

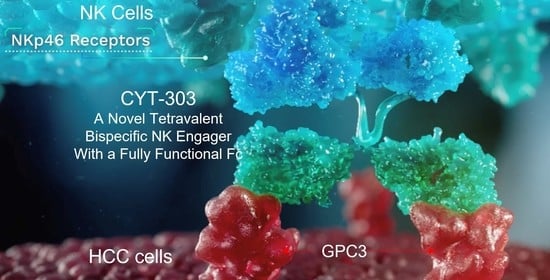
Derivation and Preclinical Characterization of CYT-303, a Novel NKp46-NK Cell Engager Targeting GPC3
 ,
,  ,
,
Abstract
:
1. Introduction
2. Materials and Methods
2.1. Cell Lines
2.2. Antibodies
2.3. Generation of Anti-NKp46 and GPC3 Monospecific Antibodies and CYT-303 Bispecific Antibodies
2.4. CYT-303 Binding to NKp46 and GPC3 Recombinant Proteins
2.5. PBNK Isolation
2.6. Anti-NKp46 mAb and hNKp46-Ig Binding Studies on NKp46 Expressing Cells and Human Tumor Cell Lines, Respectively
2.7. CYT-303 Binding PBNK and Tumor Cells
2.8. PBNK Cytolysis of HCC Tumor Cells
2.9. PBNK Degranulation in the Presence of HCC Tumor Cells
2.10. ADCP and CDC against Hep3B Tumor Cells
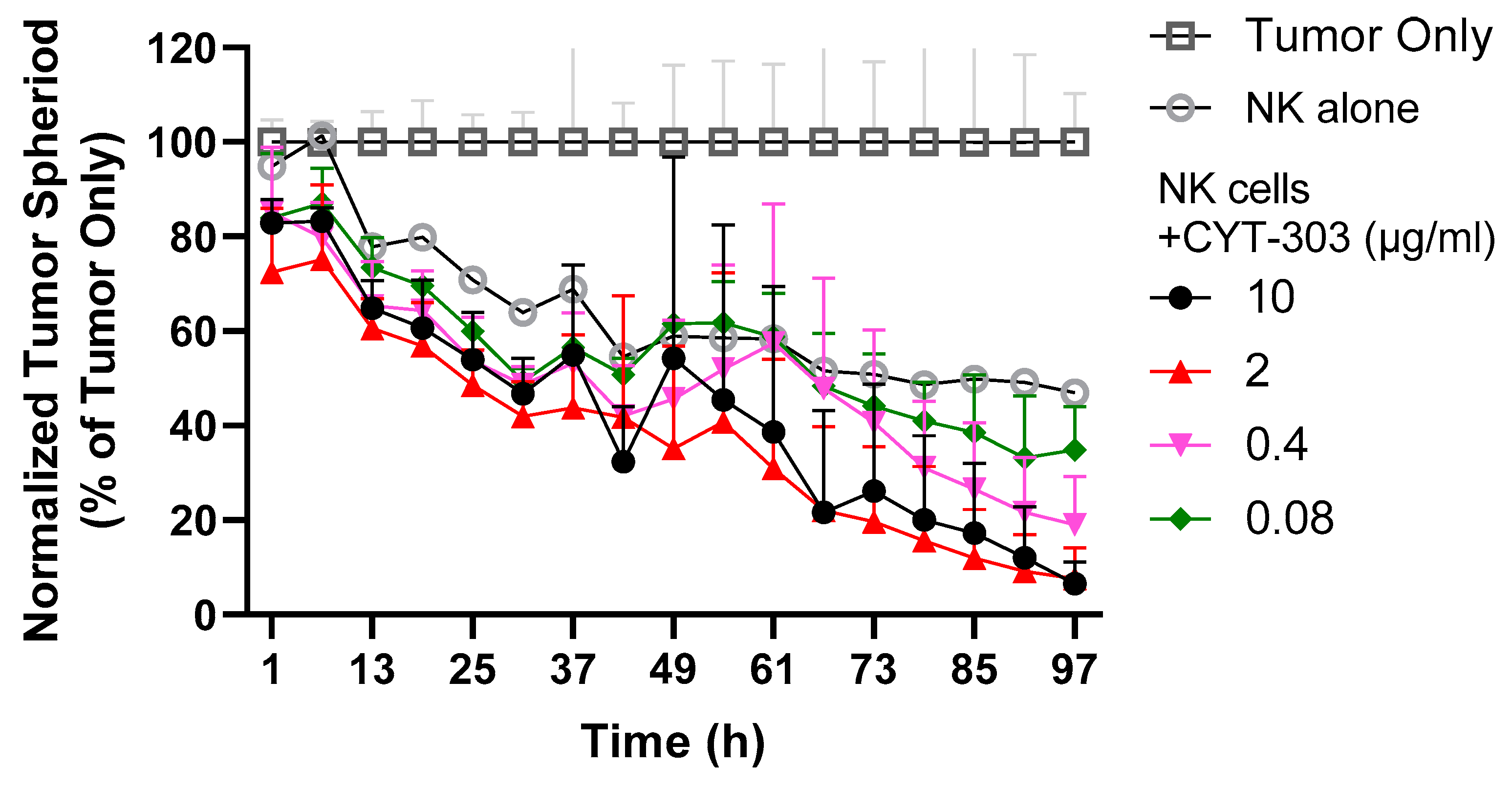
2.11. PBNK Cytolysis of HCC Tumor Spheroids
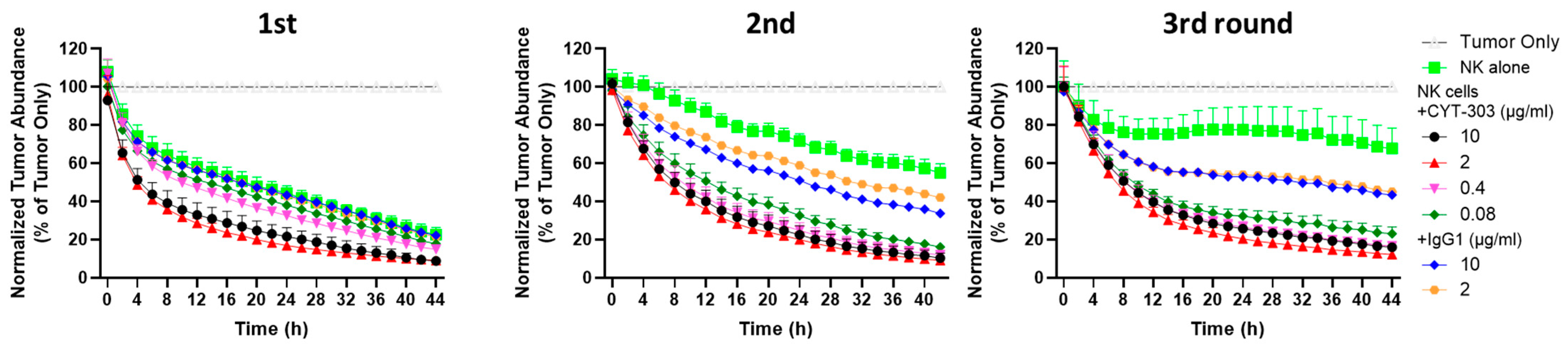
2.12. PBNK Serial Killing of HCC Tumor Cells
2.13. In Vitro PBMC Cytokine Release Assay
2.14. In Vitro Fratricide and Immune-Cell Depletion Assessments
2.15. CYT-303 Pharmacokinetics in Cynomolgus Monkeys
2.16. CYT-303 Repeat Dose 4-Week GLP Toxicology Study in Cynomolgus Monkeys
2.17. Pharmacokinetic and Anti-Drug Antibody Assays
3. Results
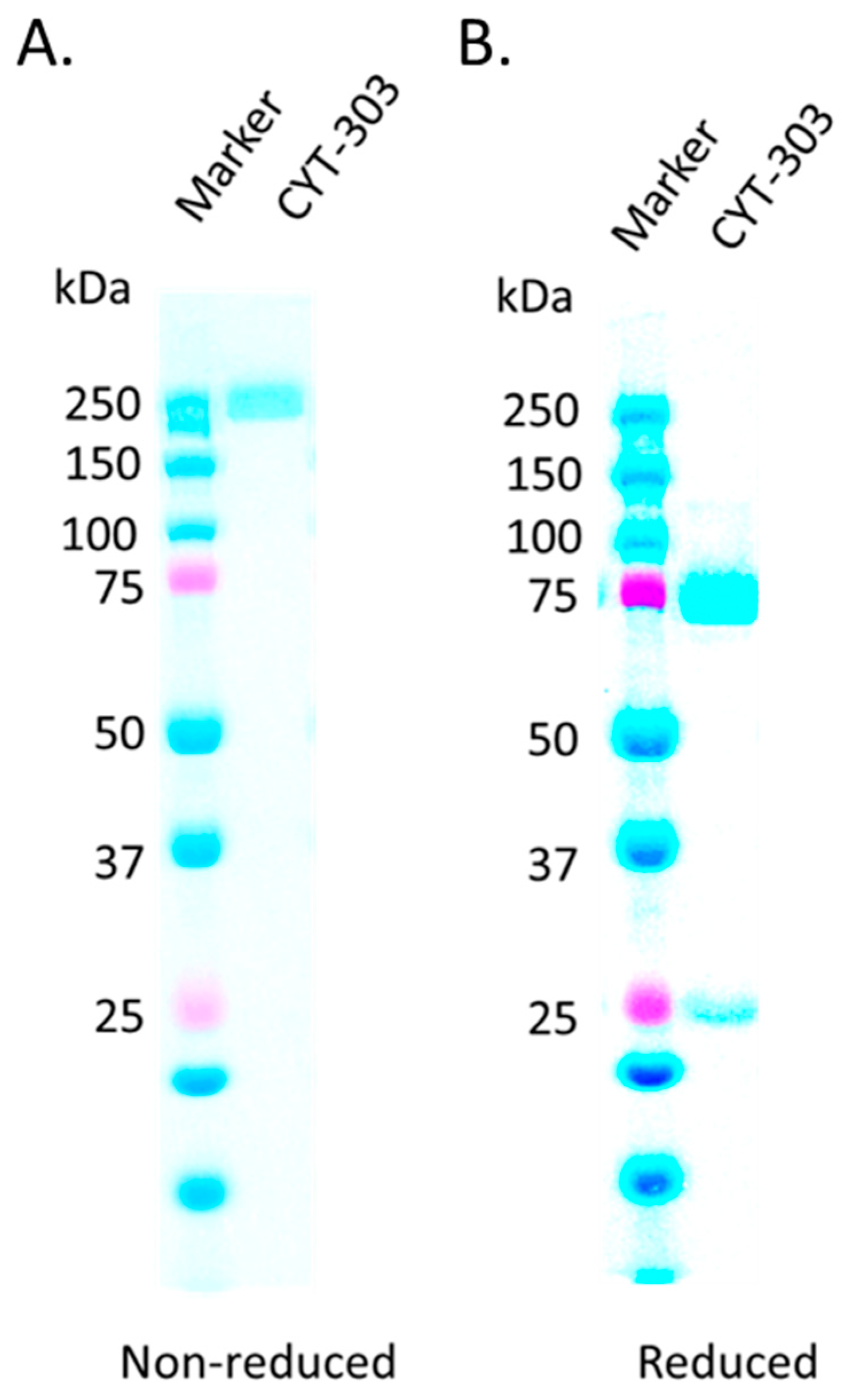
3.1. CYT-303 Design and Biochemical Characterization
