
The Multifaceted Role of WNT Signaling in Alzheimer’s Disease Onset and Age-Related Progression
Abstract
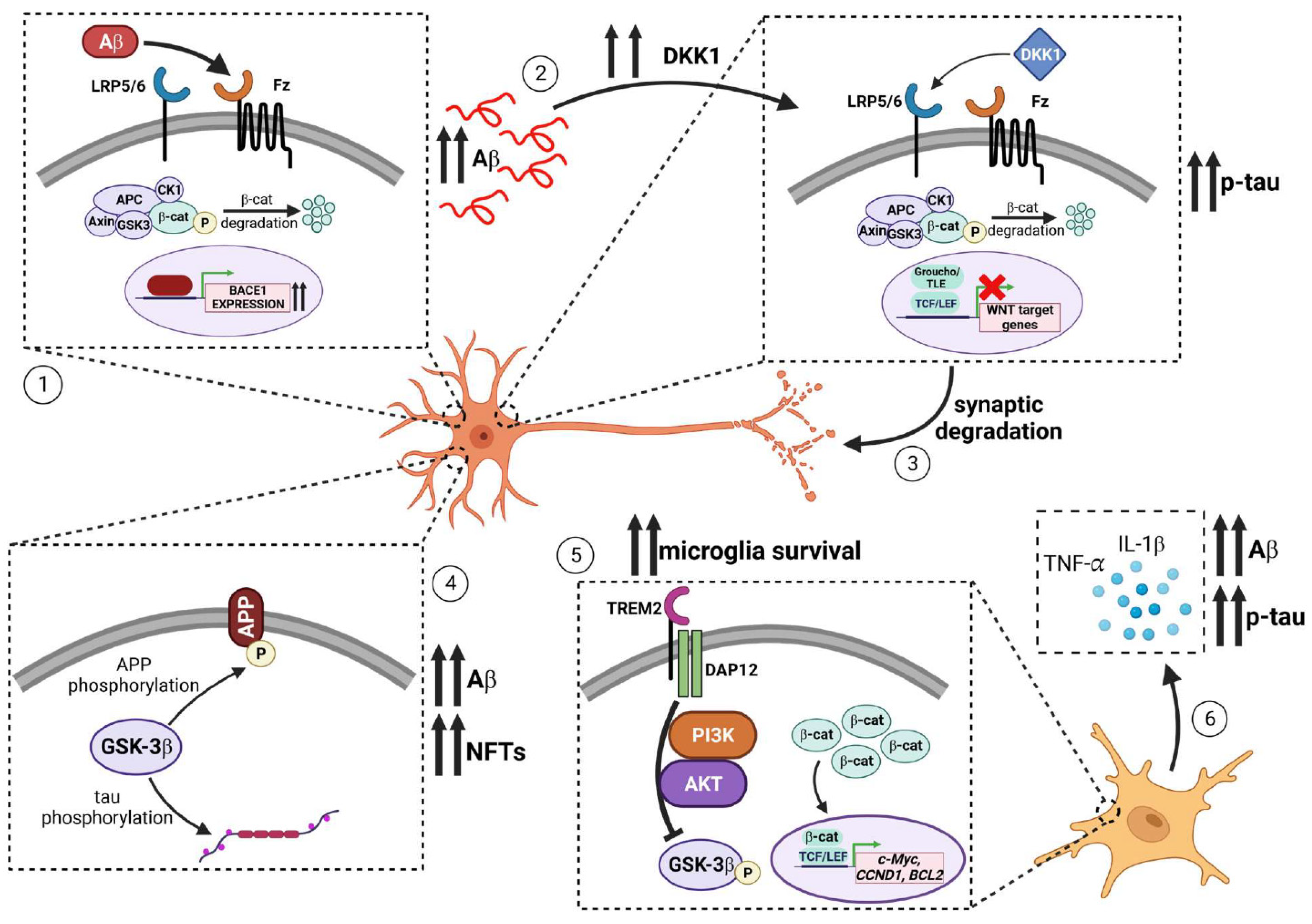
:1. Introduction
2. Alzheimer’s Disease
2.1. Amyloid-β (Aβ) Induced Pathologies in AD
2.2. Tau-Related Pathways in AD
2.3. Inflammatory Processes in AD
2.4. Synaptic Loss in AD
3. WNT Signaling
3.1. The Role of WNT Signaling in Neurogenesis
3.2. WNT Signaling in Synaptic Health and Plasticity
3.3. WNT Signaling in Memory and Learning
4. WNT Signaling and Alzheimer’s Disease
4.1. Evidence for the Role of WNT Signaling in Alzheimer’s Disease
4.2. Amyloid-Centric Mechanisms by which WNT Modulates AD-Related Phenotypes
4.2.1. WNT and APP Post-Translational Modification
4.2.2. WNT Signaling and APP Endocytosis
4.2.3. Regulation of APP processing by WNT Signaling
4.2.4. Interaction of APP and PSEN1 with Components of the WNT Signaling Pathway
4.3. Role of WNT Signaling in Modulating Tau-Related Phenotypes in AD
4.4. WNT and Inflammatory Responses in AD
4.5. The Role of DKK1 and LRP6 in Synaptic Loss in AD
5. Future Trends: Improvement in Model Systems
Human Induced Pluripotent Stem Cell (hiPSC)-Based Approaches
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Harper, L.C. 2022 Alzheimer’s Disease Facts and Figures. Alzheimers Dement. 2022, 18, 700–789. [Google Scholar]
- Serrano-Pozo, A.; Frosch, M.P.; Masliah, E.; Hyman, B.T. Neuropathological Alterations in Alzheimer Disease. Cold Spring Harb. Perspect. Med. 2011, 1, a006189. [Google Scholar] [CrossRef] [PubMed]
- Mehta, R.I.; Mehta, R.I. The Vascular-Immune Hypothesis of Alzheimer’s Disease. Biomedicines 2023, 11, 408. [Google Scholar] [CrossRef] [PubMed]
- Swerdlow, R.H. Alzheimer’s Disease Pathologic Cascades: Who Comes First, What Drives What. Neurotox. Res. 2012, 22, 182–194. [Google Scholar] [CrossRef]
- Liu, C.C.; Kanekiyo, T.; Xu, H.; Bu, G. Apolipoprotein E and Alzheimer Disease: Risk, Mechanisms and Therapy. Nat. Rev. Neurol. 2013, 9, 106–118. [Google Scholar] [CrossRef]
- Zhang, Y.W.; Thompson, R.; Zhang, H.; Xu, H. APP Processing in Alzheimer’s Disease. Mol. Brain 2011, 4, 3. [Google Scholar] [CrossRef]
- Chow, V.W.; Mattson, M.P.; Wong, P.C.; Gleichmann, M. An Overview of APP Processing Enzymes and Products. Neuromol. Med. 2010, 12, 1–12. [Google Scholar] [CrossRef]
- Lesne, S.E.; Sherman, M.A.; Grant, M.; Kuskowski, M.; Schneider, J.A.; Bennett, D.A.; Ashe, K.H. Brain Amyloid-Beta Oligomers in Ageing and Alzheimer’s Disease. Brain 2013, 136, 1383–1398. [Google Scholar] [CrossRef]
- Yatin, S.M.; Yatin, M.; Aulick, T.; Ain, K.B.; Butterfield, D.A. Alzheimer’s amyloid Beta-Peptide Associated Free Radicals Increase Rat Embryonic Neuronal Polyamine Uptake and Ornithine Decarboxylase Activity: Protective Effect of Vitamin E. Neurosci. Lett. 1999, 263, 17–20. [Google Scholar] [CrossRef]
- Mattson, M.P. Pathways towards and away from Alzheimer’s Disease. Nature 2004, 430, 631–639. [Google Scholar] [CrossRef]
- Aksenov, M.; Aksenova, M.V.; Harris, M.E.; Hensley, K.; Butterfield, D.A.; Carney, J.M. Enhancement of β-Amyloid Peptide Aβ(1-40)-Mediated Neurotoxicity by Glutamine Synthetase. J. Neurochem. 1995, 65, 1899–1902. [Google Scholar] [CrossRef] [PubMed]
- Willnow, T.E.; Andersen, O.M. Sorting Receptor SORLA—A Trafficking Path to Avoid Alzheimer Disease. J. Cell Sci. 2013, 126, 2751–2760. [Google Scholar] [CrossRef] [PubMed]
- Eggert, S.; Thomas, C.; Kins, S.; Hermey, G. Trafficking in Alzheimer’s Disease: Modulation of APP Transport and Processing by the Transmembrane Proteins LRP1, SorLA, SorCS1c, Sortilin, and Calsyntenin. Mol. Neurobiol. 2018, 55, 5809–5829. [Google Scholar] [CrossRef] [PubMed]
- Baranello, R.J.; Bharani, K.L.; Padmaraju, V.; Chopra, N.; Lahiri, D.K.; Greig, N.H.; Pappolla, M.A.; Sambamurti, K. Amyloid-Beta Protein Clearance and Degradation (ABCD) Pathways and Their Role in Alzheimer’s Disease. Curr. Alzheimer Res. 2015, 12, 32–46. [Google Scholar] [CrossRef] [PubMed]
- Haass, C.; Kaether, C.; Thinakaran, G.; Sisodia, S. Trafficking and proteolytic processing of APP. Cold Spring Harb. Perspect. Med. 2012, 2, a006270. [Google Scholar] [CrossRef]
- Thinakaran, G.; Koo, E.H. Amyloid Precursor Protein Trafficking, Processing, and Function. J. Biol. Chem. 2008, 283, 29615–29619. [Google Scholar] [CrossRef]
- Goldstein, L.S.B.; Das, U. The Cellular Machinery of Post-Endocytic APP Trafficking in Alzheimer’s Disease: A Future Target for Therapeutic Intervention? Prog. Mol. Biol. Transl. Sci. 2021, 177, 109–122. [Google Scholar] [CrossRef]
- Chen, G.F.; Xu, T.H.; Yan, Y.; Zhou, Y.R.; Jiang, Y.; Melcher, K.; Xu, H.E. Amyloid Beta: Structure, Biology and Structure-Based Therapeutic Development. Acta Pharmacol. Sin. 2017, 38, 1205–1235. [Google Scholar] [CrossRef] [PubMed]
- De Strooper, B. Loss-of-Function Presenilin Mutations in Alzheimer Disease. Talking Point on the Role of Presenilin Mutations in Alzheimer Disease. EMBO Rep. 2007, 8, 141–146. [Google Scholar] [CrossRef]
- Woodruff, G.; Reyna, S.M.; Dunlap, M.; Van Der Kant, R.; Callender, J.A.; Young, J.E.; Roberts, E.A.; Goldstein, L.S. Defective Transcytosis of APP and Lipoproteins in Human iPSC-Derived Neurons with Familial Alzheimer’s Disease Mutations. Cell Rep. 2016, 17, 759–773. [Google Scholar] [CrossRef]
- Hung, C.O.Y.; Livesey, F.J. Altered γ-Secretase Processing of APP Disrupts Lysosome and Autophagosome Function in Monogenic Alzheimer’s Disease. Cell Rep. 2018, 25, 3647–3660.e2. [Google Scholar] [CrossRef] [PubMed]
- Grimm, M.O.; Mett, J.; Stahlmann, C.P.; Haupenthal, V.J.; Zimmer, V.C.; Hartmann, T. Neprilysin and Aβ Clearance: Impact of the APP Intracellular Domain in NEP Regulation and Implications in Alzheimer’s Disease. Front. Aging Neurosci. 2013, 5, 98. [Google Scholar] [CrossRef] [PubMed]
- Nixon, R.A.; Yang, D.S. Autophagy Failure in Alzheimer’s Disease—Locating the Primary Defect. Neurobiol. Dis. 2011, 43, 38–45. [Google Scholar] [CrossRef] [PubMed]
- McBrayer, M.; Nixon, R.A. Lysosome and Calcium Dysregulation in Alzheimer’s Disease: Partners in Crime. Biochem. Soc. Trans. 2013, 41, 1495–1502. [Google Scholar] [CrossRef]
- Iwata, N.; Tsubuki, S.; Takaki, Y.; Watanabe, K.; Sekiguchi, M.; Hosoki, E.; Kawashima-Morishima, M.; Lee, H.J.; Hama, E.; Sekine-Aizawa, Y.; et al. Identification of the Major Aβ1–42-Degrading Catabolic Pathway in Brain Parenchyma: Suppression Leads to Biochemical and Pathological Deposition. Nat. Med. 2000, 6, 143–150. [Google Scholar] [CrossRef]
- Ries, M.; Sastre, M. Mechanisms of Aβ Clearance and Degradation by Glial Cells. Front. Aging Neurosci. 2016, 8, 160. [Google Scholar] [CrossRef]
- Stopschinski, B.E.; Holmes, B.B.; Miller, G.M.; Manon, V.A.; Vaquer-Alicea, J.; Prueitt, W.L.; Hsieh-Wilson, L.C.; Diamond, M.I. Specific Glycosaminoglycan Chain Length and Sulfation Patterns Are Required for Cell Uptake of Tau versus α-Synuclein and Beta-Amyloid Aggregates. J. Biol. Chem. 2018, 293, 10826–10840. [Google Scholar] [CrossRef]
- Kanekiyo, T.; Zhang, J.; Liu, Q.; Liu, C.C.; Zhang, L.; Bu, G. Heparan Sulphate Proteoglycan and the low-Density Lipoprotein Receptor-Related Protein 1 Constitute Major Pathways for Neuronal Amyloid-β Uptake. J. Neurosci. 2011, 31, 1644–1651. [Google Scholar] [CrossRef]
- Bu, G.; Cam, J.; Zerbinatti, C. LRP in Amyloid-β Production and Metabolism. Ann. N. Y. Acad. Sci. 2006, 1086, 35–53. [Google Scholar] [CrossRef]
- Yeh, F.L.; Wang, Y.; Tom, I.; Gonzalez, L.C.; Sheng, M. TREM2 Binds to Apolipoproteins, Including APOE and CLU/APOJ, and Thereby Facilitates Uptake of Amyloid-Beta by Microglia. Neuron 2016, 91, 328–340. [Google Scholar] [CrossRef]
- Li, J.; Kanekiyo, T.; Shinohara, M.; Zhang, Y.; LaDu, M.J.; Xu, H.; Bu, G. Differential Regulation of Amyloid-β Endocytic Trafficking and Lysosomal Degradation by Apolipoprotein E Isoforms. J. Biol. Chem. 2012, 287, 44593–44601. [Google Scholar] [CrossRef] [PubMed]
- Kanekiyo, T.; Xu, H.; Bu, G. ApoE and Aβ in Alzheimer’s Disease: Accidental Encounters or Partners? Neuron 2014, 81, 740–754. [Google Scholar] [CrossRef] [PubMed]
- Deane, R.; Du Yan, S.; Submamaryan, R.K.; LaRue, B.; Jovanovic, S.; Hogg, E.; Welch, D.; Manness, L.; Lin, C.; Yu, J.; et al. RAGE Mediates Amyloid-β Peptide Transport across the Blood-Brain Barrier and Accumulation in Brain. Nat. Med. 2003, 9, 907–913. [Google Scholar] [CrossRef] [PubMed]
- Shibata, M.; Yamada, S.; Kumar, S.R.; Calero, M.; Bading, J.; Frangione, B.; Holtzman, D.M.; Miller, C.A.; Strickland, D.K.; Ghiso, J.; et al. Clearance of Alzheimer’s Amyloid-β1-40 Peptide from Brain by LDL Receptor-Related Protein-1 at the Blood-Brain Barrier. J. Clin. Investig. 2000, 106, 1489–1499. [Google Scholar] [CrossRef]
- Prasad, H.; Rao, R. Amyloid Clearance Defect in ApoE4 Astrocytes Is Reversed by Epigenetic Correction of Endosomal pH. Proc. Natl. Acad. Sci. USA 2018, 115, E6640–E6649. [Google Scholar] [CrossRef]
- Narayan, P.; Sienski, G.; Bonner, J.M.; Lin, Y.T.; Seo, J.; Baru, V.; Haque, A.; Milo, B.; Akay, L.A.; Graziosi, A.; et al. PICALM Rescues Endocytic Defects Caused by the Alzheimer’s Disease Risk Factor APOE4. Cell Rep. 2020, 33, 108224. [Google Scholar] [CrossRef]
