffet, S.; Delmar, M. Plakophilin-2 and the migration, differentiation and transformation of cells derived from the epicardium of neonatal rat hearts. Cell Commun. Adhes. 2011, 18, 73–84. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.; Pu, W.; Li, G.; Huang, X.; He, L.; Tian, X.; Liu, Q.; Zhang, L.; Wu, S.M.; Sucov, H.M. Endocardium minimally contributes to coronary endothelium in the embryonic ventricular free walls. Circ. Res. 2016, 118, 1880–1893. [Google Scholar] [CrossRef] [Green Version]
- Sommariva, E.; Brambilla, S.; Carbucicchio, C.; Gambini, E.; Meraviglia, V.; Dello Russo, A.; Farina, F.; Casella, M.; Catto, V.; Pontone, G. Cardiac mesenchymal stromal cells are a source of adipocytes in arrhythmogenic cardiomyopathy. Eur. Heart J. 2016, 37, 1835–1846. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, H.; Lui, K.O.; Zhou, B. Endocardial Cell Plasticity in Cardiac Development, Diseases and Regeneration. Circ. Res. 2018, 122, 774–789. [Google Scholar] [CrossRef] [PubMed]
- Arbustini, E.; Favalli, V.; Narula, N.; Serio, A.; Grasso, M. Left Ventricular Noncompaction: A Distinct Genetic Cardiomyopathy? J. Am. Coll. Cardiol. 2016, 68, 949–966. [Google Scholar] [CrossRef] [PubMed]
- Kulikova, O.; Brodehl, A.; Kiseleva, A.; Myasnikov, R.; Meshkov, A.; Stanasiuk, C.; Gartner, A.; Divashuk, M.; Sotnikova, E.; Koretskiy, S.; et al. The Desmin (DES) Mutation p.A337P Is Associated with Left-Ventricular Non-Compaction Cardiomyopathy. Genes 2021, 12, 121. [Google Scholar] [CrossRef]
- Probst, S.; Oechslin, E.; Schuler, P.; Greutmann, M.; Boyé, P.; Knirsch, W.; Berger, F.; Thierfelder, L.; Jenni, R.; Klaassen, S. Sarcomere gene mutations in isolated left ventricular noncompaction cardiomyopathy do not predict clinical phenotype. Circ. Cardiovasc. Genet. 2011, 4, 367–374. [Google Scholar] [CrossRef] [Green Version]
- Sedaghat-Hamedani, F.; Haas, J.; Zhu, F.; Geier, C.; Kayvanpour, E.; Liss, M.; Lai, A.; Frese, K.; Pribe-Wolferts, R.; Amr, A. Clinical genetics and outcome of left ventricular non-compaction cardiomyopathy. Eur. Heart J. 2017, 38, 3449–3460. [Google Scholar] [CrossRef]
- Tian, Y.; Morrisey, E.E. Importance of myocyte-nonmyocyte interactions in cardiac development and disease. Circ. Res. 2012, 110, 1023–1034. [Google Scholar] [CrossRef]
- Muchtar, E.; Blauwet, L.A.; Gertz, M.A. Restrictive cardiomyopathy: Genetics, pathogenesis, clinical manifestations, diagnosis, and therapy. Circ. Res. 2017, 121, 819–837. [Google Scholar] [CrossRef]
- Nihoyannopoulos, P.; Dawson, D. Restrictive cardiomyopathies. Eur. J. Echocardiogr. 2009, 10, iii23–iii33. [Google Scholar] [CrossRef] [Green Version]
- Mogensen, J.; Arbustini, E. Restrictive cardiomyopathy. Curr. Opin. Cardiol. 2009, 24, 214–220. [Google Scholar] [CrossRef]
- Brodehl, A.; Gerull, B. Genetic Insights into Primary Restrictive Cardiomyopathy. J. Clin. Med. 2022, 11, 2094. [Google Scholar] [CrossRef] [PubMed]
- Peled, Y.; Gramlich, M.; Yoskovitz, G.; Feinberg, M.S.; Afek, A.; Polak-Charcon, S.; Pras, E.; Sela, B.A.; Konen, E.; Weissbrod, O.; et al. Titin mutation in familial restrictive cardiomyopathy. Int. J. Cardiol. 2014, 171, 24–30. [Google Scholar] [CrossRef] [PubMed]
- Kostareva, A.; Gudkova, A.; Sjöberg, G.; Mörner, S.; Semernin, E.; Krutikov, A.; Shlyakhto, E.; Sejersen, T. Deletion in TNNI3 gene is associated with restrictive cardiomyopathy. Int. J. Cardiol. 2009, 131, 410–412. [Google Scholar] [CrossRef]