3.2. Characterization of CYT-303 Binding to Recombinant Target Proteins and Cells
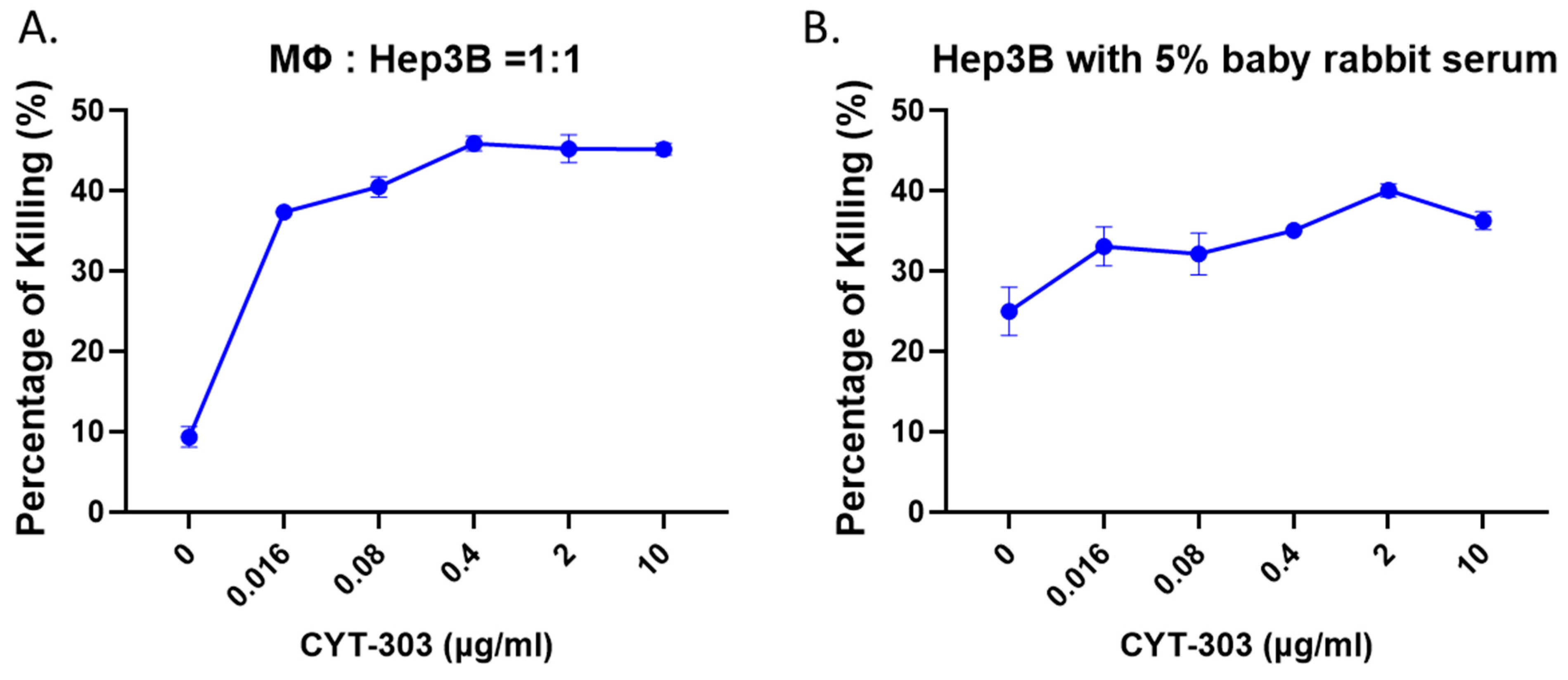
3.3. CYT-303 Redirected Human PBNK Cytotoxicity against HCC Tumor Cells
3.4. CYT-303 Mediated ADCP and CDC against HCC Tumor Cells
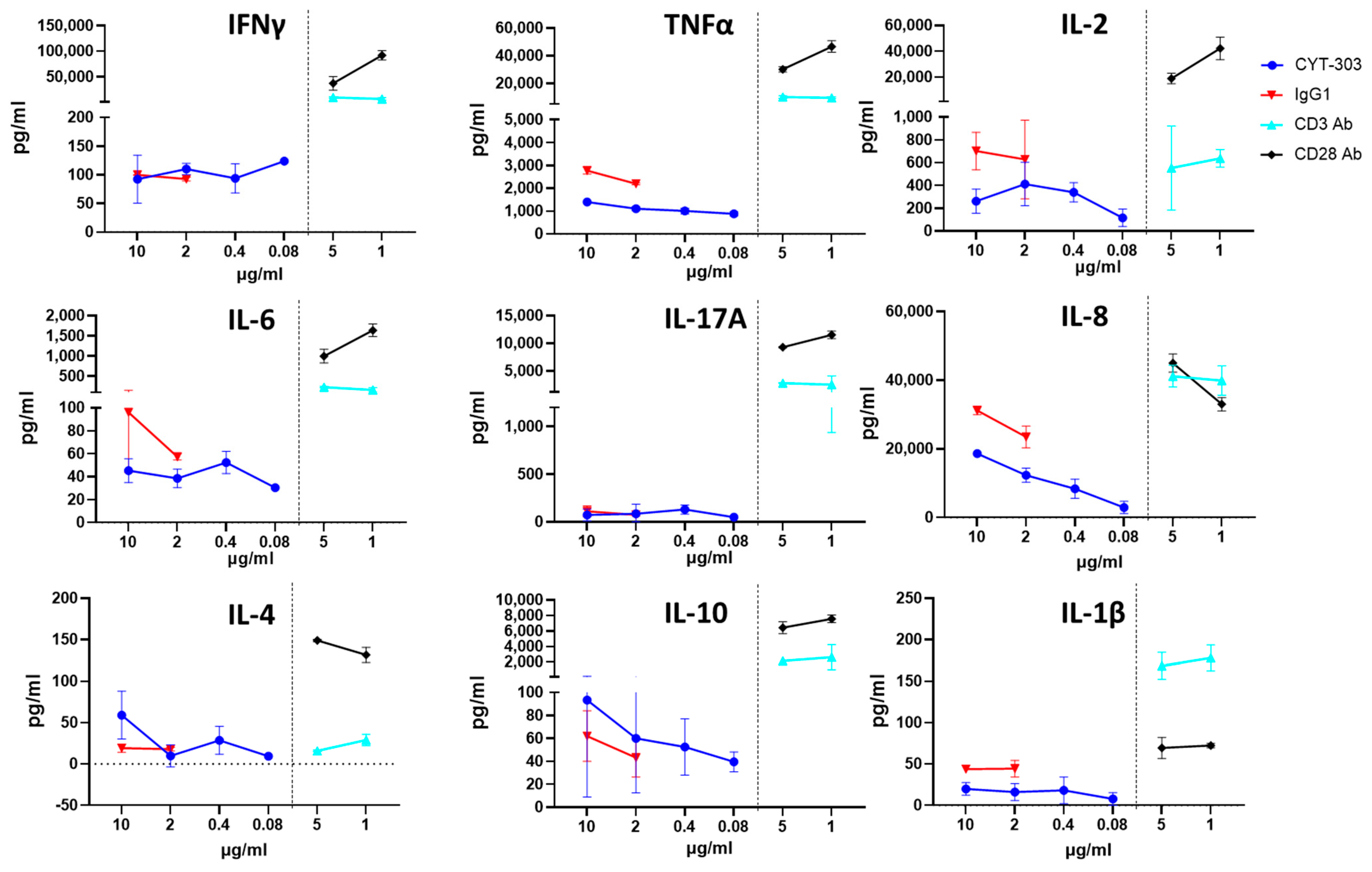
3.5. In Vitro Safety Characterization of CYT-303
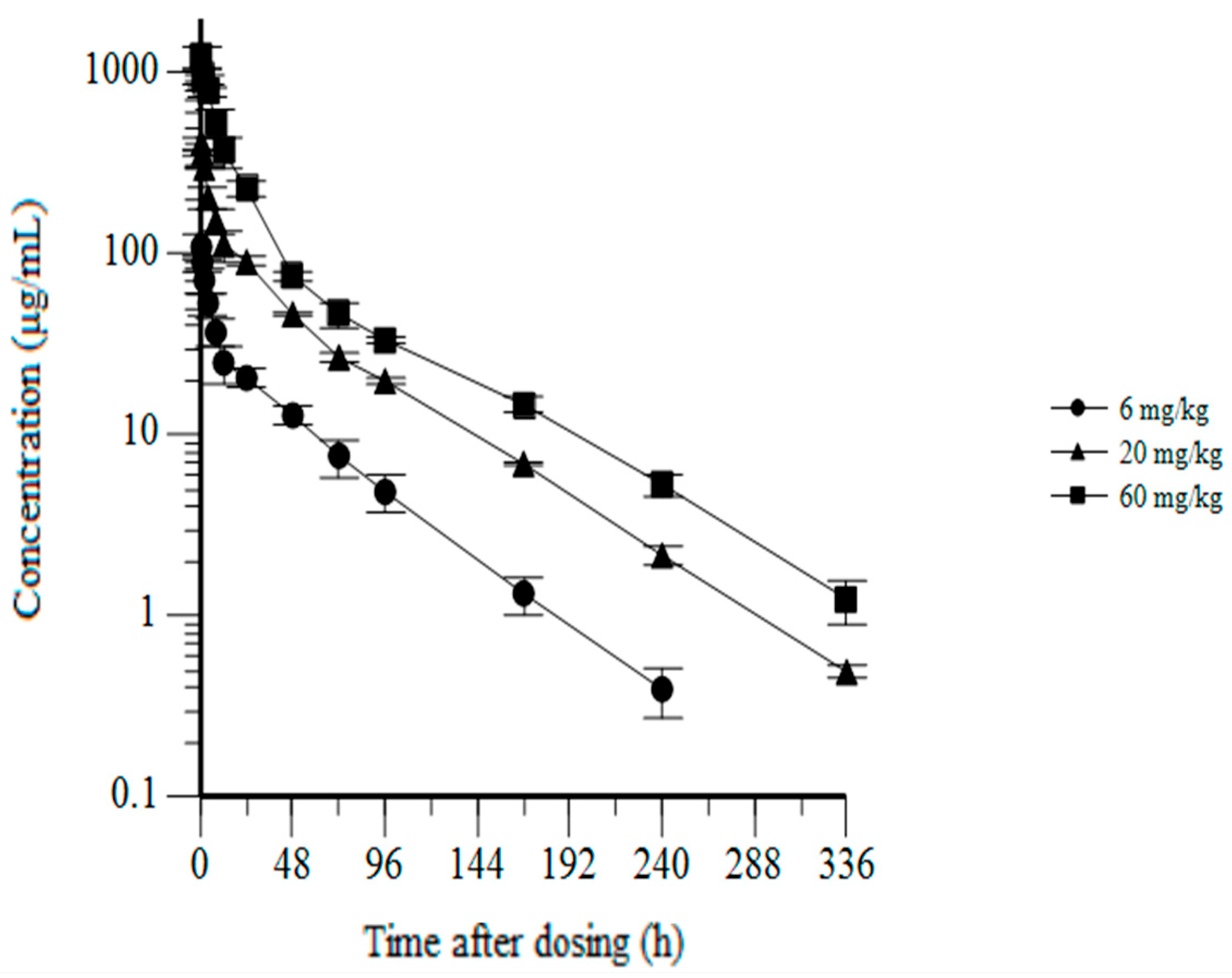
3.6. CYT-303 Pharmacokinetics and Toxicology in Cynomolgus Monkeys
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Birch, G.C.; Fehniger, T.F.; Romee, R. Biology of NK cells and NK cells in clinic. In Gene and Cellular Immunotherapy for Cancer; Ghobadi, A., DiPersio, J.F., Eds.; Springer International Publishing: Cham, Switzerland, 2022; pp. 293–325. [Google Scholar]
- Liu, E.; Marin, D.; Banerjee, P.; Macapinlac, H.A.; Thompson, P.; Basar, R.; Nassif Kerbauy, L.; Overman, B.; Thall, P.; Kaplan, M.; et al. Use of CAR-Transduced Natural Killer Cells in CD19-Positive Lymphoid Tumors. N. Engl. J. Med. 2020, 382, 545–553. [Google Scholar] [CrossRef]
- Gazit, R.; Gruda, R.; Elboim, M.; Arnon, T.I.; Katz, G.; Achdout, H.; Hanna, J.; Qimron, U.; Landau, G.; Greenbaum, E.; et al. Lethal influenza infection in the absence of the natural killer cell receptor gene Ncr1. Nat. Immunol. 2006, 7, 517–523. [Google Scholar] [CrossRef] [PubMed]
