



Residual Foci of DNA Damage Response Proteins in Relation to Cellular Senescence and Autophagy in X-Ray Irradiated Fibroblasts

 , ,
, ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. Irradiation
2.3. Immunocytochemistry
2.4. Analysis of Senescence Associated β-galactosidase Positive Cells
2.5. Statistical Analysis
3. Results
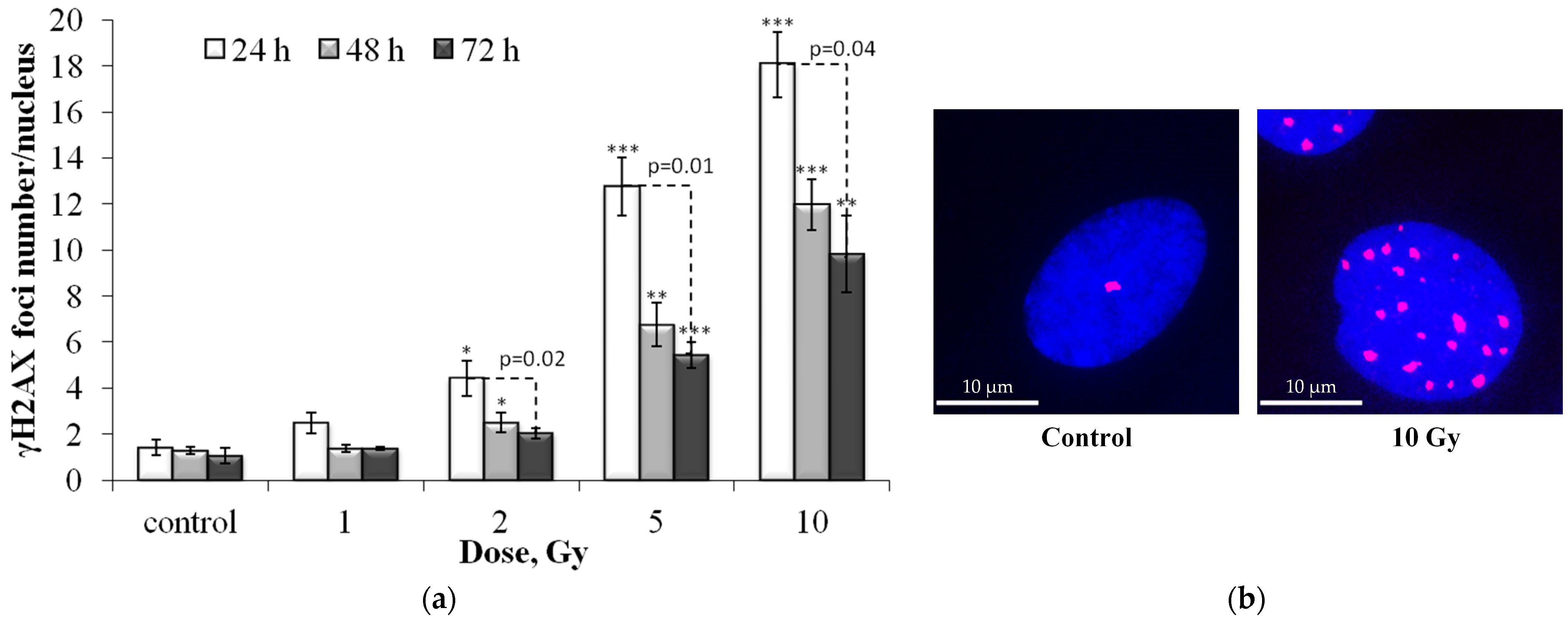
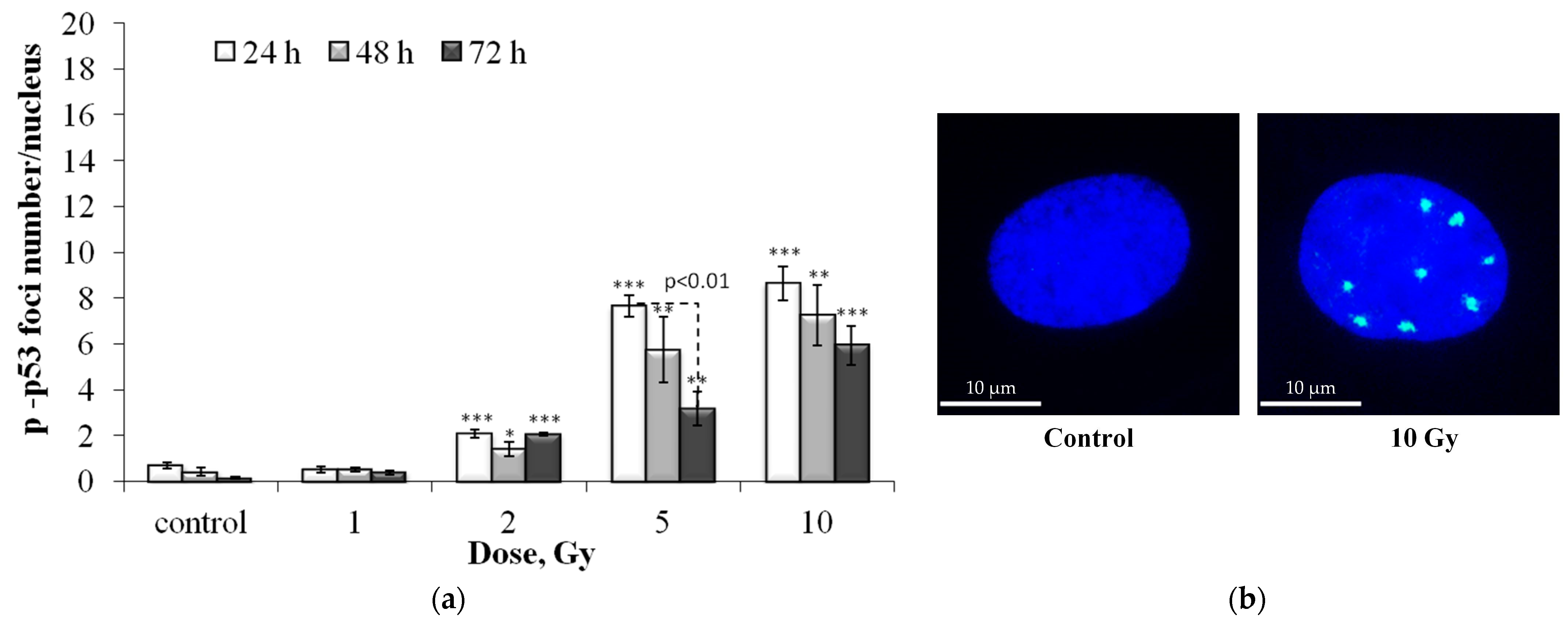
3.1. Dose and Post-Irradiation Tim- Dependent Changes in the Residual Foci of DNA Damage Response Proteins
3.2. Colocalization of pATM, 53BP1 and p-p53 Foci with γH2AX Foci
3.3. Dose and Post-Irradiation Time-Dependent Changes in the Senescent Cell Proportion
3.4. Dose and Post-Irradiation Time–Dependent Changes in the Ki-67 Negative Cell Fraction and Differential Scoring of γH2AX Foci in the Ki-67 Negative Cells
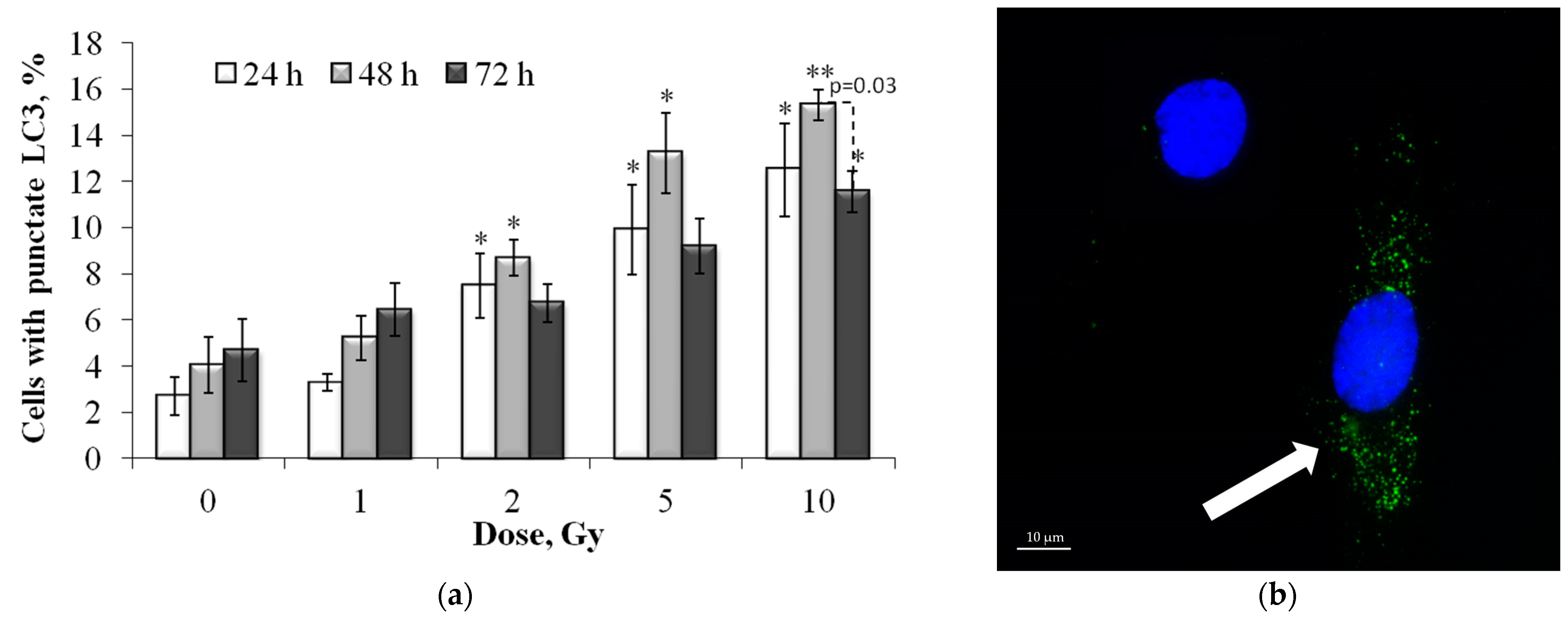
3.5. Dose and Post-Irradiation Time-Dependent Changes in the LC3-II Positive Cell Fraction
3.6. Dose and Post-Irradiation Time-Dependent Changes in the Caspase-3 Positive Cell Fraction
3.7. Correlation and Clustering Analysis
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Appendix A

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Protein | Time after X-ray Exposure, h | ||
|---|---|---|---|
| 24 | 48 | 72 | |
| γH2AX | 1.76 ± 0.20 | 1.13 ± 0.07 | 0.92 ± 0.05 |
| pATM | 0.99 ± 0.10 | 0.90 ± 0.07 | 0.62 ± 0.04 |
| 53BP1 | 1.31 ± 0.13 | 0.97 ± 0.05 | 0.76 ± 0.03 |
| p-p53 | 0.90 ± 0.21 | 0.76 ± 0.14 | 0.58 ± 0.06 |
| Protein | Time after X-ray Exposure, h | ||
|---|---|---|---|
| 24 | 48 | 72 | |
| pATM | 0.88 ± 0.08 | 0.82 ± 0.04 | 0.57 ± 0.04 |
| 53BP1 | 1.20 ± 0.12 | 0.89 ± 0.05 | 0.69 ± 0.03 |
| p-p53 | 0.80 ± 0.13 | 0.73 ± 0.08 | 0.51 ± 0.04 |
| γH2AX | 53BP1 | pATM | p-p53 | caspase-3 | LC3-II | SA-β-gal | Ki-67 | |
|---|---|---|---|---|---|---|---|---|
| 24 h | ||||||||
| γH2AX | - | 0.998 * | 0.998 * | 0.983 * | 0.877 | 0.952 * | 0.939 * | 0.928 * |
| 53BP1 | 0.998 * | - | 0.999 * | 0.977 * | 0.853 | 0.950 * | 0.926 * | 0.913 * |
| pATM | 0.998 * | 0.999 * | - | 0.978 * | 0.872 | 0.963 * | 0.941 * | 0.929 * |