- Knauer, M.F.; Soreghan, B.; Burdick, D.; Kosmoski, J.; Glabe, C.G. Intracellular Accumulation and Resistance to Degradation of the Alzheimer Amyloid A4/Beta Protein. Proc. Natl. Acad. Sci. USA 1992, 89, 7437–7441. [Google Scholar] [CrossRef]
- Hu, X.; Crick, S.L.; Bu, G.; Frieden, C.; Pappu, R.V.; Lee, J.M. Amyloid Seeds Formed by Cellular Uptake, Concentration, and Aggregation of the Amyloid-Beta Peptide. Proc. Natl. Acad. Sci. USA 2009, 106, 20324–20329. [Google Scholar] [CrossRef]
- Wolfe, D.M.; Lee, J.H.; Kumar, A.; Lee, S.; Orenstein, S.J.; Nixon, R.A. Autophagy Failure in Alzheimer’s Disease and the Role of Defective Lysosomal Acidification. Eur. J. Neurosci. 2013, 37, 1949–1961. [Google Scholar] [CrossRef]
- Lee, J.H.; Yu, W.H.; Kumar, A.; Lee, S.; Mohan, P.S.; Peterhoff, C.M.; Wolfe, D.M.; Martinez-Vicente, M.; Massey, A.C.; Sovak, G.; et al. Lysosomal Proteolysis and Autophagy Require Presenilin 1 and Are Disrupted by Alzheimer-Related PS1 Mutations. Cell 2010, 141, 1146–1158. [Google Scholar] [CrossRef]
- Cuervo, A.M.; Dice, J.F. When Lysosomes Get Old. Exp. Gerontol. 2000, 35, 119–131. [Google Scholar] [CrossRef] [PubMed]
- Coen, K.; Flannagan, R.S.; Baron, S.; Carraro-Lacroix, L.R.; Wang, D.; Vermeire, W.; Michiels, C.; Munck, S.; Baert, V.; Sugita, S.; et al. Lysosomal Calcium Homeostasis Defects, Not Proton Pump Defects, Cause Endo-Lysosomal Dysfunction in PSEN-Deficient Cells. J. Cell Biol. 2012, 198, 23–35. [Google Scholar] [CrossRef] [PubMed]
- Basak, J.M.; Verghese, P.B.; Yoon, H.; Kim, J.; Holtzman, D.M. Low-Density Lipoprotein Receptor Represents An Apolipoprotein E-Independent Pathway of Aβ Uptake and Degradation by Astrocytes. J. Biol. Chem. 2012, 287, 13959–13971. [Google Scholar] [CrossRef] [PubMed]
- Yepes, M.; Sandkvist, M.; Moore, E.G.; Bugge, T.H.; Strickland, D.K.; Lawrence, D.A. Tissue-type Plasminogen Activator Induces Opening of the Blood-Brain Barrier via the LDL Receptor-Related Protein. J. Clin. Investig. 2003, 112, 1533–1540. [Google Scholar] [CrossRef] [PubMed]
- Neve, R.L.; Harris, P.; Kosik, K.S.; Kurnit, D.M.; Donlon, T.A. Identification of cDNA Clones for the Human Microtubule-Associated Protein Tau and Chromosomal Localization of the Genes for Tau and Microtubule-Associated Protein 2. Brain Res. 1986, 387, 271–280. [Google Scholar] [CrossRef]
- Kempf, M.; Clement, A.; Faissner, A.; Lee, G.; Brandt, R. Tau Binds to the Distal Axon Early in Development of Polarity in a Microtubule- and Microfilament-Dependent Manner. J. Neurosci. 1996, 16, 5583–5592. [Google Scholar] [CrossRef]
- Himmler, A.; Drechsel, D.; Kirschner, M.W.; Martin, D.W., Jr. Tau Consists of a Set of Proteins with Repeated C-Terminal Microtubule-Binding Domains and Variable N-Terminal Domains. Mol. Cell. Biol. 1989, 9, 1381–1388. [Google Scholar] [CrossRef]
- Himmler, A. Structure of the Bovine Tau Gene: Alternatively Spliced Transcripts Generate a Protein Family. Mol. Cell. Biol. 1989, 9, 1389–1396. [Google Scholar] [CrossRef]
- Weingarten, M.D.; Lockwood, A.H.; Hwo, S.Y.; Kirschner, M.W. A Protein Factor Essential for Microtubule Assembly. Proc. Natl. Acad. Sci. USA 1975, 72, 1858–1862. [Google Scholar] [CrossRef]
- Vossel, K.A.; Zhang, K.; Brodbeck, J.; Daub, A.C.; Sharma, P.; Finkbeiner, S.; Cui, B.; Mucke, L. Tau Reduction Prevents Aβ-Induced Defects in Axonal Transport. Science 2010, 330, 198. [Google Scholar] [CrossRef]
- Qiang, L.; Yu, W.; Andreadis, A.; Luo, M.; Baas, P.W. Tau Protects Microtubules in the Axon from Severing by Katanin. J. Neurosci. 2006, 26, 3120–3129. [Google Scholar] [CrossRef] [PubMed]
- King, M.E.; Kan, H.M.; Baas, P.W.; Erisir, A.; Glabe, C.G.; Bloom, G.S. Tau-Dependent Microtubule Disassembly Initiated by Prefibrillar β-Amyloid. J. Cell Biol. 2006, 175, 541–546. [Google Scholar] [CrossRef] [PubMed]
- Harada, A.; Oguchi, K.; Okabe, S.; Kuno, J.; Terada, S.; Ohshima, T.; Sato-Yoshitake, R.; Takei, Y.; Noda, T.; Hirokawa, N. Altered Microtubule Organization in Small-Calibre Axons of Mice Lacking Tau Protein. Nature 1994, 369, 488–491. [Google Scholar] [CrossRef] [PubMed]
- Fujio, K.; Sato, M.; Uemura, T.; Sato, T.; Sato-Harada, R.; Harada, A. 14-3-3 Proteins and Protein Phosphatases Are Not Reduced in Tau-Deficient Mice. Neuroreport 2007, 18, 1049–1052. [Google Scholar] [CrossRef]
- Dawson, H.N.; Ferreira, A.; Eyster, M.V.; Ghoshal, N.; Binder, L.I.; Vitek, M.P. Inhibition of Neuronal Maturation in Primary Hippocampal Neurons from Tau Deficient Mice. J. Cell Sci. 2001, 114, 1179–1187. [Google Scholar] [CrossRef]
- Saleheen, D.; Natarajan, P.; Armean, I.M.; Zhao, W.; Rasheed, A.; Khetarpal, S.A.; Won, H.H.; Karczewski, K.J.; O’Donnell-Luria, A.H.; Samocha, K.E.; et al. Human Knockouts and Phenotypic Analysis in a Cohort with a High Rate of Consanguinity. Nature 2017, 544, 235–239. [Google Scholar] [CrossRef]
- Ittner, A.; Ittner, L.M. Dendritic Tau in Alzheimer’s Disease. Neuron 2018, 99, 13–27. [Google Scholar] [CrossRef]
- Frandemiche, M.L.; De Seranno, S.; Rush, T.; Borel, E.; Elie, A.; Arnal, I.; Lante, F.; Buisson, A. Activity-dependent Tau Protein Translocation to Excitatory Synapse Is Disrupted by Exposure to Amyloid-Beta Oligomers. J. Neurosci. 2014, 34, 6084–6097. [Google Scholar] [CrossRef]
- Selkoe, D.J. Alzheimer’s Disease Is a Synaptic Failure. Science 2002, 298, 789–791. [Google Scholar] [CrossRef]
- Nagy, Z.; Jobst, K.A.; Esiri, M.M.; Morris, J.H.; King, E.M.; MacDonald, B.; Litchfield, S.; Barnetson, L.; Smith, A.D. Hippocampal Pathology Reflects Memory Deficit and Brain Imaging Measurements in Alzheimer’s Disease: Clinicopathologic Correlations Using Three Sets of Pathologic Diagnostic Criteria. Dementia 1996, 7, 76–81. [Google Scholar] [CrossRef]
- Hyman, B.T.; Van Hoesen, G.W.; Damasio, A.R.; Barnes, C.L. Alzheimer’s Disease: Cell-Specific Pathology Isolates the Hippocampal Formation. Science 1984, 225, 1168–1170. [Google Scholar] [CrossRef]
- Ahmed, T.; Van der Jeugd, A.; Blum, D.; Galas, M.C.; D’Hooge, R.; Buee, L.; Balschun, D. Cognition and Hippocampal Synaptic Plasticity in Mice with a Homozygous Tau Deletion. Neurobiol. Aging 2014, 35, 2474–2478. [Google Scholar] [CrossRef]
- Regan, P.; Piers, T.; Yi, J.H.; Kim, D.H.; Huh, S.; Park, S.J.; Ryu, J.H.; Whitcomb, D.J.; Cho, K. Tau Phosphorylation at Serine 396 Residue Is Required for Hippocampal LTD. J. Neurosci. 2015, 35, 4804–4812. [Google Scholar] [CrossRef]
- Velazquez, R.; Ferreira, E.; Tran, A.; Turner, E.C.; Belfiore, R.; Branca, C.; Oddo, S. Acute Tau Knockdown in the Hippocampus of Adult Mice Causes Learning and Memory Deficits. Aging Cell 2018, 17, e12775. [Google Scholar] [CrossRef]
- Brion, J.P.; Anderton, B.H.; Authelet, M.; Dayanandan, R.; Leroy, K.; Lovestone, S.; Octave, J.N.; Pradier, L.; Touchet, N.; Tremp, G. Neurofibrillary Tangles and Tau Phosphorylation. Biochem. Soc. Symp. 2001, 67, 81–88. [Google Scholar] [CrossRef]
- Iqbal, K.; Liu, F.; Gong, C.X.; Grundke-Iqbal, I. Tau in Alzheimer Disease and Related Tauopathies. Curr. Alzheimer Res. 2010, 7, 656–664. [Google Scholar] [CrossRef]
- Bramblett, G.T.; Goedert, M.; Jakes, R.; Merrick, S.E.; Trojanowski, J.Q.; Lee, V.M. Abnormal Tau Phosphorylation at Ser396 in Alzheimer’s Disease Recapitulates Development and Contributes to Reduced Microtubule Binding. Neuron 1993, 10, 1089–1099. [Google Scholar] [CrossRef]
- Kellogg, E.H.; Hejab, N.M.A.; Poepsel, S.; Downing, K.H.; DiMaio, F.; Nogales, E. Near-Atomic Model of Microtubule-Tau Interactions. Science 2018, 360, 1242–1246. [Google Scholar] [CrossRef]
- Lindwall, G.; Cole, R.D. Phosphorylation Affects the Ability of Tau Protein to Promote Microtubule Assembly. J. Biol. Chem. 1984, 259, 5301–5305. [Google Scholar] [CrossRef]
- Yoshida, H.; Ihara, Y. Tau in Paired Helical Filaments Is Functionally Distinct from Fetal Tau: Assembly Incompetence of Paired Helical Filament-Tau. J. Neurochem. 1993, 61, 1183–1186. [Google Scholar] [CrossRef]
- Alonso, A.; Zaidi, T.; Novak, M.; Grundke-Iqbal, I.; Iqbal, K. Hyperphosphorylation Induces Self-Assembly of Tau into Tangles of Paired Helical Filaments/Straight Filaments. Proc. Natl. Acad. Sci. USA 2001, 98, 6923–6928. [Google Scholar] [CrossRef]
- Ittner, L.M.; Ke, Y.D.; Delerue, F.; Bi, M.; Gladbach, A.; van Eersel, J.; Wolfing, H.; Chieng, B.C.; Christie, M.J.; Napier, I.A.; et al. Dendritic Function of Tau Mediates Amyloid-β Toxicity in Alzheimer’s Disease Mouse Models. Cell 2010, 142, 387–397. [Google Scholar] [CrossRef]
- Shentu, Y.P.; Huo, Y.; Feng, X.L.; Gilbert, J.; Zhang, Q.; Liuyang, Z.Y.; Wang, X.L.; Wang, G.; Zhou, H.; Wang, X.C.; et al. CIP2A Causes Tau/APP Phosphorylation, Synaptopathy, and Memory Deficits in Alzheimer’s Disease. Cell Rep. 2018, 24, 713–723. [Google Scholar] [CrossRef]
- Tashiro, K.; Hasegawa, M.; Ihara, Y.; Iwatsubo, T. Somatodendritic Localization of Phosphorylated Tau in Neonatal and Adult Rat Cerebral Cortex. Neuroreport 1997, 8, 2797–2801. [Google Scholar] [CrossRef]