- Sivori, S.; Vitale, M.; Morelli, L.; Sanseverino, L.; Augugliaro, R.; Bottino, C.; Moretta, L.; Moretta, A. p46, a novel natural killer cell-specific surface molecule that mediates cell activation. J. Exp. Med. 1997, 186, 1129–1136. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gauthier, L.; Morel, A.; Anceriz, N.; Rossi, B.; Blanchard-Alvarez, A.; Grondin, G.; Trichard, S.; Cesari, C.; Sapet, M.; Bosco, F.; et al. Multifunctional Natural Killer Cell Engagers Targeting NKp46 Trigger Protective Tumor Immunity. Cell 2019, 177, 1701–1713.e16. [Google Scholar] [CrossRef] [PubMed]
- Pessino, A.; Sivori, S.; Bottino, C.; Malaspina, A.; Morelli, L.; Moretta, L.; Biassoni, R.; Moretta, A. Molecular cloning of NKp46: A novel member of the immunoglobulin superfamily involved in triggering of natural cytotoxicity. J. Exp. Med. 1998, 188, 953–960. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bottino, C.; Moretta, L.; Moretta, A. NK cell activating receptors and tumor recognition in humans. Curr. Top. Microbiol. Immunol. 2006, 298, 175–182. [Google Scholar] [CrossRef]
- Golay, J.; Choblet, S.; Iwaszkiewicz, J.; Cerutti, P.; Ozil, A.; Loisel, S.; Pugniere, M.; Ubiali, G.; Zoete, V.; Michielin, O.; et al. Design and Validation of a Novel Generic Platform for the Production of Tetravalent IgG1-like Bispecific Antibodies. J. Immunol. 2016, 196, 3199–3211. [Google Scholar] [CrossRef] [Green Version]