| p-p53 | 0.983 * | 0.977 * | 0.978 * | - | 0.889 * | 0.944 * | 0.920 * | 0.914 * |
| caspase-3 | 0.877 | 0.853 | 0.872 | 0.889 * | - | 0.934 * | 0.976 * | 0.984 * |
| LC3-II | 0.952 * | 0.950 * | 0.963 * | 0.944 * | 0.934 * | - | 0.974 * | 0.972 * |
| SA-β-gal | 0.939 * | 0.926 * | 0.941 * | 0.920 * | 0.976 * | 0.974 * | - | 0.999 * |
| Ki-67 | 0.928 * | 0.913 * | 0.929 * | 0.914 * | 0.984 * | 0.972 * | 0.999 * | - |
| 48 h | ||||||||
| γH2AX | - | 0.998 * | 0.996 * | 0.970 * | 0.218 | 0.938 * | 0.916 * | 0.851 |
| 53BP1 | 0.998 * | - | 0.991 * | 0.956 * | 0.200 | 0.923 * | 0.909 * | 0.837 |
| pATM | 0.996 * | 0.991 * | - | 0.984 * | 0.286 | 0.964 * | 0.940 * | 0.889 * |
| p-p53 | 0.970 * | 0.956 * | 0.984 * | - | 0.312 | 0.972 * | 0.932 * | 0.901 * |
| caspase-3 | 0.218 | 0.200 | 0.286 | 0.312 | - | 0.513 | 0.569 | 0.682 |
| LC3-II | 0.938 * | 0.923 * | 0.964 * | 0.972 * | 0.513 | - | 0.976 * | 0.970 * |
| SA-β-gal | 0.916 * | 0.909 * | 0.940 * | 0.932 * | 0.569 | 0.976 * | - | 0.984 * |
| Ki-67 | 0.851 | 0.837 | 0.889 * | 0.901 * | 0.682 | 0.970 * | 0.984 * | - |
| 72 h | ||||||||
| γH2AX | - | 0.997 * | 0.994 * | 0.974 * | −0.143 | 0.970 * | 0.827 | 0.799 |
| 53BP1 | 0.997 * | - | 0.990 * | 0.979 * | −0.150 | 0.965 * | 0.835 | 0.790 |
| pATM | 0.994 * | 0.990 * | - | 0.988 * | −0.063 | 0.981 * | 0.866 | 0.854 |
| p-p53 | 0.974 * | 0.979 * | 0.988 * | - | −0.036 | 0.966 * | 0.895 * | 0.864 |
| caspase-3 | −0.143 | −0.150 | −0.063 | −0.036 | - | 0.102 | 0.397 | 0.449 |
| LC3-II | 0.970 * | 0.965 * | 0.981 * | 0.966 * | 0.102 | - | 0.926 * | 0.908 * |
| SA-β-gal | 0.827 | 0.835 | 0.866 | 0.895 * | 0.397 | 0.926 * | - | 0.962 * |
| Ki-67 | 0.799 | 0.790 | 0.854 | 0.864 | 0.449 | 0.908 * | 0.962 * | - |
References
- Adamus-Gorka, M.; Mavroidis, P.; Lind, B.K.; Brahme, A. Comparison of dose response models for predicting normal tissue complications from cancer radiotherapy: Application in rat spinal cord. Cancers 2011, 3, 2421–2443. [Google Scholar] [CrossRef] [PubMed]