- Bories, C.; Arsenault, D.; Lemire, M.; Tremblay, C.; De Koninck, Y.; Calon, F. Transgenic Autoinhibition of p21-Activated Kinase Exacerbates Synaptic Impairments and Fronto-Dependent Behavioral Deficits in an Animal Model of Alzheimer’s Disease. Aging 2017, 9, 1386–1403. [Google Scholar] [CrossRef]
- Hoover, B.R.; Reed, M.N.; Su, J.; Penrod, R.D.; Kotilinek, L.A.; Grant, M.K.; Pitstick, R.; Carlson, G.A.; Lanier, L.M.; Yuan, L.L.; et al. Tau Mislocalization to Dendritic Spines Mediates Synaptic Dysfunction Independently of Neurodegeneration. Neuron 2010, 68, 1067–1081. [Google Scholar] [CrossRef]
- Fein, J.A.; Sokolow, S.; Miller, C.A.; Vinters, H.V.; Yang, F.; Cole, G.M.; Gylys, K.H. Co-Localization of Amyloid Beta and Tau Pathology in Alzheimer’s Disease Synaptosomes. Am. J. Pathol. 2008, 172, 1683–1692. [Google Scholar] [CrossRef]
- Henkins, K.M.; Sokolow, S.; Miller, C.A.; Vinters, H.V.; Poon, W.W.; Cornwell, L.B.; Saing, T.; Gylys, K.H. Extensive p-Tau Pathology and SDS-Stable p-Tau Oligomers in Alzheimer’s Cortical Synapses. Brain Pathol. 2012, 22, 826–833. [Google Scholar] [CrossRef]
- McInnes, J.; Wierda, K.; Snellinx, A.; Bounti, L.; Wang, Y.C.; Stancu, I.C.; Apostolo, N.; Gevaert, K.; Dewachter, I.; Spires-Jones, T.L.; et al. Synaptogyrin-3 Mediates Presynaptic Dysfunction Induced by Tau. Neuron 2018, 97, 823–835.e8. [Google Scholar] [CrossRef]
- Tai, H.C.; Serrano-Pozo, A.; Hashimoto, T.; Frosch, M.P.; Spires-Jones, T.L.; Hyman, B.T. The Synaptic Accumulation of Hyperphosphorylated Tau Oligomers in Alzheimer Disease Is Associated with Dysfunction of the Ubiquitin-Proteasome System. Am. J. Pathol. 2012, 181, 1426–1435. [Google Scholar] [CrossRef]
- Clavaguera, F.; Bolmont, T.; Crowther, R.A.; Abramowski, D.; Frank, S.; Probst, A.; Fraser, G.; Stalder, A.K.; Beibel, M.; Staufenbiel, M.; et al. Transmission and Spreading of Tauopathy in Transgenic Mouse Brain. Nat. Cell Biol. 2009, 11, 909–913. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.L.; Narasimhan, S.; Changolkar, L.; He, Z.; Stieber, A.; Zhang, B.; Gathagan, R.J.; Iba, M.; McBride, J.D.; Trojanowski, J.Q.; et al. Unique Pathological Tau Conformers from Alzheimer’s Brains Transmit Tau Pathology in Nontransgenic Mice. J. Exp. Med. 2016, 213, 2635–2654. [Google Scholar] [CrossRef] [PubMed]
- Lasagna-Reeves, C.A.; Castillo-Carranza, D.L.; Sengupta, U.; Guerrero-Munoz, M.J.; Kiritoshi, T.; Neugebauer, V.; Jackson, G.R.; Kayed, R. Alzheimer Brain-Derived Tau Oligomers Propagate Pathology from Endogenous Tau. Sci. Rep. 2012, 2, 700. [Google Scholar] [CrossRef]
- Rauch, J.N.; Chen, J.J.; Sorum, A.W.; Miller, G.M.; Sharf, T.; See, S.K.; Hsieh-Wilson, L.C.; Kampmann, M.; Kosik, K.S. Tau Internalization Is Regulated by 6-O Sulfation on Heparan Sulfate Proteoglycans (HSPGs). Sci. Rep. 2018, 8, 6382. [Google Scholar] [CrossRef]
- Rauch, J.N.; Luna, G.; Guzman, E.; Audouard, M.; Challis, C.; Sibih, Y.E.; Leshuk, C.; Hernandez, I.; Wegmann, S.; Hyman, B.T.; et al. LRP1 Is a Master Regulator of Tau Uptake and Spread. Nature 2020, 580, 381–385. [Google Scholar] [CrossRef]
- Dourlen, P.; Fernandez-Gomez, F.J.; Dupont, C.; Grenier-Boley, B.; Bellenguez, C.; Obriot, H.; Caillierez, R.; Sottejeau, Y.; Chapuis, J.; Bretteville, A.; et al. Functional Screening of Alzheimer Risk Loci Identifies PTK2B as an In Vivo Modulator and Early Marker of Tau Pathology. Mol. Psychiatry 2017, 22, 874–883. [Google Scholar] [CrossRef]
- Malki, I.; Cantrelle, F.X.; Sottejeau, Y.; Lippens, G.; Lambert, J.C.; Landrieu, I. Regulation of the Interaction between the Neuronal BIN1 Isoform 1 and Tau Proteins—Role of the SH3 Domain. FEBS J. 2017, 284, 3218–3229. [Google Scholar] [CrossRef]
- Sartori, M.; Mendes, T.; Desai, S.; Lasorsa, A.; Herledan, A.; Malmanche, N.; Makinen, P.; Marttinen, M.; Malki, I.; Chapuis, J.; et al. BIN1 Recovers Tauopathy-Induced Long-Term Memory Deficits in Mice and Interacts with Tau through Thr348 Phosphorylation. Acta Neuropathol. 2019, 138, 631–652. [Google Scholar] [CrossRef]
- Ando, K.; Houben, S.; Homa, M.; de Fisenne, M.A.; Potier, M.C.; Erneux, C.; Brion, J.P.; Leroy, K. Alzheimer’s Disease: Tau Pathology and Dysfunction of Endocytosis. Front. Mol. Neurosci. 2020, 13, 583755. [Google Scholar] [CrossRef]
- Frost, B.; Jacks, R.L.; Diamond, M.I. Propagation of Tau Misfolding from the Outside to the Inside of a Cell. J. Biol. Chem. 2009, 284, 12845–12852. [Google Scholar] [CrossRef]
- Wu, J.W.; Herman, M.; Liu, L.; Simoes, S.; Acker, C.M.; Figueroa, H.; Steinberg, J.I.; Margittai, M.; Kayed, R.; Zurzolo, C.; et al. Small Misfolded Tau Species Are Internalized via Bulk Endocytosis and Anterogradely and Retrogradely Transported in Neurons. J. Biol. Chem. 2013, 288, 1856–1870. [Google Scholar] [CrossRef] [PubMed]
- Saman, S.; Kim, W.; Raya, M.; Visnick, Y.; Miro, S.; Saman, S.; Jackson, B.; McKee, A.C.; Alvarez, V.E.; Lee, N.C.; et al. Exosome-Associated Tau Is Secreted in Tauopathy Models and Is Selectively Phosphorylated in Cerebrospinal Fluid in Early Alzheimer Disease. J. Biol. Chem. 2012, 287, 3842–3849. [Google Scholar] [CrossRef] [PubMed]
- Simon, D.; Garcia-Garcia, E.; Royo, F.; Falcon-Perez, J.M.; Avila, J. Proteostasis of Tau. Tau Overexpression Results in Its Secretion via Membrane Vesicles. FEBS Lett. 2012, 586, 47–54. [Google Scholar] [CrossRef] [PubMed]
- Calafate, S.; Flavin, W.; Verstreken, P.; Moechars, D. Loss of Bin1 Promotes the Propagation of Tau Pathology. Cell Rep. 2016, 17, 931–940. [Google Scholar] [CrossRef]
- Bennett, R.E.; DeVos, S.L.; Dujardin, S.; Corjuc, B.; Gor, R.; Gonzalez, J.; Roe, A.D.; Frosch, M.P.; Pitstick, R.; Carlson, G.A.; et al. Enhanced Tau Aggregation in the Presence of Amyloid β. Am. J. Pathol. 2017, 187, 1601–1612. [Google Scholar] [CrossRef]
- Hanseeuw, B.J.; Betensky, R.A.; Jacobs, H.I.L.; Schultz, A.P.; Sepulcre, J.; Becker, J.A.; Cosio, D.M.O.; Farrell, M.; Quiroz, Y.T.; Mormino, E.C.; et al. Association of Amyloid and Tau with Cognition in Preclinical Alzheimer Disease: A Longitudinal Study. JAMA Neurol. 2019, 76, 915–924. [Google Scholar] [CrossRef]
- Takeda, S.; Wegmann, S.; Cho, H.; DeVos, S.L.; Commins, C.; Roe, A.D.; Nicholls, S.B.; Carlson, G.A.; Pitstick, R.; Nobuhara, C.K.; et al. Neuronal Uptake and Propagation of a Rare Phosphorylated High-Molecular-Weight Tau Derived from Alzheimer’s Disease Brain. Nat. Commun. 2015, 6, 8490. [Google Scholar] [CrossRef]
- Crary, J.F.; Trojanowski, J.Q.; Schneider, J.A.; Abisambra, J.F.; Abner, E.L.; Alafuzoff, I.; Arnold, S.E.; Attems, J.; Beach, T.G.; Bigio, E.H.; et al. Primary Age-Related Tauopathy (PART): A Common Pathology Associated with Human Aging. Acta Neuropathol. 2014, 128, 755–766. [Google Scholar] [CrossRef]
- Oddo, S.; Caccamo, A.; Shepherd, J.D.; Murphy, M.P.; Golde, T.E.; Kayed, R.; Metherate, R.; Mattson, M.P.; Akbari, Y.; LaFerla, F.M. Triple-Transgenic Model of Alzheimer’s Disease with Plaques and Tangles: Intracellular Aβ and Synaptic Dysfunction. Neuron 2003, 39, 409–421. [Google Scholar] [CrossRef]
- Choi, S.H.; Kim, Y.H.; Hebisch, M.; Sliwinski, C.; Lee, S.; D’Avanzo, C.; Chen, H.; Hooli, B.; Asselin, C.; Muffat, J.; et al. A Three-Dimensional Human Neural Cell Culture Model of Alzheimer’s Disease. Nature 2014, 515, 274–278. [Google Scholar] [CrossRef]
- Israel, M.A.; Yuan, S.H.; Bardy, C.; Reyna, S.M.; Mu, Y.; Herrera, C.; Hefferan, M.P.; Van Gorp, S.; Nazor, K.L.; Boscolo, F.S.; et al. Probing Sporadic and Familial Alzheimer’s Disease Using Induced Pluripotent Stem Cells. Nature 2012, 482, 216–220. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.K.; Velazquez Sanchez, C.; Chen, M.; Morin, P.J.; Wells, J.M.; Hanlon, E.B.; Xia, W. Three Dimensional Human Neuro-Spheroid Model of Alzheimer’s Disease Based on Differentiated Induced Pluripotent Stem Cells. PLoS ONE 2016, 11, e0163072. [Google Scholar] [CrossRef] [PubMed]
- van der Kant, R.; Goldstein, L.S.B.; Ossenkoppele, R. Amyloid-β-Independent Regulators of Tau Pathology in Alzheimer Disease. Nat. Rev. Neurosci. 2020, 21, 21–35. [Google Scholar] [CrossRef]
- Moreau, K.; Fleming, A.; Imarisio, S.; Lopez Ramirez, A.; Mercer, J.L.; Jimenez-Sanchez, M.; Bento, C.F.; Puri, C.; Zavodszky, E.; Siddiqi, F.; et al. PICALM Modulates Autophagy Activity and Tau Accumulation. Nat. Commun. 2014, 5, 4998. [Google Scholar] [CrossRef] [PubMed]
- Perea, J.R.; Bolos, M.; Avila, J. Microglia in Alzheimer’s Disease in the Context of Tau Pathology. Biomolecules 2020, 10, 1439. [Google Scholar] [CrossRef] [PubMed]
- Wong, C.O. Endosomal-Lysosomal Processing of Neurodegeneration-Associated Proteins in Astrocytes. Int. J. Mol. Sci. 2020, 21, 5149. [Google Scholar] [CrossRef]
- Chan, R.B.; Oliveira, T.G.; Cortes, E.P.; Honig, L.S.; Duff, K.E.; Small, S.A.; Wenk, M.R.; Shui, G.; Di Paolo, G. Comparative Lipidomic Analysis of Mouse and Human Brain with Alzheimer Disease. J. Biol. Chem. 2012, 287, 2678–2688. [Google Scholar] [CrossRef]
- van der Kant, R.; Langness, V.F.; Herrera, C.M.; Williams, D.A.; Fong, L.K.; Leestemaker, Y.; Steenvoorden, E.; Rynearson, K.D.; Brouwers, J.F.; Helms, J.B.; et al. Cholesterol Metabolism Is a Druggable Axis that Independently Regulates Tau and Amyloid-β in iPSC-Derived Alzheimer’s Disease Neurons. Cell Stem Cell 2019, 24, 363–375.e9. [Google Scholar] [CrossRef]