- Capurro, M.; Wanless, I.R.; Sherman, M.; Deboer, G.; Shi, W.; Miyoshi, E.; Filmus, J. Glypican-3: A novel serum and histochemical marker for hepatocellular carcinoma. Gastroenterology 2003, 125, 89–97. [Google Scholar] [CrossRef]
- Baj, J.; Brylinski, L.; Wolinski, F.; Granat, M.; Kostelecka, K.; Duda, P.; Flieger, J.; Teresinski, G.; Buszewicz, G.; Furtak-Niczyporuk, M.; et al. Biomarkers and Genetic Markers of Hepatocellular Carcinoma and Cholangiocarcinoma-What Do We Already Know. Cancers 2022, 14, 1493. [Google Scholar] [CrossRef]
- Haruyama, Y.; Kataoka, H. Glypican-3 is a prognostic factor and an immunotherapeutic target in hepatocellular carcinoma. World J. Gastroenterol. 2016, 22, 275–283. [Google Scholar] [CrossRef]
- Shirakawa, H.; Suzuki, H.; Shimomura, M.; Kojima, M.; Gotohda, N.; Takahashi, S.; Nakagohri, T.; Konishi, M.; Kobayashi, N.; Kinoshita, T.; et al. Glypican-3 expression is correlated with poor prognosis in hepatocellular carcinoma. Cancer Sci. 2009, 100, 1403–1407. [Google Scholar] [CrossRef] [PubMed]