- Greenway, S.C.; Wilson, G.J.; Wilson, J.; George, K.; Kantor, P.F. Sudden death in an infant with angina, restrictive cardiomyopathy, and coronary artery bridging: An unusual phenotype for a β-myosin heavy chain (MYH7) sarcomeric protein mutation. Circ. Heart Fail. 2012, 5, e92–e93. [Google Scholar] [CrossRef] [Green Version]
- Kaski, J.P.; Syrris, P.; Burch, M.; Tome-Esteban, M.-T.; Fenton, M.; Christiansen, M.; Andersen, P.S.; Sebire, N.; Ashworth, M.; Deanfield, J.E. Idiopathic restrictive cardiomyopathy in children is caused by mutations in cardiac sarcomere protein genes. Heart 2008, 94, 1478–1484. [Google Scholar] [CrossRef] [PubMed]
- Brodehl, A.; Pour Hakimi, S.A.; Stanasiuk, C.; Ratnavadivel, S.; Hendig, D.; Gaertner, A.; Gerull, B.; Gummert, J.; Paluszkiewicz, L.; Milting, H. Restrictive Cardiomyopathy is Caused by a Novel Homozygous Desmin (DES) Mutation p.Y122H Leading to a Severe Filament Assembly Defect. Genes 2019, 10, 918. [Google Scholar] [CrossRef] [Green Version]
- Yu, H.-C.; Coughlin, C.R.; Geiger, E.A.; Salvador, B.J.; Elias, E.R.; Cavanaugh, J.L.; Chatfield, K.C.; Miyamoto, S.D.; Shaikh, T.H. Discovery of a potentially deleterious variant in TMEM87B in a patient with a hemizygous 2q13 microdeletion suggests a recessive condition characterized by congenital heart disease and restrictive cardiomyopathy. Mol. Case Stud. 2016, 2, a000844. [Google Scholar] [CrossRef] [Green Version]
- Brodehl, A.; Ferrier, R.A.; Hamilton, S.J.; Greenway, S.C.; Brundler, M.-A.; Yu, W.; Gibson, W.T.; McKinnon, M.L.; McGillivray, B.C.; Alvarez, N.; et al. Mutations in FLNC are Associated with Familial Restrictive Cardiomyopathy. Hum. Mutat. 2016, 37, 269–279. [Google Scholar] [CrossRef]
- Brayson, D.; Shanahan, C.M. Current insights into LMNA cardiomyopathies: Existing models and missing LINCs. Nucleus 2017, 8, 17–33. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cho, S.; Vashisth, M.; Abbas, A.; Majkut, S.; Vogel, K.; Xia, Y.; Ivanovska, I.L.; Irianto, J.; Tewari, M.; Zhu, K. Mechanosensing by the lamina protects against nuclear rupture, DNA damage, and cell-cycle arrest. Dev. Cell 2019, 49, 920–935.e5. [Google Scholar] [CrossRef] [PubMed]
- Chai, R.J.; Werner, H.; Li, P.Y.; Lee, Y.L.; Nyein, K.T.; Solovei, I.; Luu, T.D.A.; Sharma, B.; Navasankari, R.; Maric, M.; et al. Disrupting the LINC complex by AAV mediated gene transduction prevents progression of Lamin induced cardiomyopathy. Nat. Commun. 2021, 12, 4722. [Google Scholar] [CrossRef] [PubMed]
- Andres, V.; Gonzalez, J.M. Role of A-type lamins in signaling, transcription, and chromatin organization. J. Cell Biol. 2009, 187, 945–957. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bernasconi, P.; Carboni, N.; Ricci, G.; Siciliano, G.; Lattanzi, G. Elevated TGF β2 serum levels in Emery-Dreifuss Muscular Dystrophy: Implications for myocyte and tenocyte differentiation and fibrogenic processes. Nucleus 2018, 9, 292–304. [Google Scholar] [CrossRef] [Green Version]
- Chatzifrangkeskou, M.; Le Dour, C.; Wu, W.; Morrow, J.P.; Joseph, L.C.; Beuvin, M.; Sera, F.; Homma, S.; Vignier, N.; Mougenot, N.; et al. ERK1/2 directly acts on CTGF/CCN2 expression to mediate myocardial fibrosis in cardiomyopathy caused by mutations in the lamin A/C gene. Hum. Mol. Genet. 2016, 25, 2220–2233. [Google Scholar] [CrossRef] [Green Version]