- Shibuya, Y.; Niu, Z.; Bryleva, E.Y.; Harris, B.T.; Murphy, S.R.; Kheirollah, A.; Bowen, Z.D.; Chang, C.C.Y.; Chang, T.Y. Acyl-Coenzyme A:Cholesterol Acyltransferase 1 Blockage Enhances Autophagy in the Neurons of Triple Transgenic Alzheimer’s Disease Mouse and Reduces Human P301L-Tau Content at the Presymptomatic Stage. Neurobiol. Aging 2015, 36, 2248–2259. [Google Scholar] [CrossRef]
- Boimel, M.; Grigoriadis, N.; Lourbopoulos, A.; Touloumi, O.; Rosenmann, D.; Abramsky, O.; Rosenmann, H. Statins Reduce the Neurofibrillary Tangle Burden in a Mouse Model of Tauopathy. J. Neuropathol. Exp. Neurol. 2009, 68, 314–325. [Google Scholar] [CrossRef]
- Beach, T.G.; Walker, R.; McGeer, E.G. Patterns of Gliosis in Alzheimer’s Disease and Aging Cerebrum. Glia 1989, 2, 420–436. [Google Scholar] [CrossRef] [PubMed]
- Itagaki, S.; McGeer, P.L.; Akiyama, H.; Zhu, S.; Selkoe, D. Relationship of Microglia and Astrocytes to Amyloid Deposits of Alzheimer Disease. J. Neuroimmunol. 1989, 24, 173–182. [Google Scholar] [CrossRef] [PubMed]
- McGeer, P.L.; Itagaki, S.; Tago, H.; McGeer, E.G. Reactive Microglia in Patients with Senile Dementia of the Alzheimer Type Are Positive for the Histocompatibility Glycoprotein HLA-DR. Neurosci. Lett. 1987, 79, 195–200. [Google Scholar] [CrossRef] [PubMed]
- Kinney, J.W.; Bemiller, S.M.; Murtishaw, A.S.; Leisgang, A.M.; Salazar, A.M.; Lamb, B.T. Inflammation as a Central Mechanism in Alzheimer’s Disease. Alzheimers Dement. 2018, 4, 575–590. [Google Scholar] [CrossRef]
- Wyss-Coray, T.; Mucke, L. Inflammation in Neurodegenerative Disease—A Double-Edged Sword. Neuron 2002, 35, 419–432. [Google Scholar] [CrossRef]
- Paresce, D.M.; Ghosh, R.N.; Maxfield, F.R. Microglial Cells Internalize Aggregates of the Alzheimer’s Disease Amyloid β-Protein via a Scavenger Receptor. Neuron 1996, 17, 553–565. [Google Scholar] [CrossRef]
- Sondag, C.M.; Dhawan, G.; Combs, C.K. Beta Amyloid Oligomers and Fibrils Stimulate Differential Activation of Primary Microglia. J. Neuroinflamm. 2009, 6, 1. [Google Scholar] [CrossRef]
- Neniskyte, U.; Neher, J.J.; Brown, G.C. Neuronal Death Induced by Nanomolar Amyloid β Is Mediated by Primary Phagocytosis of Neurons by Microglia. J. Biol. Chem. 2011, 286, 39904–39913. [Google Scholar] [CrossRef]
- Akiyama, H.; Barger, S.; Barnum, S.; Bradt, B.; Bauer, J.; Cole, G.M.; Cooper, N.R.; Eikelenboom, P.; Emmerling, M.; Fiebich, B.L.; et al. Inflammation and Alzheimer’s Disease. Neurobiol. Aging 2000, 21, 383–421. [Google Scholar] [CrossRef]
- Calsolaro, V.; Edison, P. Neuroinflammation in Alzheimer’s Disease: Current Evidence and Future Directions. Alzheimers Dement. 2016, 12, 719–732. [Google Scholar] [CrossRef]
- Rozemuller, A.J.; Eikelenboom, P.; Theeuwes, J.W.; Jansen Steur, E.N.; de Vos, R.A. Activated Microglial Cells and Complement Factors Are Unrelated to Cortical Lewy Bodies. Acta Neuropathol. 2000, 100, 701–708. [Google Scholar] [CrossRef] [PubMed]
- Yoshiyama, Y.; Higuchi, M.; Zhang, B.; Huang, S.M.; Iwata, N.; Saido, T.C.; Maeda, J.; Suhara, T.; Trojanowski, J.Q.; Lee, V.M. Synapse Loss and Microglial Activation Precede Tangles in a P301S Tauopathy Mouse Model. Neuron 2007, 53, 337–351. [Google Scholar] [CrossRef] [PubMed]
- Tarkowski, E.; Andreasen, N.; Tarkowski, A.; Blennow, K. Intrathecal Inflammation Precedes Development of Alzheimer’s Disease. J. Neurol. Neurosurg. Psychiatry 2003, 74, 1200–1205. [Google Scholar] [CrossRef] [PubMed]
- Bhaskar, K.; Konerth, M.; Kokiko-Cochran, O.N.; Cardona, A.; Ransohoff, R.M.; Lamb, B.T. Regulation of Tau Pathology by the Microglial Fractalkine Receptor. Neuron 2010, 68, 19–31. [Google Scholar] [CrossRef]
- Gorlovoy, P.; Larionov, S.; Pham, T.T.; Neumann, H. Accumulation of Tau Induced in Neurites by Microglial Proinflammatory Mediators. FASEB J. 2009, 23, 2502–2513. [Google Scholar] [CrossRef]
- Kitazawa, M.; Oddo, S.; Yamasaki, T.R.; Green, K.N.; LaFerla, F.M. Lipopolysaccharide-Induced Inflammation Exacerbates Tau Pathology by a Cyclin-Dependent Kinase 5-Mediated Pathway in a Transgenic Model of Alzheimer’s Disease. J. Neurosci. 2005, 25, 8843–8853. [Google Scholar] [CrossRef]
- Maphis, N.; Xu, G.; Kokiko-Cochran, O.N.; Jiang, S.; Cardona, A.; Ransohoff, R.M.; Lamb, B.T.; Bhaskar, K. Reactive Microglia Drive Tau Pathology and Contribute to the Spreading of Pathological Tau in the Brain. Brain 2015, 138, 1738–1755. [Google Scholar] [CrossRef]
- Ising, C.; Venegas, C.; Zhang, S.; Scheiblich, H.; Schmidt, S.V.; Vieira-Saecker, A.; Schwartz, S.; Albasset, S.; McManus, R.M.; Tejera, D.; et al. NLRP3 Inflammasome Activation Drives Tau Pathology. Nature 2019, 575, 669–673. [Google Scholar] [CrossRef]
- de Wilde, M.C.; Overk, C.R.; Sijben, J.W.; Masliah, E. Meta-Analysis of Synaptic Pathology in Alzheimer’s Disease Reveals Selective Molecular Vesicular Machinery Vulnerability. Alzheimers Dement. 2016, 12, 633–644. [Google Scholar] [CrossRef]
- DeKosky, S.T.; Scheff, S.W.; Styren, S.D. Structural Correlates of Cognition in Dementia: Quantification and Assessment of Synapse Change. Neurodegeneration 1996, 5, 417–421. [Google Scholar] [CrossRef]
- Gomez-Isla, T.; Price, J.L.; McKeel, D.W., Jr.; Morris, J.C.; Growdon, J.H.; Hyman, B.T. Profound Loss of Layer II Entorhinal Cortex Neurons Occurs in very Mild Alzheimer’s Disease. J. Neurosci. 1996, 16, 4491–4500. [Google Scholar] [CrossRef] [PubMed]
- Scheff, S.W.; Price, D.A. Alzheimer’s Disease-Related Alterations in Synaptic Density: Neocortex and Hippocampus. J. Alzheimers Dis. 2006, 9, 101–115. [Google Scholar] [CrossRef] [PubMed]
- Scheff, S.W.; Price, D.A.; Schmitt, F.A.; DeKosky, S.T.; Mufson, E.J. Synaptic Alterations in CA1 in Mild Alzheimer Disease and Mild Cognitive Impairment. Neurology 2007, 68, 1501–1508. [Google Scholar] [CrossRef] [PubMed]
- Terry, R.D.; Masliah, E.; Salmon, D.P.; Butters, N.; DeTeresa, R.; Hill, R.; Hansen, L.A.; Katzman, R. Physical Basis of Cognitive Alterations in Alzheimer’s Disease: Synapse Loss Is the Major Correlate of Cognitive Impairment. Ann. Neurol. 1991, 30, 572–580. [Google Scholar] [CrossRef] [PubMed]
- Forner, S.; Baglietto-Vargas, D.; Martini, A.C.; Trujillo-Estrada, L.; LaFerla, F.M. Synaptic Impairment in Alzheimer’s Disease: A Dysregulated Symphony. Trends Neurosci. 2017, 40, 347–357. [Google Scholar] [CrossRef] [PubMed]
- Knobloch, M.; Mansuy, I.M. Dendritic Spine Loss and Synaptic Alterations in Alzheimer’s Disease. Mol. Neurobiol. 2008, 37, 73–82. [Google Scholar] [CrossRef]
- Pozueta, J.; Lefort, R.; Shelanski, M.L. Synaptic Changes in Alzheimer’s Disease and Its Models. Neuroscience 2013, 251, 51–65. [Google Scholar] [CrossRef]
- Spires-Jones, T.; Knafo, S. Spines, Plasticity, and Cognition in Alzheimer’s Model Mice. Neural Plast. 2012, 2012, 319836. [Google Scholar] [CrossRef]
- Yu, W.; Lu, B. Synapses and Dendritic Spines as Pathogenic Targets in Alzheimer’s Disease. Neural Plast. 2012, 2012, 247150. [Google Scholar] [CrossRef]
- Cirrito, J.R.; Kang, J.E.; Lee, J.; Stewart, F.R.; Verges, D.K.; Silverio, L.M.; Bu, G.; Mennerick, S.; Holtzman, D.M. Endocytosis Is Required for Synaptic Activity-Dependent Release of Amyloid-β In Vivo. Neuron 2008, 58, 42–51. [Google Scholar] [CrossRef]
- Cirrito, J.R.; Yamada, K.A.; Finn, M.B.; Sloviter, R.S.; Bales, K.R.; May, P.C.; Schoepp, D.D.; Paul, S.M.; Mennerick, S.; Holtzman, D.M. Synaptic Activity Regulates Interstitial Fluid Amyloid-β Levels In Vivo. Neuron 2005, 48, 913–922. [Google Scholar] [CrossRef] [PubMed]
- Verges, D.K.; Restivo, J.L.; Goebel, W.D.; Holtzman, D.M.; Cirrito, J.R. Opposing Synaptic Regulation of Amyloid-β Metabolism by NMDA Receptors In Vivo. J. Neurosci. 2011, 31, 11328–11337. [Google Scholar] [CrossRef] [PubMed]
- Long, J.M.; Holtzman, D.M. Alzheimer Disease: An Update on Pathobiology and Treatment Strategies. Cell 2019, 179, 312–339. [Google Scholar] [CrossRef]
- Hong, S.; Beja-Glasser, V.F.; Nfonoyim, B.M.; Frouin, A.; Li, S.; Ramakrishnan, S.; Merry, K.M.; Shi, Q.; Rosenthal, A.; Barres, B.A.; et al. Complement and Microglia Mediate Early Synapse Loss in Alzheimer Mouse Models. Science 2016, 352, 712–716. [Google Scholar] [CrossRef] [PubMed]
- Edwards, F.A. A Unifying Hypothesis for Alzheimer’s Disease: From Plaques to Neurodegeneration. Trends Neurosci. 2019, 42, 310–322. [Google Scholar] [CrossRef]
- Robinson, J.L.; Molina-Porcel, L.; Corrada, M.M.; Raible, K.; Lee, E.B.; Lee, V.M.; Kawas, C.H.; Trojanowski, J.Q. Perforant Path Synaptic Loss Correlates with Cognitive Impairment and Alzheimer’s Disease in the Oldest-Old. Brain 2014, 137, 2578–2587. [Google Scholar] [CrossRef] [PubMed]
- Turano, E.; Busetto, G.; Marconi, S.; Guzzo, F.; Farinazzo, A.; Commisso, M.; Bistaffa, E.; Angiari, S.; Musumeci, S.; Sotgiu, S.; et al. Neurotoxicity and Synaptic Plasticity Impairment of N-Acetylglucosamine Polymers: Implications for Alzheimer’s Disease. Neurobiol. Aging 2015, 36, 1780–1791. [Google Scholar] [CrossRef]