- Falk, M.; Hausmann, M. A Paradigm Revolution or Just Better Resolution-Will Newly Emerging Superresolution Techniques Identify Chromatin Architecture as a Key Factor in Radiation-Induced DNA Damage and Repair Regulation? Cancers 2020, 13, 18. [Google Scholar] [CrossRef] [PubMed]
- Shibata, A.; Jeggo, P.A. DNA double-strand break repair in a cellular context. Clin. Oncol. (R Coll. Radiol.) 2014, 26, 243–249. [Google Scholar] [CrossRef] [PubMed]
- Oh, J.M.; Myung, K. Crosstalk between different DNA repair pathways for DNA double strand break repairs. Mutat. Res. Genet. Toxicol. Environ. Mutagen. 2022, 873, 503438. [Google Scholar] [CrossRef] [PubMed]
- White, R.R.; Vijg, J. Do DNA Double-Strand Breaks Drive Aging? Mol. Cell 2016, 63, 729–738. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y. Contribution of Microhomology to Genome Instability: Connection between DNA Repair and Replication Stress. Int. J. Mol. Sci. 2022, 23, 2937. [Google Scholar] [CrossRef]
- Sishc, B.J.; Davis, A.J. The Role of the Core Non-Homologous End Joining Factors in Carcinogenesis and Cancer. Cancers 2017, 9, 81. [Google Scholar] [CrossRef]
- Rothkamm, K.; Barnard, S.; Moquet, J.; Ellender, M.; Rana, Z.; Burdak-Rothkamm, S. DNA damage foci: Meaning and significance. Environ. Mol. Mutagen. 2015, 56, 491–504. [Google Scholar] [CrossRef]
- Bushmanov, A.; Vorobyeva, N.; Molodtsova, D.; Osipov, A.N. Utilization of DNA double-strand breaks for biodosimetry of ionizing radiation exposure. Environ. Adv. 2022, 8, 100207. [Google Scholar] [CrossRef]
- Rahmanian, N.; Shokrzadeh, M.; Eskandani, M. Recent advances in gammaH2AX biomarker-based genotoxicity assays: A marker of DNA damage and repair. DNA Repair 2021, 108, 103243. [Google Scholar] [CrossRef]
- Raavi, V.; Perumal, V.; Paul, S.F.D. Potential application of gamma-H2AX as a biodosimetry tool for radiation triage. Mutat. Res. Rev. Mutat. Res. 2021, 787, 108350. [Google Scholar] [CrossRef] [PubMed]
- Osipov, A.N.; Pustovalova, M.; Grekhova, A.; Eremin, P.; Vorobyova, N.; Pulin, A.; Zhavoronkov, A.; Roumiantsev, S.; Klokov, D.Y.; Eremin, I. Low doses of X-rays induce prolonged and ATM-independent persistence of gammaH2AX foci in human gingival mesenchymal stem cells. Oncotarget 2015, 6, 27275–27287. [Google Scholar] [CrossRef]