- Wu, W.; Muchir, A.; Shan, J.; Bonne, G.; Worman, H.J. Mitogen-activated protein kinase inhibitors improve heart function and prevent fibrosis in cardiomyopathy caused by mutation in lamin A/C gene. Circulation 2011, 123, 53–61. [Google Scholar] [CrossRef] [Green Version]
- Muchir, A.; Pavlidis, P.; Decostre, V.; Herron, A.J.; Arimura, T.; Bonne, G.; Worman, H.J. Activation of MAPK pathways links LMNA mutations to cardiomyopathy in Emery-Dreifuss muscular dystrophy. J. Clin. Investig. 2007, 117, 1282–1293. [Google Scholar] [CrossRef]
- Wu, W.; Shan, J.; Bonne, G.; Worman, H.J.; Muchir, A. Pharmacological inhibition of c-Jun N-terminal kinase signaling prevents cardiomyopathy caused by mutation in LMNA gene. Biochim. Biophys. Acta (BBA)-Mol. Basis Dis. 2010, 1802, 632–638. [Google Scholar] [CrossRef] [Green Version]
- Umbarkar, P.; Tousif, S.; Singh, A.P. Fibroblast GSK-3α Promotes Fibrosis via RAF-MEK-ERK Pathway in the Injured Heart. Circ. Res. 2022, 131, 620–636. [Google Scholar] [CrossRef]
- Cattin, M.E.; Muchir, A.; Bonne, G. ‘State-of-the-heart’ of cardiac laminopathies. Curr. Opin. Cardiol. 2013, 28, 297–304. [Google Scholar] [CrossRef] [PubMed]
- Bertero, A.; Fields, P.A.; Smith, A.S.; Leonard, A.; Beussman, K.; Sniadecki, N.J.; Kim, D.-H.; Tse, H.-F.; Pabon, L.; Shendure, J. Chromatin compartment dynamics in a haploinsufficient model of cardiac laminopathy. J. Cell Biol. 2019, 218, 2919–2944. [Google Scholar] [CrossRef] [PubMed] [Green Version]
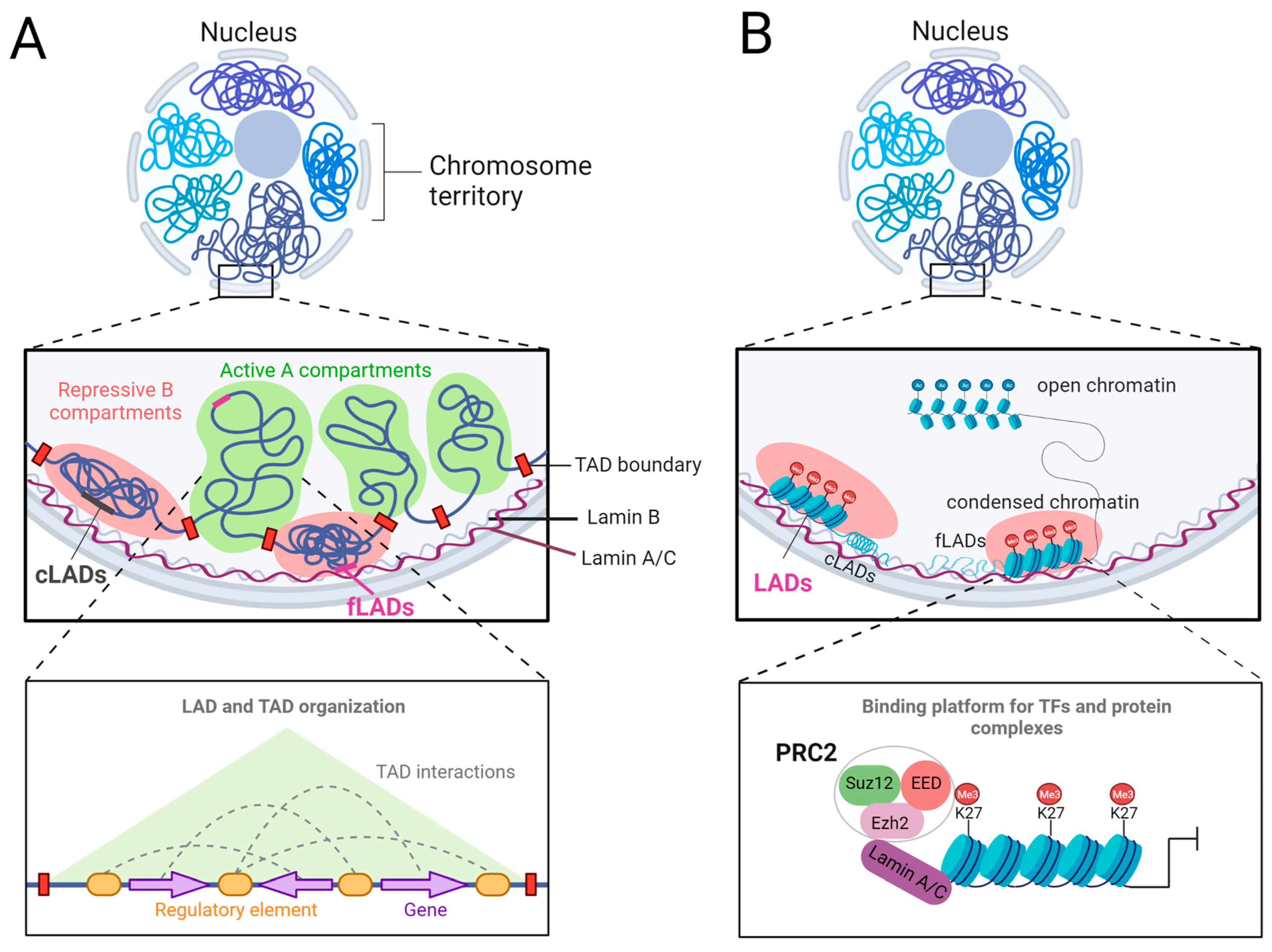
- Meuleman, W.; Peric-Hupkes, D.; Kind, J.; Beaudry, J.B.; Pagie, L.; Kellis, M.; Reinders, M.; Wessels, L.; van Steensel, B. Constitutive nuclear lamina-genome interactions are highly conserved and associated with A/T-rich sequence. Genome Res. 2013, 23, 270–280. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peric-Hupkes, D.; Meuleman, W.; Pagie, L.; Bruggeman, S.W.; Solovei, I.; Brugman, W.; Graf, S.; Flicek, P.; Kerkhoven, R.M.; van Lohuizen, M.; et al. Molecular maps of the reorganization of genome-nuclear lamina interactions during differentiation. Mol. Cell 2010, 38, 603–613. [Google Scholar] [CrossRef] [PubMed]
- Van Bortle, K.; Corces, V.G. Spinning the web of cell fate. Cell 2013, 152, 1213–1217. [Google Scholar] [CrossRef] [Green Version]
- Poleshko, A.; Shah, P.P.; Gupta, M.; Babu, A.; Morley, M.P.; Manderfield, L.J.; Ifkovits, J.L.; Calderon, D.; Aghajanian, H.; Sierra-Pagán, J.E. Genome-nuclear lamina interactions regulate cardiac stem cell lineage restriction. Cell 2017, 171, 573–587.e14. [Google Scholar] [CrossRef] [Green Version]
- Oldenburg, A.; Briand, N.; Sørensen, A.L.; Cahyani, I.; Shah, A.; Moskaug, J.Ø.; Collas, P. A lipodystrophy-causing lamin A mutant alters conformation and epigenetic regulation of the anti-adipogenic MIR335 locus. J. Cell Biol. 2017, 216, 2731–2743. [Google Scholar] [CrossRef] [Green Version]
- Pellegrini, C.; Columbaro, M.; Schena, E.; Prencipe, S.; Andrenacci, D.; Iozzo, P.; Angela Guzzardi, M.; Capanni, C.; Mattioli, E.; Loi, M. Altered adipocyte differentiation and unbalanced autophagy in type 2 Familial Partial Lipodystrophy: An in vitro and in vivo study of adipose tissue browning. Exp. Mol. Med. 2019, 51, 1–17. [Google Scholar] [CrossRef] [Green Version]