- Gao, C.; Chen, Y.G. Dishevelled: The Hub of Wnt Signaling. Cell. Signal. 2010, 22, 717–727. [Google Scholar] [CrossRef]
- Nusse, R.; Clevers, H. Wnt/β-Catenin Signaling, Disease, and Emerging Therapeutic Modalities. Cell 2017, 169, 985–999. [Google Scholar] [CrossRef]
- Song, Z.; Wang, H.; Zhang, S. Negative Regulators of Wnt Signaling in Non-Small Cell Lung Cancer: Theoretical Basis and Therapeutic Potency. Biomed. Pharmacother. 2019, 118, 109336. [Google Scholar] [CrossRef]
- Luo, X.; Ye, S.; Jiang, Q.; Gong, Y.; Yuan, Y.; Hu, X.; Su, X.; Zhu, W. Wnt Inhibitory Factor-1-Mediated Autophagy Inhibits Wnt/β-Catenin Signaling by Downregulating Dishevelled-2 Expression in Non-Small Cell Lung Cancer Cells. Int. J. Oncol. 2018, 53, 904–914. [Google Scholar] [CrossRef] [PubMed]
- Fukui, T.; Kondo, M.; Ito, G.; Maeda, O.; Sato, N.; Yoshioka, H.; Yokoi, K.; Ueda, Y.; Shimokata, K.; Sekido, Y. Transcriptional Silencing of Secreted Frizzled Related Protein 1 (SFRP 1) by Promoter Hypermethylation in Non-Small-Cell Lung Cancer. Oncogene 2005, 24, 6323–6327. [Google Scholar] [CrossRef] [PubMed]
- Schlensog, M.; Magnus, L.; Heide, T.; Eschenbruch, J.; Steib, F.; Tator, M.; Kloten, V.; Rose, M.; Noetzel, E.; Gaisa, N.T.; et al. Epigenetic Loss of Putative Tumor Suppressor SFRP3 Correlates with Poor Prognosis of Lung Adenocarcinoma Patients. Epigenetics 2018, 13, 214–227. [Google Scholar] [CrossRef] [PubMed]
- Ghoshal, A.; Ghosh, S.S. Antagonizing Canonical Wnt Signaling Pathway by Recombinant Human sFRP4 Purified from E. coli and Its Implications in Cancer Therapy. Mol. Cell. Biochem. 2016, 418, 119–135. [Google Scholar] [CrossRef] [PubMed]
- Kagey, M.H.; He, X. Rationale for Targeting the Wnt Signalling Modulator Dickkopf-1 for Oncology. Br. J. Pharmacol. 2017, 174, 4637–4650. [Google Scholar] [CrossRef]
- Yue, W.; Sun, Q.; Dacic, S.; Landreneau, R.J.; Siegfried, J.M.; Yu, J.; Zhang, L. Downregulation of Dkk3 Activates β-Catenin/TCF-4 Signaling in Lung Cancer. Carcinogenesis 2008, 29, 84–92. [Google Scholar] [CrossRef]
- Seifert, J.R.; Mlodzik, M. Frizzled/PCP Signalling: A Conserved Mechanism Regulating Cell Polarity and Directed Motility. Nat. Rev. Genet. 2007, 8, 126–138. [Google Scholar] [CrossRef]
- De, A. Wnt/Ca2+ Signaling Pathway: A Brief Overview. Acta Biochim. Biophys. Sin. 2011, 43, 745–756. [Google Scholar] [CrossRef]
- Green, J.; Nusse, R.; van Amerongen, R. The Role of Ryk and Ror Receptor Tyrosine Kinases in Wnt Signal Transduction. Cold Spring Harb. Perspect. Biol. 2014, 6, a009175. [Google Scholar] [CrossRef]
- Mikels, A.; Minami, Y.; Nusse, R. Ror2 Receptor Requires Tyrosine Kinase Activity to Mediate Wnt5A Signaling. J. Biol. Chem. 2009, 284, 30167–30176. [Google Scholar] [CrossRef]
- Brafman, D.; Willert, K. Wnt/β-Catenin Signaling during Early Vertebrate Neural Development. Dev. Neurobiol. 2017, 77, 1239–1259. [Google Scholar] [CrossRef] [PubMed]
- Paganoni, S.; Bernstein, J.; Ferreira, A. Ror1-Ror2 Complexes Modulate Synapse Formation in Hippocampal Neurons. Neuroscience 2010, 165, 1261–1274. [Google Scholar] [CrossRef] [PubMed]
- Ming, G.L.; Song, H. Adult Neurogenesis in the Mammalian Brain: Significant Answers and Significant Questions. Neuron 2011, 70, 687–702. [Google Scholar] [CrossRef]
- Lie, D.C.; Colamarino, S.A.; Song, H.J.; Desire, L.; Mira, H.; Consiglio, A.; Lein, E.S.; Jessberger, S.; Lansford, H.; Dearie, A.R.; et al. Wnt Signalling Regulates Adult Hippocampal Neurogenesis. Nature 2005, 437, 1370–1375. [Google Scholar] [CrossRef] [PubMed]
- Miranda, C.J.; Braun, L.; Jiang, Y.; Hester, M.E.; Zhang, L.; Riolo, M.; Wang, H.; Rao, M.; Altura, R.A.; Kaspar, B.K. Aging Brain Microenvironment Decreases Hippocampal Neurogenesis through Wnt-Mediated Survivin Signaling. Aging Cell 2012, 11, 542–552. [Google Scholar] [CrossRef]
- Song, H.; Stevens, C.F.; Gage, F.H. Astroglia Induce Neurogenesis from Adult Neural Stem Cells. Nature 2002, 417, 39–44. [Google Scholar] [CrossRef] [PubMed]
- Kalani, M.Y.; Cheshier, S.H.; Cord, B.J.; Bababeygy, S.R.; Vogel, H.; Weissman, I.L.; Palmer, T.D.; Nusse, R. Wnt-Mediated Self-Renewal of Neural Stem/Progenitor Cells. Proc. Natl. Acad. Sci. USA 2008, 105, 16970–16975. [Google Scholar] [CrossRef]
- Qu, Q.; Sun, G.; Murai, K.; Ye, P.; Li, W.; Asuelime, G.; Cheung, Y.T.; Shi, Y. Wnt7a Regulates Multiple Steps of Neurogenesis. Mol. Cell. Biol. 2013, 33, 2551–2559. [Google Scholar] [CrossRef]
- Garcia, A.; Steiner, B.; Kronenberg, G.; Bick-Sander, A.; Kempermann, G. Age-Dependent Expression of Glucocorticoid- and Mineralocorticoid Receptors on Neural Precursor Cell Populations in the Adult Murine Hippocampus. Aging Cell 2004, 3, 363–371. [Google Scholar] [CrossRef]
- Luo, J.; Daniels, S.B.; Lennington, J.B.; Notti, R.Q.; Conover, J.C. The Aging Neurogenic Subventricular Zone. Aging Cell 2006, 5, 139–152. [Google Scholar] [CrossRef]
- Okamoto, M.; Inoue, K.; Iwamura, H.; Terashima, K.; Soya, H.; Asashima, M.; Kuwabara, T. Reduction in Paracrine Wnt3 Factors during Aging Causes Impaired Adult Neurogenesis. FASEB J. 2011, 25, 3570–3582. [Google Scholar] [CrossRef] [PubMed]
- Buechler, J.; Salinas, P.C. Deficient Wnt Signaling and Synaptic Vulnerability in Alzheimer’s Disease: Emerging Roles for the LRP6 Receptor. Front. Synaptic Neurosci. 2018, 10, 38. [Google Scholar] [CrossRef] [PubMed]
- McLeod, F.; Salinas, P.C. Wnt Proteins as Modulators of Synaptic Plasticity. Curr. Opin. Neurobiol. 2018, 53, 90–95. [Google Scholar] [CrossRef] [PubMed]
- Oliva, C.A.; Inestrosa, N.C. A Novel Function for Wnt Signaling Modulating Neuronal Firing Activity and the temporal Structure of Spontaneous Oscillation in the Entorhinal-Hippocampal Circuit. Exp. Neurol. 2015, 269, 43–55. [Google Scholar] [CrossRef] [PubMed]
- Varela-Nallar, L.; Alfaro, I.E.; Serrano, F.G.; Parodi, J.; Inestrosa, N.C. Wingless-Type Family Member 5A (Wnt-5a) Stimulates Synaptic Differentiation and Function of Glutamatergic Synapses. Proc. Natl. Acad. Sci. USA 2010, 107, 21164–21169. [Google Scholar] [CrossRef] [PubMed]
- McLeod, F.; Bossio, A.; Marzo, A.; Ciani, L.; Sibilla, S.; Hannan, S.; Wilson, G.A.; Palomer, E.; Smart, T.G.; Gibb, A.; et al. Wnt Signaling Mediates LTP-Dependent Spine Plasticity and AMPAR Localization through Frizzled-7 Receptors. Cell Rep. 2018, 23, 1060–1071. [Google Scholar] [CrossRef]
- Cerpa, W.; Gambrill, A.; Inestrosa, N.C.; Barria, A. Regulation of NMDA-Receptor Synaptic Transmission by Wnt Signaling. J. Neurosci. 2011, 31, 9466–9471. [Google Scholar] [CrossRef]
- Ciani, L.; Marzo, A.; Boyle, K.; Stamatakou, E.; Lopes, D.M.; Anane, D.; McLeod, F.; Rosso, S.B.; Gibb, A.; Salinas, P.C. Wnt Signalling Tunes Neurotransmitter Release by Directly Targeting Synaptotagmin-1. Nat. Commun. 2015, 6, 8302. [Google Scholar] [CrossRef]
- Sahores, M.; Gibb, A.; Salinas, P.C. Frizzled-5, a Receptor for the Synaptic Organizer Wnt7a, Regulates Activity-Mediated Synaptogenesis. Development 2010, 137, 2215–2225. [Google Scholar] [CrossRef]
- Avila, M.E.; Sepulveda, F.J.; Burgos, C.F.; Moraga-Cid, G.; Parodi, J.; Moon, R.T.; Aguayo, L.G.; Opazo, C.; De Ferrari, G.V. Canonical Wnt3a Modulates Intracellular Calcium and Enhances Excitatory Neurotransmission in Hippocampal Neurons. J. Biol. Chem. 2010, 285, 18939–18947. [Google Scholar] [CrossRef]
- Beaumont, V.; Thompson, S.A.; Choudhry, F.; Nuthall, H.; Glantschnig, H.; Lipfert, L.; David, G.R.; Swain, C.J.; McAllister, G.; Munoz-Sanjuan, I. Evidence for an Enhancement of Excitatory Transmission in Adult CNS by Wnt Signaling Pathway Modulation. Mol. Cell. Neurosci. 2007, 35, 513–524. [Google Scholar] [CrossRef] [PubMed]
- Cerpa, W.; Godoy, J.A.; Alfaro, I.; Farias, G.G.; Metcalfe, M.J.; Fuentealba, R.; Bonansco, C.; Inestrosa, N.C. Wnt-7a Modulates the Synaptic Vesicle Cycle and Synaptic Transmission in Hippocampal Neurons. J. Biol. Chem. 2008, 283, 5918–5927. [Google Scholar] [CrossRef]
- Ciani, L.; Boyle, K.A.; Dickins, E.; Sahores, M.; Anane, D.; Lopes, D.M.; Gibb, A.J.; Salinas, P.C. Wnt7a Signaling Promotes Dendritic Spine Growth and Synaptic Strength through Ca2+/Calmodulin-Dependent Protein Kinase II. Proc. Natl. Acad. Sci. USA 2011, 108, 10732–10737. [Google Scholar] [CrossRef] [PubMed]
- Tabatadze, N.; Tomas, C.; McGonigal, R.; Lin, B.; Schook, A.; Routtenberg, A. Wnt Transmembrane Signaling and Long-Term Spatial Memory. Hippocampus 2012, 22, 1228–1241. [Google Scholar] [CrossRef] [PubMed]
- Fortress, A.M.; Schram, S.L.; Tuscher, J.J.; Frick, K.M. Canonical Wnt Signaling Is Necessary for Object Recognition Memory Consolidation. J. Neurosci. 2013, 33, 12619–12626. [Google Scholar] [CrossRef] [PubMed]
- Maguschak, K.A.; Ressler, K.J. Wnt Signaling in Amygdala-Dependent Learning and Memory. J. Neurosci. 2011, 31, 13057–13067. [Google Scholar] [CrossRef]
- Aghaizu, N.D.; Jin, H.; Whiting, P.J. Dysregulated Wnt Signalling in the Alzheimer’s Brain. Brain Sci. 2020, 10, 902. [Google Scholar] [CrossRef]
- Liu, L.; Wan, W.; Xia, S.; Kalionis, B.; Li, Y. Dysfunctional Wnt/β-Catenin Signaling Contributes to Blood-Brain Barrier Breakdown in Alzheimer’s Disease. Neurochem. Int. 2014, 75, 19–25. [Google Scholar] [CrossRef]