- Ulyanenko, S.; Pustovalova, M.; Koryakin, S.; Beketov, E.; Lychagin, A.; Ulyanenko, L.; Kaprin, A.; Grekhova, A.; Ozerova, A.M.; Ozerov, I.O.; et al. Formation of gammaH2AX and pATM Foci in Human Mesenchymal Stem Cells Exposed to Low Dose-Rate Gamma-Radiation. Int. J. Mol. Sci. 2019, 20, 2645. [Google Scholar] [CrossRef]
- Markova, E.; Vasilyev, S.; Belyaev, I. 53BP1 foci as a marker of tumor cell radiosensitivity. Neoplasma 2015, 62, 770–776. [Google Scholar] [CrossRef] [PubMed]
- Shibata, A.; Jeggo, P.A. Roles for 53BP1 in the repair of radiation-induced DNA double strand breaks. DNA Repair 2020, 93, 102915. [Google Scholar] [CrossRef] [PubMed]
- Al Rashid, S.T.; Dellaire, G.; Cuddihy, A.; Jalali, F.; Vaid, M.; Coackley, C.; Folkard, M.; Xu, Y.; Chen, B.P.; Chen, D.J.; et al. Evidence for the direct binding of phosphorylated p53 to sites of DNA breaks in vivo. Cancer Res. 2005, 65, 10810–10821. [Google Scholar] [CrossRef] [PubMed]
- Grekhova, A.K.; Pustovalova, M.V.; Eremin, P.S.; Ozerov, I.V.; Maksimova, O.A.; Gordeev, A.V.; Vorobyeva, N.Y.; Osipov, A.N. Evaluation of the Contribution of Homologous Recombination in DNA Double-Strand Break Repair in Human Fibroblasts after Exposure to Low and Intermediate Doses of X-ray Radiation. Biol. Bull. 2020, 46, 1496–1502. [Google Scholar] [CrossRef]
- Belyaev, I.Y. Radiation-induced DNA repair foci: Spatio-temporal aspects of formation, application for assessment of radiosensitivity and biological dosimetry. Mutat. Res. 2010, 704, 132–141. [Google Scholar] [CrossRef]
- Sorokin, M.; Kholodenko, R.; Grekhova, A.; Suntsova, M.; Pustovalova, M.; Vorobyeva, N.; Kholodenko, I.; Malakhova, G.; Garazha, A.; Nedoluzhko, A.; et al. Acquired resistance to tyrosine kinase inhibitors may be linked with the decreased sensitivity to X-ray irradiation. Oncotarget 2017, 9, 5111–5124. [Google Scholar] [CrossRef]
- Banath, J.P.; Klokov, D.; MacPhail, S.H.; Banuelos, C.A.; Olive, P.L. Residual gammaH2AX foci as an indication of lethal DNA lesions. BMC Cancer 2010, 10, 4. [Google Scholar] [CrossRef]