- Czapiewski, R.; Batrakou, D.G.; de Las Heras, J.I.; Carter, R.N.; Sivakumar, A.; Sliwinska, M.; Dixon, C.R.; Webb, S.; Lattanzi, G.; Morton, N.M. Genomic loci mispositioning in Tmem120a knockout mice yields latent lipodystrophy. Nat. Commun. 2022, 13, 321. [Google Scholar] [CrossRef]
- Ramirez-Martinez, A.; Zhang, Y.; Chen, K.; Kim, J.; Cenik, B.K.; McAnally, J.R.; Cai, C.; Shelton, J.M.; Huang, J.; Brennan, A.; et al. The nuclear envelope protein Net39 is essential for muscle nuclear integrity and chromatin organization. Nat. Commun. 2021, 12, 690. [Google Scholar] [CrossRef]
- Shimi, T.; Pfleghaar, K.; Kojima, S.; Pack, C.G.; Solovei, I.; Goldman, A.E.; Adam, S.A.; Shumaker, D.K.; Kinjo, M.; Cremer, T.; et al. The A- and B-type nuclear lamin networks: Microdomains involved in chromatin organization and transcription. Genes Dev. 2008, 22, 3409–3421. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Han, L.; Choudhury, S.; Mich-Basso, J.D.; Ammanamanchi, N.; Ganapathy, B.; Suresh, S.; Khaladkar, M.; Singh, J.; Maehr, R.; Zuppo, D.A.; et al. Lamin B2 Levels Regulate Polyploidization of Cardiomyocyte Nuclei and Myocardial Regeneration. Dev. Cell 2020, 53, 42–59.e11. [Google Scholar] [CrossRef] [PubMed]
- Soufi, A.; Garcia, M.F.; Jaroszewicz, A.; Osman, N.; Pellegrini, M.; Zaret, K.S. Pioneer transcription factors target partial DNA motifs on nucleosomes to initiate reprogramming. Cell 2015, 161, 555–568. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spitz, F.; Furlong, E.E. Transcription factors: From enhancer binding to developmental control. Nat. Rev. Genet. 2012, 13, 613–626. [Google Scholar] [CrossRef]
- Swinstead, E.E.; Miranda, T.B.; Paakinaho, V.; Baek, S.; Goldstein, I.; Hawkins, M.; Karpova, T.S.; Ball, D.; Mazza, D.; Lavis, L.D.; et al. Steroid Receptors Reprogram FoxA1 Occupancy through Dynamic Chromatin Transitions. Cell 2016, 165, 593–605. [Google Scholar] [CrossRef] [Green Version]
- Zaret, K.S.; Carroll, J.S. Pioneer transcription factors: Establishing competence for gene expression. Genes Dev. 2011, 25, 2227–2241. [Google Scholar] [CrossRef] [Green Version]
- Hatta, M.; Cirillo, L.A. Chromatin opening and stable perturbation of core histone:DNA contacts by FoxO1. J. Biol. Chem. 2007, 282, 35583–35593. [Google Scholar] [CrossRef] [Green Version]
- Auguste, G.; Gurha, P.; Lombardi, R.; Coarfa, C.; Willerson, J.T.; Marian, A.J. Suppression of Activated FOXO Transcription Factors in the Heart Prolongs Survival in a Mouse Model of Laminopathies. Circ. Res. 2018, 122, 678–692. [Google Scholar] [CrossRef]
- Glass, C.A.; Glass, J.R.; Taniura, H.; Hasel, K.W.; Blevitt, J.M.; Gerace, L. The alpha-helical rod domain of human lamins A and C contains a chromatin binding site. EMBO J. 1993, 12, 4413–4424. [Google Scholar] [CrossRef]
- Dechat, T.; Korbei, B.; Vaughan, O.A.; Vlcek, S.; Hutchison, C.J.; Foisner, R. Lamina-associated polypeptide 2alpha binds intranuclear A-type lamins. J. Cell Sci. 2000, 113 Pt 19, 3473–3484. [Google Scholar] [CrossRef]
- Lee, K.K.; Haraguchi, T.; Lee, R.S.; Koujin, T.; Hiraoka, Y.; Wilson, K.L. Distinct functional domains in emerin bind lamin A and DNA-bridging protein BAF. J. Cell Sci. 2001, 114, 4567–4573. [Google Scholar] [CrossRef]