- Llovet, J.M.; Montal, R.; Villanueva, A. Randomized trials and endpoints in advanced HCC: Role of PFS as a surrogate of survival. J. Hepatol. 2019, 70, 1262–1277. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sarveazad, A.; Agah, S.; Babahajian, A.; Amini, N.; Bahardoust, M. Predictors of 5 year survival rate in hepatocellular carcinoma patients. J. Res. Med. Sci. 2019, 24, 86. [Google Scholar] [CrossRef] [PubMed]
- Cai, L.; Zhang, Z.; Zhou, L.; Wang, H.; Fu, J.; Zhang, S.; Shi, M.; Zhang, H.; Yang, Y.; Wu, H.; et al. Functional impairment in circulating and intrahepatic NK cells and relative mechanism in hepatocellular carcinoma patients. Clin. Immunol. 2008, 129, 428–437. [Google Scholar] [CrossRef]
- Hao, X.; Sun, G.; Zhang, Y.; Kong, X.; Rong, D.; Song, J.; Tang, W.; Wang, X. Targeting Immune Cells in the Tumor Microenvironment of HCC: New Opportunities and Challenges. Front. Cell Dev. Biol. 2021, 9, 775462. [Google Scholar] [CrossRef]
- Berhani, O.; Glasner, A.; Kahlon, S.; Duev-Cohen, A.; Yamin, R.; Horwitz, E.; Enk, J.; Moshel, O.; Varvak, A.; Porgador, A.; et al. Human anti-NKp46 antibody for studies of NKp46-dependent NK cell function and its applications for type 1 diabetes and cancer research. Eur. J. Immunol. 2019, 49, 228–241. [Google Scholar] [CrossRef]