- Vorobyeva, N.Y.; Babayan, N.S.; Grigoryan, B.A.; Sargsyan, A.A.; Khondkaryan, L.G.; Apresyan, L.S.; Chigasova, A.K.; Yashkina, E.I.; Guryev, D.V.; Rodneva, S.M.; et al. Increased Yield of Residual γH2AX Foci in p53-Deficient Human Lung Carcinoma Cells Exposed to Subpicosecond Beams of Accelerated Electrons. Bull. Exp. Biol. Med. 2022, 172, 756–759. [Google Scholar] [CrossRef] [PubMed]
- Olive, P.L. Retention of gammaH2AX foci as an indication of lethal DNA damage. Radiother. Oncol. 2011, 101, 18–23. [Google Scholar] [CrossRef] [PubMed]
- Babayan, N.; Vorobyeva, N.; Grigoryan, B.; Grekhova, A.; Pustovalova, M.; Rodneva, S.; Fedotov, Y.; Tsakanova, G.; Aroutiounian, R.; Osipov, A. Low Repair Capacity of DNA Double-Strand Breaks Induced by Laser-Driven Ultrashort Electron Beams in Cancer Cells. Int. J. Mol. Sci. 2020, 21, 9488. [Google Scholar] [CrossRef] [PubMed]
- Pustovalova, M.; Astrelina capital Te, C.; Grekhova, A.; Vorobyeva, N.; Tsvetkova, A.; Blokhina, T.; Nikitina, V.; Suchkova, Y.; Usupzhanova, D.; Brunchukov, V.; et al. Residual gammaH2AX foci induced by low dose x-ray radiation in bone marrow mesenchymal stem cells do not cause accelerated senescence in the progeny of irradiated cells. Aging 2017, 9, 2397–2410. [Google Scholar] [CrossRef]
- Zorin, V.; Zorina, A.; Smetanina, N.; Kopnin, P.; Ozerov, I.V.; Leonov, S.; Isaev, A.; Klokov, D.; Osipov, A.N. Diffuse colonies of human skin fibroblasts in relation to cellular senescence and proliferation. Aging 2017, 9, 1404–1413. [Google Scholar] [CrossRef]
- Maier, A.B.; Westendorp, R.G.; Heemst, D.V. Beta-galactosidase activity as a biomarker of replicative senescence during the course of human fibroblast cultures. Ann. N. Y. Acad. Sci. 2007, 1100, 323–332. [Google Scholar] [CrossRef]
- Miller, I.; Min, M.; Yang, C.; Tian, C.; Gookin, S.; Carter, D.; Spencer, S.L. Ki-67 is a Graded Rather than a Binary Marker of Proliferation versus Quiescence. Cell Rep. 2018, 24, 1105–1112.e5. [Google Scholar] [CrossRef]