- Ye, Q.; Worman, H.J. Interaction between an integral protein of the nuclear envelope inner membrane and human chromodomain proteins homologous to Drosophila HP1. J. Biol. Chem. 1996, 271, 14653–14656. [Google Scholar] [CrossRef] [Green Version]
- Lieberman-Aiden, E.; van Berkum, N.L.; Williams, L.; Imakaev, M.; Ragoczy, T.; Telling, A.; Amit, I.; Lajoie, B.R.; Sabo, P.J.; Dorschner, M.O.; et al. Comprehensive mapping of long-range interactions reveals folding principles of the human genome. Science 2009, 326, 289–293. [Google Scholar] [CrossRef] [Green Version]
- Rao, S.S.; Huntley, M.H.; Durand, N.C.; Stamenova, E.K.; Bochkov, I.D.; Robinson, J.T.; Sanborn, A.L.; Machol, I.; Omer, A.D.; Lander, E.S. A 3D map of the human genome at kilobase resolution reveals principles of chromatin looping. Cell 2014, 159, 1665–1680. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lund, E.; Oldenburg, A.R.; Delbarre, E.; Freberg, C.T.; Duband-Goulet, I.; Eskeland, R.; Buendia, B.; Collas, P. Lamin A/C-promoter interactions specify chromatin state–dependent transcription outcomes. Genome Res. 2013, 23, 1580–1589. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cesarini, E.; Mozzetta, C.; Marullo, F.; Gregoretti, F.; Gargiulo, A.; Columbaro, M.; Cortesi, A.; Antonelli, L.; Di Pelino, S.; Squarzoni, S. Lamin A/C sustains PcG protein architecture, maintaining transcriptional repression at target genes. J. Cell Biol. 2015, 211, 533–551. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schuettengruber, B.; Chourrout, D.; Vervoort, M.; Leblanc, B.; Cavalli, G. Genome regulation by polycomb and trithorax proteins. Cell 2007, 128, 735–745. [Google Scholar] [CrossRef] [Green Version]
- Margueron, R.; Reinberg, D. The Polycomb complex PRC2 and its mark in life. Nature 2011, 469, 343–349. [Google Scholar] [CrossRef] [Green Version]
- He, A.; Ma, Q.; Cao, J.; Von Gise, A.; Zhou, P.; Xie, H.; Zhang, B.; Hsing, M.; Christodoulou, D.C.; Cahan, P. Polycomb repressive complex 2 regulates normal development of the mouse heart. Circ. Res. 2012, 110, 406–415. [Google Scholar] [CrossRef] [Green Version]
- Delgado-Olguín, P.; Huang, Y.; Li, X.; Christodoulou, D.; Seidman, C.E.; Seidman, J.; Tarakhovsky, A.; Bruneau, B.G. Epigenetic repression of cardiac progenitor gene expression by Ezh2 is required for postnatal cardiac homeostasis. Nat. Genet. 2012, 44, 343. [Google Scholar] [CrossRef] [Green Version]
- Bianchi, A.; Mozzetta, C.; Pegoli, G.; Lucini, F.; Valsoni, S.; Rosti, V.; Petrini, C.; Cortesi, A.; Gregoretti, F.; Antonelli, L. Dysfunctional polycomb transcriptional repression contributes to Lamin A/C dependent muscular dystrophy. J. Clin. Investig. 2020, 130, 2408–2421. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hershberger, R.E.; Morales, A.; Siegfried, J.D. Clinical and genetic issues in dilated cardiomyopathy: A review for genetics professionals. Genet. Med. 2010, 12, 655–667. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hershberger, R.E.; Jordan, E. LMNA-Related Dilated Cardiomyopathy; University of Washington: Seattle, WA, USA, 2022. [Google Scholar]