- Mandelboim, O.; Lieberman, N.; Lev, M.; Paul, L.; Arnon, T.I.; Bushkin, Y.; Davis, D.M.; Strominger, J.L.; Yewdell, J.W.; Porgador, A. Recognition of haemagglutinins on virus-infected cells by NKp46 activates lysis by human NK cells. Nature 2001, 409, 1055–1060. [Google Scholar] [CrossRef]
- Phung, Y.; Gao, W.; Man, Y.G.; Nagata, S.; Ho, M. High-affinity monoclonal antibodies to cell surface tumor antigen glypican-3 generated through a combination of peptide immunization and flow cytometry screening. MAbs 2012, 4, 592–599. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.F.; Ho, M. Humanization of high-affinity antibodies targeting glypican-3 in hepatocellular carcinoma. Sci. Rep. 2016, 6, 33878. [Google Scholar] [CrossRef] [Green Version]
- Li, N.; Wei, L.; Liu, X.; Bai, H.; Ye, Y.; Li, D.; Li, N.; Baxa, U.; Wang, Q.; Lv, L.; et al. A Frizzled-Like Cysteine-Rich Domain in Glypican-3 Mediates Wnt Binding and Regulates Hepatocellular Carcinoma Tumor Growth in Mice. Hepatology 2019, 70, 1231–1245. [Google Scholar] [CrossRef]
- Feng, M.; Gao, W.; Wang, R.; Chen, W.; Man, Y.G.; Figg, W.D.; Wang, X.W.; Dimitrov, D.S.; Ho, M. Therapeutically targeting glypican-3 via a conformation-specific single-domain antibody in hepatocellular carcinoma. Proc. Natl. Acad. Sci. USA 2013, 110, E1083–E1091. [Google Scholar] [CrossRef] [Green Version]