- Sobecki, M.; Mrouj, K.; Camasses, A.; Parisis, N.; Nicolas, E.; Lleres, D.; Gerbe, F.; Prieto, S.; Krasinska, L.; David, A.; et al. The cell proliferation antigen Ki-67 organises heterochromatin. Elife 2016, 5, e13722. [Google Scholar] [CrossRef]
- Sobecki, M.; Mrouj, K.; Colinge, J.; Gerbe, F.; Jay, P.; Krasinska, L.; Dulic, V.; Fisher, D. Cell-Cycle Regulation Accounts for Variability in Ki-67 Expression Levels. Cancer Res. 2017, 77, 2722–2734. [Google Scholar] [CrossRef]
- Hansen, T.E.; Johansen, T. Following autophagy step by step. BMC Biol 2011, 9, 39. [Google Scholar] [CrossRef]
- Tanida, I.; Ueno, T.; Kominami, E. LC3 and Autophagy. Methods Mol. Biol. 2008, 445, 77–88. [Google Scholar] [CrossRef] [PubMed]
- Boice, A.; Bouchier-Hayes, L. Targeting apoptotic caspases in cancer. Biochim Biophys. Acta Mol. Cell Res. 2020, 1867, 118688. [Google Scholar] [CrossRef] [PubMed]
- Tsapras, P.; Nezis, I.P. Caspase involvement in autophagy. Cell Death Differ. 2017, 24, 1369–1379. [Google Scholar] [CrossRef] [PubMed]
- Panneer Selvam, S.; Roth, B.M.; Nganga, R.; Kim, J.; Cooley, M.A.; Helke, K.; Smith, C.D.; Ogretmen, B. Balance between senescence and apoptosis is regulated by telomere damage-induced association between p16 and caspase-3. J. Biol. Chem. 2018, 293, 9784–9800. [Google Scholar] [CrossRef] [PubMed]
- Eskandari, E.; Eaves, C.J. Paradoxical roles of caspase-3 in regulating cell survival, proliferation, and tumorigenesis. J. Cell Biol. 2022, 221, e202201159. [Google Scholar] [CrossRef]
- di Masi, A.; Cilli, D.; Berardinelli, F.; Talarico, A.; Pallavicini, I.; Pennisi, R.; Leone, S.; Antoccia, A.; Noguera, N.I.; Lo-Coco, F.; et al. PML nuclear body disruption impairs DNA double-strand break sensing and repair in APL. Cell Death Dis. 2016, 7, e2308. [Google Scholar] [CrossRef]
- Scully, R.; Xie, A. Double strand break repair functions of histone H2AX. Mutat. Res. 2013, 750, 5–14. [Google Scholar] [CrossRef]
- Lamarche, B.J.; Orazio, N.I.; Weitzman, M.D. The MRN complex in double-strand break repair and telomere maintenance. FEBS Lett. 2010, 584, 3682–3695. [Google Scholar] [CrossRef]