- Ramos, F.J.; Chen, S.C.; Garelick, M.G.; Dai, D.F.; Liao, C.Y.; Schreiber, K.H.; MacKay, V.L.; An, E.H.; Strong, R.; Ladiges, W.C.; et al. Rapamycin reverses elevated mTORC1 signaling in lamin A/C-deficient mice, rescues cardiac and skeletal muscle function, and extends survival. Sci. Transl. Med. 2012, 4, 144ra103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- MacRae, C.; Taylor, M.; Mestroni, L.; Moses, J.; Ashley, E.; Wheeler, M.; Lakdawala, N.; Hershberger, R.; Ptaszynski, M.; Sandor, V. Phase 2 study of a797, an oral, selective p38 mitogen-activated protein kinase inhibitor, in patients with lamin a/c-related dilated cardiomyopathy. In Proceedings of European Heart Journal; Oxford Univ Press: Oxford, UK, 2016; p. 1011. [Google Scholar]
- Santiago-Fernández, O.; Osorio, F.G.; Quesada, V.; Rodríguez, F.; Basso, S.; Maeso, D.; Rolas, L.; Barkaway, A.; Nourshargh, S.; Folgueras, A.R. Development of a CRISPR/Cas9-based therapy for Hutchinson–Gilford progeria syndrome. Nat. Med. 2019, 25, 423–426. [Google Scholar] [CrossRef] [PubMed]
- Beyret, E.; Liao, H.K.; Yamamoto, M.; Hernandez-Benitez, R.; Fu, Y.; Erikson, G. Single-dose CRISPR-Cas9 therapy extends lifespan of mice with Hutchinson-Gilford progeria syndrome. Nat. Med. 2019, 25, 419–422. [Google Scholar] [CrossRef] [PubMed]
- Koblan, L.W.; Erdos, M.R.; Wilson, C.; Cabral, W.A.; Levy, J.M.; Xiong, Z.-M.; Tavarez, U.L.; Davison, L.M.; Gete, Y.G.; Mao, X.; et al. In vivo base editing rescues Hutchinson Gilford progeria syndrome in mice. Nature 2021, 589, 608–614. [Google Scholar] [CrossRef] [PubMed]
- Anzalone, A.V.; Koblan, L.W.; Liu, D.R. Genome editing with CRISPR–Cas nucleases, base editors, transposases and prime editors. Nat. Biotechnol. 2020, 38, 824–844. [Google Scholar] [CrossRef]
- Gaudelli, N.M.; Komor, A.C.; Rees, H.A.; Packer, M.S.; Liu, D.R. Programmable base editing of AT to GC in genomic DNA without DNA cleavage. Nature 2017, 551, 464–471. [Google Scholar] [CrossRef] [Green Version]
- Zuris, J.A.; Liu, D.R.; Packer, M.S.; Komor, A.C.; Kim, Y.B. Programmable editing of a target base in genomic DNA without double-stranded DNA cleavage. Nature 2016, 533, 420–424. [Google Scholar]
- Anzalone, A.V.; Randolph, P.B.; Davis, J.R.; Sousa, A.A.; Koblan, L.W.; Levy, J.M.; Chen, P.J.; Christopher, W.; Newby, G.A.; Aditya, R. Search-and-replace genome editing without double-strand breaks or donor DNA. Nature 2019, 576, 149–157. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| LMNA Mutation | Disease | Clinical Features | References |
|---|---|---|---|
| p. N195K | DCM | Heart dilatation, fibrosis, arrhythmia, sinus bradycardia, atrioventricular conduction block, and atrial arrhythmias | [26,35] |
| p. R225X | DCM | Atrial fibrillation, complete atrioventricular block, ventricular tachyarrhythmia, and heart failure | [36,37] |
| p. K117fs | DCM | Atrioventricular block, ventricular tachycardia, atrial fibrillation, arrhythmias at the single-cell level | [38] |
| p.c.908_909 delCT | DCM | Atrial fibrillation, sick sinus syndrome, dilated cardiomyopathy | [39] |
| p.28insA | DCM | Dilated cardiomyopathy with conduction defects | [40] |
| p. T101 | DCM, lipodystrophy, atypical progeroid syndrome | Hypertriglyceridemia, diabetes mellitus, insulin resistance, left ventricular myocyte hypertrophy, interstitial fibrosis | [41,42,43,44] |
| p. R377H | EDMD, LGMD, DCM | Muscular dystrophy, atrial fibrillation or flutter; conduction defects | [45,46,47,48] |
| p. S143p | DCM | Atrioventricular conduction defects, left ventricular systolic dysfunction and dilatation | [49,50] |
| p.H222P | EDMD | Muscle weakness, cardiac arrhythmias | [51] |
| p. K219T | DCM | Heart dilatation, atrial fibrillation, atrioventricular block | [52,53] |