- Wang, Q.; Huang, X.; Su, Y.; Yin, G.; Wang, S.; Yu, B.; Li, H.; Qi, J.; Chen, H.; Zeng, W.; et al. Activation of Wnt/β-Catenin Pathway Mitigates Blood-Brain Barrier Dysfunction in Alzheimer’s Disease. Brain 2022, 145, 4474–4488. [Google Scholar] [CrossRef]
- Acevedo, K.M.; Opazo, C.M.; Norrish, D.; Challis, L.M.; Li, Q.X.; White, A.R.; Bush, A.I.; Camakaris, J. Phosphorylation of Amyloid Precursor Protein at Threonine 668 Is Essential for Its Copper-Responsive Trafficking in SH-SY5Y Neuroblastoma Cells. J. Biol. Chem. 2014, 289, 11007–11019. [Google Scholar] [CrossRef]
- Aplin, A.E.; Gibb, G.M.; Jacobsen, J.S.; Gallo, J.M.; Anderton, B.H. In Vitro Phosphorylation of the Cytoplasmic Domain of the Amyloid Precursor Protein by Glycogen Synthase Kinase-3β. J. Neurochem. 1996, 67, 699–707. [Google Scholar] [CrossRef] [PubMed]
- Scheinfeld, M.H.; Ghersi, E.; Davies, P.; D’Adamio, L. Amyloid β Protein Precursor Is Phosphorylated by JNK-1 Independent of, Yet Facilitated by, JNK-Interacting Protein (JIP)-1. J. Biol. Chem. 2003, 278, 42058–42063. [Google Scholar] [CrossRef] [PubMed]
- Standen, C.L.; Brownlees, J.; Grierson, A.J.; Kesavapany, S.; Lau, K.F.; McLoughlin, D.M.; Miller, C.C. Phosphorylation of thr668 in the Cytoplasmic Domain of the Alzheimer’s Disease Amyloid Precursor Protein by Stress-Activated Protein Kinase 1b (Jun N-Terminal Kinase-3). J. Neurochem. 2001, 76, 316–320. [Google Scholar] [CrossRef] [PubMed]
- Taru, H.; Iijima, K.; Hase, M.; Kirino, Y.; Yagi, Y.; Suzuki, T. Interaction of Alzheimer’s β-Amyloid Precursor Family Proteins with Scaffold Proteins of the JNK Signaling Cascade. J. Biol. Chem. 2002, 277, 20070–20078. [Google Scholar] [CrossRef]
- Liu, C.C.; Tsai, C.W.; Deak, F.; Rogers, J.; Penuliar, M.; Sung, Y.M.; Maher, J.N.; Fu, Y.; Li, X.; Xu, H.; et al. Deficiency in LRP6-Mediated Wnt Signaling Contributes to Synaptic Abnormalities and Amyloid Pathology in Alzheimer’s Disease. Neuron 2014, 84, 63–77. [Google Scholar] [CrossRef]
- Liu, T.; Zhang, T.; Nicolas, M.; Boussicault, L.; Rice, H.; Soldano, A.; Claeys, A.; Petrova, I.; Fradkin, L.; De Strooper, B.; et al. The Amyloid Precursor Protein Is a Conserved Wnt Receptor. eLife 2021, 10, e69199. [Google Scholar] [CrossRef]
- LaChance, L.R.; Ramsey, D. Antidepressant Foods: An Evidence-Based Nutrient Profiling System for Depression. World J. Psychiatry 2018, 8, 97–104. [Google Scholar] [CrossRef]
- Tapia-Rojas, C.; Burgos, P.V.; Inestrosa, N.C. Inhibition of Wnt Signaling Induces Amyloidogenic Processing of Amyloid Precursor Protein and the Production and Aggregation of Amyloid-β (Aβ)42 Peptides. J. Neurochem. 2016, 139, 1175–1191. [Google Scholar] [CrossRef]
- Parr, C.; Carzaniga, R.; Gentleman, S.M.; Van Leuven, F.; Walter, J.; Sastre, M. Glycogen Synthase Kinase 3 Inhibition Promotes Lysosomal Biogenesis and Autophagic Degradation of the Amyloid-β Precursor Protein. Mol. Cell. Biol. 2012, 32, 4410–4418. [Google Scholar] [CrossRef]
- Takashima, A.; Murayama, M.; Murayama, O.; Kohno, T.; Honda, T.; Yasutake, K.; Nihonmatsu, N.; Mercken, M.; Yamaguchi, H.; Sugihara, S.; et al. Presenilin 1 Associates with Glycogen Synthase Kinase-3β and Its Substrate Tau. Proc. Natl. Acad. Sci. USA 1998, 95, 9637–9641. [Google Scholar] [CrossRef]
- Hernandez, F.; Lucas, J.J.; Avila, J. GSK3 and Tau: Two Convergence Points in Alzheimer’s Disease. J. Alzheimers Dis. 2013, 33 (Suppl. 1), S141–S144. [Google Scholar] [CrossRef] [PubMed]
- Kremer, A.; Louis, J.V.; Jaworski, T.; Van Leuven, F. GSK3 and Alzheimer’s Disease: Facts and Fiction. Front. Mol. Neurosci. 2011, 4, 17. [Google Scholar] [CrossRef] [PubMed]
- Van Steenwinckel, J.; Schang, A.L.; Krishnan, M.L.; Degos, V.; Delahaye-Duriez, A.; Bokobza, C.; Csaba, Z.; Verdonk, F.; Montane, A.; Sigaut, S.; et al. Decreased Microglial Wnt/β-Catenin Signalling Drives Microglial Pro-Inflammatory Activation in the Developing Brain. Brain 2019, 142, 3806–3833. [Google Scholar] [CrossRef] [PubMed]
- Halleskog, C.; Mulder, J.; Dahlstrom, J.; Mackie, K.; Hortobagyi, T.; Tanila, H.; Kumar Puli, L.; Farber, K.; Harkany, T.; Schulte, G. WNT Signaling in Activated Microglia Is Proinflammatory. Glia 2011, 59, 119–131. [Google Scholar] [CrossRef] [PubMed]
- Purro, S.A.; Dickins, E.M.; Salinas, P.C. The Secreted Wnt Antagonist Dickkopf-1 Is Required for Amyloid β-Mediated Synaptic Loss. J. Neurosci. 2012, 32, 3492–3498. [Google Scholar] [CrossRef]
- Sellers, K.J.; Elliott, C.; Jackson, J.; Ghosh, A.; Ribe, E.; Rojo, A.I.; Jarosz-Griffiths, H.H.; Watson, I.A.; Xia, W.; Semenov, M.; et al. Amyloid β Synaptotoxicity Is Wnt-PCP Dependent and Blocked by Fasudil. Alzheimers Dement. 2018, 14, 306–317. [Google Scholar] [CrossRef]
- Killick, R.; Ribe, E.M.; Al-Shawi, R.; Malik, B.; Hooper, C.; Fernandes, C.; Dobson, R.; Nolan, P.M.; Lourdusamy, A.; Furney, S.; et al. Clusterin Regulates β-Amyloid Toxicity via Dickkopf-1-Driven Induction of the wnt-PCP-JNK Pathway. Mol. Psychiatry 2014, 19, 88–98. [Google Scholar] [CrossRef]
- Vargas, J.Y.; Ahumada, J.; Arrazola, M.S.; Fuenzalida, M.; Inestrosa, N.C. WASP-1, a Canonical Wnt Signaling Potentiator, Rescues Hippocampal Synaptic Impairments Induced by Aβ Oligomers. Exp. Neurol. 2015, 264, 14–25. [Google Scholar] [CrossRef]
- Hooper, C.; Killick, R.; Lovestone, S. The GSK3 Hypothesis of Alzheimer’s Disease. J. Neurochem. 2008, 104, 1433–1439. [Google Scholar] [CrossRef]
- Kawamura, Y.; Kikuchi, A.; Takada, R.; Takada, S.; Sudoh, S.; Shibamoto, S.; Yanagisawa, K.; Komano, H. Inhibitory Effect of a Presenilin 1 Mutation on the Wnt Signalling Pathway by Enhancement of β-Catenin Phosphorylation. Eur. J. Biochem. 2001, 268, 3036–3041. [Google Scholar] [CrossRef]
- Llorens-Martin, M.; Jurado, J.; Hernandez, F.; Avila, J. GSK-3β, a Pivotal Kinase in Alzheimer Disease. Front. Mol. Neurosci. 2014, 7, 46. [Google Scholar] [CrossRef] [PubMed]
- Pei, J.J.; Braak, E.; Braak, H.; Grundke-Iqbal, I.; Iqbal, K.; Winblad, B.; Cowburn, R.F. Distribution of Active Glycogen Synthase Kinase 3β (GSK-3β) in Brains Staged for Alzheimer Disease Neurofibrillary Changes. J. Neuropathol. Exp. Neurol. 1999, 58, 1010–1019. [Google Scholar] [CrossRef] [PubMed]
- Caricasole, A.; Copani, A.; Caraci, F.; Aronica, E.; Rozemuller, A.J.; Caruso, A.; Storto, M.; Gaviraghi, G.; Terstappen, G.C.; Nicoletti, F. Induction of Dickkopf-1, a Negative Modulator of the Wnt Pathway, Is Associated with Neuronal Degeneration in Alzheimer’s Brain. J. Neurosci. 2004, 24, 6021–6027. [Google Scholar] [CrossRef] [PubMed]
- Folke, J.; Pakkenberg, B.; Brudek, T. Impaired Wnt Signaling in the Prefrontal Cortex of Alzheimer’s Disease. Mol. Neurobiol. 2019, 56, 873–891. [Google Scholar] [CrossRef] [PubMed]
- Forlenza, O.V.; de Paula, V.J.; Machado-Vieira, R.; Diniz, B.S.; Gattaz, W.F. Does Lithium Prevent Alzheimer’s Disease? Drugs Aging 2012, 29, 335–342. [Google Scholar] [CrossRef]
- Palomer, E.; Buechler, J.; Salinas, P.C. Wnt Signaling Deregulation in the Aging and Alzheimer’s Brain. Front. Cell. Neurosci. 2019, 13, 227. [Google Scholar] [CrossRef]
- Hofmann, J.W.; McBryan, T.; Adams, P.D.; Sedivy, J.M. The Effects of Aging on the Expression of Wnt Pathway Genes in Mouse Tissues. Age 2014, 36, 9618. [Google Scholar] [CrossRef]
- Bayod, S.; Felice, P.; Andres, P.; Rosa, P.; Camins, A.; Pallas, M.; Canudas, A.M. Downregulation of Canonical Wnt Signaling in Hippocampus of SAMP8 Mice. Neurobiol. Aging 2015, 36, 720–729. [Google Scholar] [CrossRef]
- Orellana, A.M.; Vasconcelos, A.R.; Leite, J.A.; de Sa Lima, L.; Andreotti, D.Z.; Munhoz, C.D.; Kawamoto, E.M.; Scavone, C. Age-Related Neuroinflammation and Changes in AKT-GSK-3β and WNT/β-CATENIN Signaling in Rat Hippocampus. Aging 2015, 7, 1094–1111. [Google Scholar] [CrossRef]
- Scott, E.L.; Zhang, Q.G.; Han, D.; Desai, B.N.; Brann, D.W. Long-Term Estrogen Deprivation Leads to Elevation of Dickkopf-1 and Dysregulation of Wnt/β-Catenin Signaling in Hippocampal CA1 Neurons. Steroids 2013, 78, 624–632. [Google Scholar] [CrossRef]
- Seib, D.R.; Corsini, N.S.; Ellwanger, K.; Plaas, C.; Mateos, A.; Pitzer, C.; Niehrs, C.; Celikel, T.; Martin-Villalba, A. Loss of Dickkopf-1 Restores Neurogenesis in Old Age and Counteracts Cognitive Decline. Cell Stem Cell 2013, 12, 204–214. [Google Scholar] [CrossRef] [PubMed]
- Tapia-Rojas, C.; Inestrosa, N.C. Wnt Signaling Loss Accelerates the Appearance of Neuropathological Hallmarks of Alzheimer’s Disease in J20-APP Transgenic and Wild-Type Mice. J. Neurochem. 2018, 144, 443–465. [Google Scholar] [CrossRef]
- Esquerda-Canals, G.; Montoliu-Gaya, L.; Guell-Bosch, J.; Villegas, S. Mouse Models of Alzheimer’s Disease. J. Alzheimers Dis. 2017, 57, 1171–1183. [Google Scholar] [CrossRef] [PubMed]
- Marchetti, C.; Marie, H. Hippocampal Synaptic Plasticity in Alzheimer’s Disease: What Have We Learned So Far from transgenic Models? Rev. Neurosci. 2011, 22, 373–402. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.; Chen, D.; Lee, T.H. Phosphorylation Signaling in APP Processing in Alzheimer’s Disease. Int. J. Mol. Sci. 2019, 21, 209. [Google Scholar] [CrossRef] [PubMed]