- Sun, L.; Gao, F.; Gao, Z.; Ao, L.; Li, N.; Ma, S.; Jia, M.; Li, N.; Lu, P.; Sun, B.; et al. Shed antigen-induced blocking effect on CAR-T cells targeting Glypican-3 in Hepatocellular Carcinoma. J. Immunother. Cancer 2021, 9, e001875. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Li, N.; Zhang, Y.F.; Fu, H.; Feng, M.; Schneider, D.; Su, L.; Wu, X.; Zhou, J.; Mackay, S.; et al. Persistent Polyfunctional Chimeric Antigen Receptor T Cells That Target Glypican 3 Eliminate Orthotopic Hepatocellular Carcinomas in Mice. Gastroenterology 2020, 158, 2250–2265.e20. [Google Scholar] [CrossRef] [PubMed]
- Laskowski, T.J.; Biederstadt, A.; Rezvani, K. Natural killer cells in antitumour adoptive cell immunotherapy. Nat. Rev. Cancer 2022, 22, 557–575. [Google Scholar] [CrossRef] [PubMed]













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| GPC3 | NKp46 | CD16α |
|---|---|---|
| KD = 0.646 nM | KD = 0.183 nM | KD = 642 nM (158 F) |
| KD = 147 nM (158 V) |
| Dose (mg/kg) | Cmax (μg/mL) | AUC0–168h (μg·h/mL) | Tmax (h) | T1/2 (h) | Vdss (mL/kg) |
|---|---|---|---|---|---|
| 6 | 109 ± 16.9 | 1910 ± 286 | 0.5 | 39.0 ± 1.48 | 136 ± 13.5 |
| 20 | 408 ± 26.9 | 7740 ± 302 | 0.5 | 44.3 ± 0.651 | 119 ± 8.39 |