| p. E161K | DCM | Dilatation, atrial fibrillation, conduction system diseases | [40,54] |
| p. R541C | DCM, LVNC | Reduced heart contractility, left ventricle dilatation, polymorphic premature ventricular contraction, diffuse ST-T change | [41,55] |
| p. R190W | DCM, LVNC, ARVC | Left ventricular noncompaction, conduction system defect, abnormal activation of ERK1/2 signaling and sarcomeric disorganization | [56,57,58] |
| p. R644C | DCM, LVNC, ARVC, | Also leads to lipodystrophy, atypical progeria, phenotypic diversity, and low penetrance associated with the R644C mutation | [59,60] |
| p. V445E | LVNC | Ventricular tachycardia/fibrillation | [58] |
| p.c.835 delG:p.Glu279ArgfsX201 | RCM | Diastolic dysfunction, biatrial enlargement, atrial fibrillation, skeletal muscle weakness | [61] |
| Mouse Model | Gene Targeting Strategy | Disease | Homozygous Phenotype | Heterozygous Phonotype | References |
|---|---|---|---|---|---|
| Lmna−/− * | Deletion of exons 8-11; a truncated lamin A protein of 54 kDa is still expressed | DCM, EDMD | Retarded postnatal growth, conduction disorders, DCM, EDMD, death by 8 weeks of age | AV conduction defects, both atrial and ventricular arrhythmias; develop DCM by 50 weeks | [64,65] |
| Lmna−/− | Deletion of exon 2 | DCM LVNC | Retarded postnatal growth, conduction disorders, DCM, noncompaction, death within 1 month, developmental defects | RV dilatation, RV noncompaction, developmental defects | [62,63] |
| Lmna GT−/− | A gene trap cassette inserted upstream of exon 2 of Lmna | N/A | Growth retardation at 2 weeks, impaired postnatal cardiac hypertrophy, skeletal muscle hypotrophy, defects in lipid metabolism | No apparent abnormalities | [66] |
| Myh6 cre -Lmna f/f | Conditional deletion of Lmna in cardiomyocytes | DCM | DCM, cardiac dysfunction, conduction defects, ventricular arrhythmias, fibrosis, apoptosis, and premature death within 4 weeks | Develop cardiac dilatation and dysfunction, cardiac arrhythmias, fibrosis in older mice | [67] |
| Lamin C only | Deletion of the last 150 nucleotides of exon 11 and the complete intron 11 | N/A | No obvious phenotype | No obvious phenotype | [68] |
| Lamin A only | Deletion of introns 10 and 11, the last 30 bp of exon 11, and the first 24 bp of exon 12 | N/A | No apparent abnormalities | N/A | [69] |
| Pre-lamin A only | Deletion of intron 10 | N/A | No apparent abnormalities | N/A | [69] |
| Lmna N195K | Missense mutation in exon 3 | DCM | DCM, conduction defects, fibrosis, minor growth retardation, increased heart weight, death at 12–14 weeks | No obvious phenotype | [35] |
| Lmna R225X | Nonsense mutation at exon 4 causing premature truncation of both lamin A and lamin C | DCM | Retarded postnatal growth, conduction disorders, dilated cardiomyopathy, AV node fibrosis, death within postnatal 2 weeks | No apparent abnormalities | [36] |
| Lmna E82K | Transgenic mice expressing Lmna E82K under the control of α-MHC promoter | DCM | N/A | DCM, conduction defects, fibrosis, increased heart weight | [70] |
| Lmna ∆K32 | Deletion of lysine 32 of lamin A/C in exon 1 | DCM | Retarded postnatal growth, striated muscle maturation delay, metabolic defects including reduced adipose tissue and hypoglycemia, death within 3 weeks | Develop a progressive cardiac dysfunction and DCM | [71,72,73] |