- Iijima, K.; Ando, K.; Takeda, S.; Satoh, Y.; Seki, T.; Itohara, S.; Greengard, P.; Kirino, Y.; Nairn, A.C.; Suzuki, T. Neuron-Specific Phosphorylation of Alzheimer’s β-Amyloid Precursor Protein by Cyclin-Dependent Kinase 5. J. Neurochem. 2000, 75, 1085–1091. [Google Scholar] [CrossRef] [PubMed]
- Sodhi, C.P.; Perez, R.G.; Gottardi-Littell, N.R. Phosphorylation of β-Amyloid Precursor Protein (APP) Cytoplasmic Tail Facilitates Amyloidogenic Processing during Apoptosis. Brain Res. 2008, 1198, 204–212. [Google Scholar] [CrossRef]
- Muresan, Z.; Muresan, V. c-Jun NH2-Terminal Kinase-Interacting Protein-3 Facilitates Phosphorylation and Controls Localization of Amyloid-β Precursor Protein. J. Neurosci. 2005, 25, 3741–3751. [Google Scholar] [CrossRef]
- Muresan, Z.; Muresan, V. The Amyloid-β Precursor Protein Is Phosphorylated via Distinct Pathways during Differentiation, Mitosis, Stress, and Degeneration. Mol. Biol. Cell 2007, 18, 3835–3844. [Google Scholar] [CrossRef]
- Salcedo-Tello, P.; Ortiz-Matamoros, A.; Arias, C. GSK3 Function in the Brain during Development, Neuronal Plasticity, and Neurodegeneration. Int. J. Alzheimers Dis. 2011, 2011, 189728. [Google Scholar] [CrossRef]
- Colombo, A.; Bastone, A.; Ploia, C.; Sclip, A.; Salmona, M.; Forloni, G.; Borsello, T. JNK Regulates APP Cleavage and Degradation in a Model of Alzheimer’s Disease. Neurobiol. Dis. 2009, 33, 518–525. [Google Scholar] [CrossRef] [PubMed]
- Marzolo, M.P.; Bu, G. Lipoprotein Receptors and Cholesterol in APP Trafficking and Proteolytic Processing, Implications for Alzheimer’s Disease. Semin. Cell Dev. Biol. 2009, 20, 191–200. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.; Suh, J.; Romano, D.; Truong, M.H.; Mullin, K.; Hooli, B.; Norton, D.; Tesco, G.; Elliott, K.; Wagner, S.L.; et al. Potential Late-Onset Alzheimer’s Disease-Associated Mutations in the ADAM10 Gene Attenuate α-Secretase Activity. Hum. Mol. Genet. 2009, 18, 3987–3996. [Google Scholar] [CrossRef] [PubMed]
- Zhang, N.; Parr, C.J.C.; Birch, A.M.; Goldfinger, M.H.; Sastre, M. The Amyloid Precursor Protein Binds to β-Catenin and Modulates Its Cellular Distribution. Neurosci. Lett. 2018, 685, 190–195. [Google Scholar] [CrossRef]
- Zhou, F.; Gong, K.; Song, B.; Ma, T.; van Laar, T.; Gong, Y.; Zhang, L. The APP Intracellular Domain (AICD) Inhibits Wnt Signalling and Promotes Neurite Outgrowth. Biochim. Biophys. Acta 2012, 1823, 1233–1241. [Google Scholar] [CrossRef]
- Capell, A.; Grunberg, J.; Pesold, B.; Diehlmann, A.; Citron, M.; Nixon, R.; Beyreuther, K.; Selkoe, D.J.; Haass, C. The Proteolytic Fragments of the Alzheimer’s Disease-Associated Presenilin-1 Form Heterodimers and Occur as a 100-150-kDa Molecular Mass Complex. J. Biol. Chem. 1998, 273, 3205–3211. [Google Scholar] [CrossRef]
- Yu, G.; Chen, F.; Levesque, G.; Nishimura, M.; Zhang, D.M.; Levesque, L.; Rogaeva, E.; Xu, D.; Liang, Y.; Duthie, M.; et al. The Presenilin 1 Protein Is a Component of a High Molecular Weight Intracellular Complex that Contains β-Catenin. J. Biol. Chem. 1998, 273, 16470–16475. [Google Scholar] [CrossRef]
- Zhang, Z.; Hartmann, H.; Do, V.M.; Abramowski, D.; Sturchler-Pierrat, C.; Staufenbiel, M.; Sommer, B.; van de Wetering, M.; Clevers, H.; Saftig, P.; et al. Destabilization of β-Catenin by Mutations in Presenilin-1 Potentiates Neuronal Apoptosis. Nature 1998, 395, 698–702. [Google Scholar] [CrossRef]
- Nishimura, M.; Yu, G.; Levesque, G.; Zhang, D.M.; Ruel, L.; Chen, F.; Milman, P.; Holmes, E.; Liang, Y.; Kawarai, T.; et al. Presenilin Mutations Associated with Alzheimer Disease Cause Defective Intracellular Trafficking of β-Catenin, a Component of the Presenilin Protein Complex. Nat. Med. 1999, 5, 164–169. [Google Scholar] [CrossRef]
- Dudek, H.; Datta, S.R.; Franke, T.F.; Birnbaum, M.J.; Yao, R.; Cooper, G.M.; Segal, R.A.; Kaplan, D.R.; Greenberg, M.E. Regulation of Neuronal Survival by the Serine-Threonine Protein Kinase Akt. Science 1997, 275, 661–665. [Google Scholar] [CrossRef]
- Hemmings, B.A. Akt Signaling: Linking Membrane Events to Life and Death Decisions. Science 1997, 275, 628–630. [Google Scholar] [CrossRef] [PubMed]
- Takashima, A.; Noguchi, K.; Michel, G.; Mercken, M.; Hoshi, M.; Ishiguro, K.; Imahori, K. Exposure of Rat Hippocampal Neurons to Amyloid β Peptide (25–35) Induces the Inactivation of Phosphatidyl Inositol-3 Kinase and the Activation of Tau Protein Kinase I/Glycogen Synthase Kinase-3β. Neurosci. Lett. 1996, 203, 33–36. [Google Scholar] [CrossRef]
- Mandelkow, E.M.; Mandelkow, E. Biochemistry and Cell Biology of Tau Protein in Neurofibrillary Degeneration. Cold Spring Harb. Perspect. Med. 2012, 2, a006247. [Google Scholar] [CrossRef] [PubMed]
- Scali, C.; Caraci, F.; Gianfriddo, M.; Diodato, E.; Roncarati, R.; Pollio, G.; Gaviraghi, G.; Copani, A.; Nicoletti, F.; Terstappen, G.C.; et al. Inhibition of Wnt Signaling, Modulation of Tau Phosphorylation and Induction of Neuronal Cell Death by DKK1. Neurobiol. Dis. 2006, 24, 254–265. [Google Scholar] [CrossRef]
- Vargas, J.Y.; Fuenzalida, M.; Inestrosa, N.C. In Vivo Activation of Wnt Signaling Pathway Enhances Cognitive Function of Adult Mice and Reverses Cognitive Deficits in an Alzheimer’s Disease Model. J. Neurosci. 2014, 34, 2191–2202. [Google Scholar] [CrossRef]
- Esposito, G.; De Filippis, D.; Carnuccio, R.; Izzo, A.A.; Iuvone, T. The Marijuana Component Cannabidiol Inhibits β-Amyloid-Induced Tau Protein Hyperphosphorylation through Wnt/β-Catenin Pathway Rescue in PC12 Cells. J. Mol. Med. (Berl.) 2006, 84, 253–258. [Google Scholar] [CrossRef]
- Hu, S.; Begum, A.N.; Jones, M.R.; Oh, M.S.; Beech, W.K.; Beech, B.H.; Yang, F.; Chen, P.; Ubeda, O.J.; Kim, P.C.; et al. GSK3 Inhibitors Show Benefits in an Alzheimer’s Disease (AD) Model of Neurodegeneration but Adverse Effects in Control Animals. Neurobiol. Dis. 2009, 33, 193–206. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, S.; Wu, M.D.; Shaftel, S.S.; Kyrkanides, S.; LaFerla, F.M.; Olschowka, J.A.; O’Banion, M.K. Sustained Interleukin-1β Overexpression Exacerbates Tau Pathology Despite Reduced Amyloid Burden in an Alzheimer’s Mouse Model. J. Neurosci. 2013, 33, 5053–5064. [Google Scholar] [CrossRef]
- Li, Y.; Liu, L.; Barger, S.W.; Griffin, W.S. Interleukin-1 Mediates Pathological Effects of Microglia on Tau Phosphorylation and on Synaptophysin Synthesis in Cortical Neurons through a p38-MAPK Pathway. J. Neurosci. 2003, 23, 1605–1611. [Google Scholar] [CrossRef]
- Bhat, S.A.; Goel, R.; Shukla, S.; Shukla, R.; Hanif, K. Angiotensin Receptor Blockade by Inhibiting Glial Activation Promotes Hippocampal Neurogenesis via Activation of Wnt/β-Catenin Signaling in Hypertension. Mol. Neurobiol. 2018, 55, 5282–5298. [Google Scholar] [CrossRef]
- Song, D.; Zhang, X.; Chen, J.; Liu, X.; Xue, J.; Zhang, L.; Lan, X. Wnt Canonical Pathway Activator TWS119 Drives Microglial Anti-Inflammatory Activation and Facilitates Neurological Recovery following Experimental Stroke. J. Neuroinflamm. 2019, 16, 256. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Lu, Z.; Man, J.; Cui, K.; Fu, X.; Yu, L.; Gao, Y.; Liao, L.; Xiao, Q.; Guo, R.; et al. Wnt-3a Alleviates Neuroinflammation after Ischemic Stroke by Modulating the Responses of Microglia/Macrophages and Astrocytes. Int. Immunopharmacol. 2019, 75, 105760. [Google Scholar] [CrossRef] [PubMed]
- Zheng, H.; Jia, L.; Liu, C.C.; Rong, Z.; Zhong, L.; Yang, L.; Chen, X.F.; Fryer, J.D.; Wang, X.; Zhang, Y.W.; et al. TREM2 Promotes Microglial Survival by Activating Wnt/β-Catenin Pathway. J. Neurosci. 2017, 37, 1772–1784. [Google Scholar] [CrossRef] [PubMed]
- Bottcher, C.; Schlickeiser, S.; Sneeboer, M.A.M.; Kunkel, D.; Knop, A.; Paza, E.; Fidzinski, P.; Kraus, L.; Snijders, G.J.L.; Kahn, R.S.; et al. Human Microglia Regional Heterogeneity and Phenotypes Determined by Multiplexed Single-Cell Mass Cytometry. Nat. Neurosci. 2019, 22, 78–90. [Google Scholar] [CrossRef]
- Halleskog, C.; Schulte, G. Pertussis Toxin-Sensitive Heterotrimeric Gαi/o Proteins Mediate WNT/β-Catenin and WNT/ERK1/2 Signaling in Mouse Primary Microglia Stimulated with Purified WNT-3A. Cell. Signal. 2013, 25, 822–828. [Google Scholar] [CrossRef]
- Mrdjen, D.; Pavlovic, A.; Hartmann, F.J.; Schreiner, B.; Utz, S.G.; Leung, B.P.; Lelios, I.; Heppner, F.L.; Kipnis, J.; Merkler, D.; et al. High-Dimensional Single-Cell Mapping of Central Nervous System Immune Cells Reveals Distinct Myeloid Subsets in Health, Aging, and Disease. Immunity 2018, 48, 599. [Google Scholar] [CrossRef]
- O’Koren, E.G.; Yu, C.; Klingeborn, M.; Wong, A.Y.W.; Prigge, C.L.; Mathew, R.; Kalnitsky, J.; Msallam, R.A.; Silvin, A.; Kay, J.N.; et al. Microglial Function Is Distinct in Different Anatomical Locations during Retinal Homeostasis and Degeneration. Immunity 2019, 50, 723–737.e7. [Google Scholar] [CrossRef]