| 60 | 1240 ± 175 | 19,800 ± 1530 | 0.5 | 47.6 ± 3.11 | 115 ± 20.6 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Arulanandam, A.; Lin, L.; Chang, H.-M.; Cerutti, M.; Choblet, S.; Gao, P.; Rath, A.; Bensussan, A.; Kadouche, J.; Teper, D.; et al. Derivation and Preclinical Characterization of CYT-303, a Novel NKp46-NK Cell Engager Targeting GPC3. Cells 2023, 12, 996. https://doi.org/10.3390/cells12070996
Arulanandam A, Lin L, Chang H-M, Cerutti M, Choblet S, Gao P, Rath A, Bensussan A, Kadouche J, Teper D, et al. Derivation and Preclinical Characterization of CYT-303, a Novel NKp46-NK Cell Engager Targeting GPC3. Cells. 2023; 12(7):996. https://doi.org/10.3390/cells12070996
Chicago/Turabian StyleArulanandam, Antonio, Liang Lin, Hao-Ming Chang, Martine Cerutti, Sylvie Choblet, Peng Gao, Armin Rath, Armand Bensussan, Jean Kadouche, Daniel Teper, and et al. 2023. "Derivation and Preclinical Characterization of CYT-303, a Novel NKp46-NK Cell Engager Targeting GPC3" Cells 12, no. 7: 996. https://doi.org/10.3390/cells12070996
APA StyleArulanandam, A., Lin, L., Chang, H. -M., Cerutti, M., Choblet, S., Gao, P., Rath, A., Bensussan, A., Kadouche, J., Teper, D., Mandelboim, O., & Li, W. (2023). Derivation and Preclinical Characterization of CYT-303, a Novel NKp46-NK Cell Engager Targeting GPC3. Cells, 12(7), 996. https://doi.org/10.3390/cells12070996