| Lmna R541C | Missense mutation in exon 10 | DCM | Ventricular dilatation and reduced systolic function | N/A | [55] |
| Lmna H222P | Missense mutation in exon 4 | EDMD DCM | Heart dilatation, conduction defects, increased fibrosis, hypertrophy defects, death by 9 months of age, developmental defects | No apparent abnormalities | [74,75,76] |
| Lmna M317K | Transgenic mice expressing Lmna M317K missense mutation under the control of α-MHC promoter | EDMD | N/A | Increased eosinophilia and fragmentation of cardiomyofibrils, nuclear pyknosis and edema without fibrosis or significant inflammation, death at 2–7 weeks of age | [77] |
| Lmna D300N | Transgenic mice expressing Lmna D300N; Myh6-tTA mice | DCM | N/A | Heart dilatation, increased heart-to-body-weight ratio, fibrosis, death within two months | [78] |
| Lmna L530P | Missense mutation in exon 9 | HGPS | Loss of subcutaneous fat, reduction in growth rate, and death by 4 weeks of age | No apparent abnormalities | [79] |
| Lmna G609G | Point mutation in exon 11 | HGPS | Shortened life span, reduced body weight, bone and cardiovascular abnormalities, death at an average of 100 days | Develop a similar phenotype to homozygotes but at an older age, average death at 242 days | [80] |
| Lmna HG | Deletion of introns 10–11 and last 150 nucleotides of exon 11 | HGPS | Growth retardation osteoporosis, micrognathia, loss of adipose tissue, death by 3–4 weeks of age | Similar phenotype to homozygotes but less severe, death by 21 weeks | [81] |
| Lmna nHG | Deletion of introns 10 and 11 and the last 150 bp of exon 11 together with an exchange of cysteine to serine in the CaaX motif | HGPS | Weight loss, reduced subcutaneous and abdominal fat by 4–8 weeks of age, death at 17 weeks of age | Similar spectrum of disease phenotypes as Lmna HG/+ mice but less severe, death by 36 weeks of age | [82] |
| csmHG | Deletion of introns 10 and 11 and the last 150 bp of exon 11, three-nucleotide deletion (the isoleucine in progerin’s CaaX motif) | HGPS | No bone phenotype, normal body weight and survival | No bone disease, normal body weight and survival | [83] |
| G608G BAC | 164-kb BAC carrying mutated G608G human LMNA | HGPS | N/A | Progressive loss of vascular smooth muscle cells | [84] |
| tetop_LAG608G | Targeted expression of the lamin A G608G minigene using the keratin 5 transactivator | HGPS | N/A | Growth retardation, skin and teeth abnormalities, fibrosis, loss of hypodermal adipocytes | [85] |
| Keratin14-progerin | Vector expressing progerin in epidermis under the control of the keratin 14 promoter | HGPS | N/A | Severe abnormalities in skin keratinocyte nuclei, including nuclear envelope lobulation and decreased nuclear circularity | [86] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, Y.; Dobreva, G. Epigenetics in LMNA-Related Cardiomyopathy. Cells 2023, 12, 783. https://doi.org/10.3390/cells12050783
Wang Y, Dobreva G. Epigenetics in LMNA-Related Cardiomyopathy. Cells. 2023; 12(5):783. https://doi.org/10.3390/cells12050783
Chicago/Turabian StyleWang, Yinuo, and Gergana Dobreva. 2023. "Epigenetics in LMNA-Related Cardiomyopathy" Cells 12, no. 5: 783. https://doi.org/10.3390/cells12050783
APA StyleWang, Y., & Dobreva, G. (2023). Epigenetics in LMNA-Related Cardiomyopathy. Cells, 12(5), 783. https://doi.org/10.3390/cells12050783