- Meilandt, W.J.; Ngu, H.; Gogineni, A.; Lalehzadeh, G.; Lee, S.H.; Srinivasan, K.; Imperio, J.; Wu, T.; Weber, M.; Kruse, A.J.; et al. Trem2 Deletion Reduces Late-Stage Amyloid Plaque Accumulation, Elevates the Aβ42:Aβ40 Ratio, and Exacerbates Axonal Dystrophy and Dendritic Spine Loss in the PS2APP Alzheimer’s Mouse Model. J. Neurosci. 2020, 40, 1956–1974. [Google Scholar] [CrossRef]
- Marzo, A.; Galli, S.; Lopes, D.; McLeod, F.; Podpolny, M.; Segovia-Roldan, M.; Ciani, L.; Purro, S.; Cacucci, F.; Gibb, A.; et al. Reversal of Synapse Degeneration by Restoring Wnt Signaling in the Adult Hippocampus. Curr. Biol. 2016, 26, 2551–2561. [Google Scholar] [CrossRef]
- Elliott, C.; Rojo, A.I.; Ribe, E.; Broadstock, M.; Xia, W.; Morin, P.; Semenov, M.; Baillie, G.; Cuadrado, A.; Al-Shawi, R.; et al. A Role for APP in Wnt Signalling Links Synapse Loss with β-Amyloid Production. Transl. Psychiatry 2018, 8, 179. [Google Scholar] [CrossRef]
- Magdesian, M.H.; Carvalho, M.M.; Mendes, F.A.; Saraiva, L.M.; Juliano, M.A.; Juliano, L.; Garcia-Abreu, J.; Ferreira, S.T. Amyloid-β Binds to the Extracellular Cysteine-Rich Domain of Frizzled and Inhibits Wnt/β-Catenin Signaling. J. Biol. Chem. 2008, 283, 9359–9368. [Google Scholar] [CrossRef] [PubMed]
- He, P.; Shen, Y. Interruption of β-Catenin Signaling Reduces Neurogenesis in Alzheimer’s Disease. J. Neurosci. 2009, 29, 6545–6557. [Google Scholar] [CrossRef] [PubMed]
- Cerpa, W.; Farias, G.G.; Godoy, J.A.; Fuenzalida, M.; Bonansco, C.; Inestrosa, N.C. Wnt-5a Occludes Aβ oligomer-Induced Depression of Glutamatergic Transmission in Hippocampal Neurons. Mol. Neurodegener. 2010, 5, 3. [Google Scholar] [CrossRef] [PubMed]
- Boonen, R.A.; van Tijn, P.; Zivkovic, D. Wnt Signaling in Alzheimer’s Disease: Up or Down, That Is the Question. Ageing Res. Rev. 2009, 8, 71–82. [Google Scholar] [CrossRef]
- Bali, J.; Gheinani, A.H.; Zurbriggen, S.; Rajendran, L. Role of Genes Linked to Sporadic Alzheimer’s Disease Risk in the Production of β-Amyloid Peptides. Proc. Natl. Acad. Sci. USA 2012, 109, 15307–15311. [Google Scholar] [CrossRef]
- Cabrera, O.; Berman, D.M.; Kenyon, N.S.; Ricordi, C.; Berggren, P.O.; Caicedo, A. The Unique Cytoarchitecture of Human Pancreatic Islets Has Implications for Islet Cell Function. Proc. Natl. Acad. Sci. USA 2006, 103, 2334–2339. [Google Scholar] [CrossRef]
- Masjosthusmann, S.; Becker, D.; Petzuch, B.; Klose, J.; Siebert, C.; Deenen, R.; Barenys, M.; Baumann, J.; Dach, K.; Tigges, J.; et al. A Transcriptome Comparison of Time-Matched Developing Human, Mouse and Rat Neural Progenitor Cells Reveals Human Uniqueness. Toxicol. Appl. Pharmacol. 2018, 354, 40–55. [Google Scholar] [CrossRef]
- Mestas, J.; Hughes, C.C. Of Mice and Not Men: Differences between Mouse and Human Immunology. J. Immunol. 2004, 172, 2731–2738. [Google Scholar] [CrossRef]
- Seok, J.; Warren, H.S.; Cuenca, A.G.; Mindrinos, M.N.; Baker, H.V.; Xu, W.; Richards, D.R.; McDonald-Smith, G.P.; Gao, H.; Hennessy, L.; et al. Genomic Responses in Mouse Models Poorly Mimic Human Inflammatory Diseases. Proc. Natl. Acad. Sci. USA 2013, 110, 3507–3512. [Google Scholar] [CrossRef]
- Bardy, C.; van den Hurk, M.; Eames, T.; Marchand, C.; Hernandez, R.V.; Kellogg, M.; Gorris, M.; Galet, B.; Palomares, V.; Brown, J.; et al. Neuronal Medium that Supports Basic Synaptic Functions and Activity of Human Neurons In Vitro. Proc. Natl. Acad. Sci. USA 2015, 112, E2725–E2734. [Google Scholar] [CrossRef]
- Begum, A.N.; Guoynes, C.; Cho, J.; Hao, J.; Lutfy, K.; Hong, Y. Rapid Generation of Sub-Type, Region-Specific Neurons and Neural Networks from Human Pluripotent Stem Cell-Derived Neurospheres. Stem Cell Res. 2015, 15, 731–741. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Pak, C.; Han, Y.; Ahlenius, H.; Zhang, Z.; Chanda, S.; Marro, S.; Patzke, C.; Acuna, C.; Covy, J.; et al. Rapid Single-Step Induction of Functional Neurons from Human Pluripotent Stem Cells. Neuron 2013, 78, 785–798. [Google Scholar] [CrossRef] [PubMed]
- Shaltouki, A.; Peng, J.; Liu, Q.; Rao, M.S.; Zeng, X. Efficient Generation of Astrocytes from Human Pluripotent Stem Cells in Defined Conditions. Stem Cells 2013, 31, 941–952. [Google Scholar] [CrossRef] [PubMed]
- Tcw, J.; Wang, M.; Pimenova, A.A.; Bowles, K.R.; Hartley, B.J.; Lacin, E.; Machlovi, S.I.; Abdelaal, R.; Karch, C.M.; Phatnani, H.; et al. An Efficient Platform for Astrocyte Differentiation from Human Induced Pluripotent Stem Cells. Stem Cell Rep. 2017, 9, 600–614. [Google Scholar] [CrossRef]
- Abud, E.M.; Ramirez, R.N.; Martinez, E.S.; Healy, L.M.; Nguyen, C.H.H.; Newman, S.A.; Yeromin, A.V.; Scarfone, V.M.; Marsh, S.E.; Fimbres, C.; et al. iPSC-Derived Human Microglia-like Cells to Study Neurological Diseases. Neuron 2017, 94, 278–293.e9. [Google Scholar] [CrossRef]
- Wang, S.; Bates, J.; Li, X.; Schanz, S.; Chandler-Militello, D.; Levine, C.; Maherali, N.; Studer, L.; Hochedlinger, K.; Windrem, M.; et al. Human iPSC-Derived Oligodendrocyte Progenitor Cells can Myelinate and Rescue a Mouse Model of Congenital Hypomyelination. Cell Stem Cell 2013, 12, 252–264. [Google Scholar] [CrossRef]
- Arber, C.; Toombs, J.; Lovejoy, C.; Ryan, N.S.; Paterson, R.W.; Willumsen, N.; Gkanatsiou, E.; Portelius, E.; Blennow, K.; Heslegrave, A.; et al. Familial Alzheimer’s Disease Patient-Derived Neurons Reveal Distinct Mutation-Specific Effects on Amyloid Beta. Mol. Psychiatry 2020, 25, 2919–2931. [Google Scholar] [CrossRef]
- Muratore, C.R.; Rice, H.C.; Srikanth, P.; Callahan, D.G.; Shin, T.; Benjamin, L.N.; Walsh, D.M.; Selkoe, D.J.; Young-Pearse, T.L. The Familial Alzheimer’s Disease APPV717I Mutation Alters APP Processing and Tau Expression in iPSC-Derived Neurons. Hum. Mol. Genet. 2014, 23, 3523–3536. [Google Scholar] [CrossRef]
- Ochalek, A.; Mihalik, B.; Avci, H.X.; Chandrasekaran, A.; Teglasi, A.; Bock, I.; Giudice, M.L.; Tancos, Z.; Molnar, K.; Laszlo, L.; et al. Neurons Derived from Sporadic Alzheimer’s Disease iPSCs Reveal Elevated TAU hyperphosphorylation, Increased Amyloid Levels, and GSK3B Activation. Alzheimers Res. Ther. 2017, 9, 90. [Google Scholar] [CrossRef]
- Raman, S.; Brookhouser, N.; Brafman, D.A. Using Human Induced Pluripotent Stem Cells (hiPSCs) to Investigate the Mechanisms by which Apolipoprotein E (APOE) Contributes to Alzheimer’s Disease (AD) Risk. Neurobiol. Dis. 2020, 138, 104788. [Google Scholar] [CrossRef]
- Sen, T.; Thummer, R.P. CRISPR and iPSCs: Recent Developments and Future Perspectives in Neurodegenerative Disease Modelling, Research, and Therapeutics. Neurotox. Res. 2022, 40, 1597–1623. [Google Scholar] [CrossRef] [PubMed]
- Li, X.F.; Zhou, Y.W.; Cai, P.F.; Fu, W.C.; Wang, J.H.; Chen, J.Y.; Yang, Q.N. CRISPR/Cas9 Facilitates Genomic Editing for Large-Scale Functional Studies in Pluripotent Stem Cell Cultures. Hum. Genet. 2019, 138, 1217–1225. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Inoue, H.; Wu, J.C.; Yamanaka, S. Induced Pluripotent Stem Cell Technology: A Decade of Progress. Nat. Rev. Drug Discov. 2017, 16, 115–130. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Effect | AD-Associated Phenotype | WNT Signaling Pathway | WNT Component | Reference |
|---|---|---|---|---|
| ↑ APP phosphorylation | ↑ Aβ | Canonical | GSK3β | [190,191] |
| ↑ APP phosphorylation | ↑ Aβ | Non-canonical (WNT/PCP) | JNK | [192,193,194] |
| ↓ APP endocytosis | ↓ Aβ | Canonical | LRP6 | [195] |
| ↓ APP endocytosis | ↓ Aβ | Non-canonical | WNT5a | [196] |
| ↓ APP endocytosis | ↓ Aβ | Canonical | WNT3a | [196] |
| ↑ non-amyloidogenic processing | ↓ Aβ | Canonical | WNT3a | [197] |
| ↑ non-amyloidogenic processing | ↓ Aβ | Non-canonical (WNT/PCP) | WNT5a | [197] |
| ↓ BACE1 expression | ↓ Aβ | Canonical | TCF4 | [198,199] |
| ↑ PSEN1 binding activity | ↑ p-tau | Canonical | GSK3β | [200] |
| ↑ GSK3β phosphorylation activity | ↑ p-tau | Canonical | GSK3β | [201,202] |
| ↑ β-catenin degradation | ↑ pro-inflammatory microglia | Canonical | AXIN2 | [203] |
| ↓ neuronal LRP6 | ↑ neuroinflammation | Canonical | LRP6 | [195] |
| ↑ pro-inflammatory gene expression | ↑ neuroinflammation | Canonical | WNT3a | [204] |
| ↑ DKK1 expression | ↑ synapse loss and Aβ | Canonical | DKK1 | [205] |
| ↑ DKK1 expression | ↑ synapse loss | Non-canonical (WNT/PCP) | DKK1 | [206,207] |
| ↓ LRP6 expression | ↑ synapse loss and Aβ | Canonical | LRP6 | [195] |
| ↓ Aβ aggregation | ↓ p-tau and synapse loss | Canonical | WASP-1 | [208] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kostes, W.W.; Brafman, D.A. The Multifaceted Role of WNT Signaling in Alzheimer’s Disease Onset and Age-Related Progression. Cells 2023, 12, 1204. https://doi.org/10.3390/cells12081204
Kostes WW, Brafman DA. The Multifaceted Role of WNT Signaling in Alzheimer’s Disease Onset and Age-Related Progression. Cells. 2023; 12(8):1204. https://doi.org/10.3390/cells12081204
Chicago/Turabian StyleKostes, William W., and David A. Brafman. 2023. "The Multifaceted Role of WNT Signaling in Alzheimer’s Disease Onset and Age-Related Progression" Cells 12, no. 8: 1204. https://doi.org/10.3390/cells12081204
APA StyleKostes, W. W., & Brafman, D. A. (2023). The Multifaceted Role of WNT Signaling in Alzheimer’s Disease Onset and Age-Related Progression. Cells, 12(8), 1204. https://doi.org/10.3390/cells12081204