- Merighi, A.; Gionchiglia, N.; Granato, A.; Lossi, L. The Phosphorylated Form of the Histone H2AX (gammaH2AX) in the Brain from Embryonic Life to Old Age. Molecules 2021, 26, 7198. [Google Scholar] [CrossRef]
- Yan, W.; Shao, Z.; Li, F.; Niu, L.; Shi, Y.; Teng, M.; Li, X. Structural basis of gammaH2AX recognition by human PTIP BRCT5-BRCT6 domains in the DNA damage response pathway. FEBS Lett. 2011, 585, 3874–3879. [Google Scholar] [CrossRef]
- Bakkenist, C.J.; Kastan, M.B. DNA damage activates ATM through intermolecular autophosphorylation and dimer dissociation. Nature 2003, 421, 499–506. [Google Scholar] [CrossRef]
- Firsanov, D.V.; Solovjeva, L.V.; Svetlova, M.P. H2AX phosphorylation at the sites of DNA double-strand breaks in cultivated mammalian cells and tissues. Clin. Epigenetics 2011, 2, 283–297. [Google Scholar] [CrossRef]
- Kinner, A.; Wu, W.; Staudt, C.; Iliakis, G. Gamma-H2AX in recognition and signaling of DNA double-strand breaks in the context of chromatin. Nucleic Acids Res. 2008, 36, 5678–5694. [Google Scholar] [CrossRef] [PubMed]
- Ward, I.M.; Chen, J. Histone H2AX is phosphorylated in an ATR-dependent manner in response to replicational stress. J. Biol. Chem. 2001, 276, 47759–47762. [Google Scholar] [CrossRef] [PubMed]
- Gelot, C.; Magdalou, I.; Lopez, B.S. Replication stress in Mammalian cells and its consequences for mitosis. Genes 2015, 6, 267–298. [Google Scholar] [CrossRef]
- Kim, K.P.; Mirkin, E.V. So similar yet so different: The two ends of a double strand break. Mutat. Res. 2017, 809, 70–80. [Google Scholar] [CrossRef] [PubMed]
- Mirman, Z.; de Lange, T. 53BP1: A DSB escort. Genes Dev. 2020, 34, 7–23. [Google Scholar] [CrossRef]
- Derbyshire, D.J.; Basu, B.P.; Serpell, L.C.; Joo, W.S.; Date, T.; Iwabuchi, K.; Doherty, A.J. Crystal structure of human 53BP1 BRCT domains bound to p53 tumour suppressor. EMBO J. 2002, 21, 3863–3872. [Google Scholar] [CrossRef]
- Zimmermann, M.; de Lange, T. 53BP1: Pro choice in DNA repair. Trends Cell Biol. 2014, 24, 108–117. [Google Scholar] [CrossRef]
- Lei, T.; Du, S.; Peng, Z.; Chen, L. Multifaceted regulation and functions of 53BP1 in NHEJmediated DSB repair (Review). Int. J. Mol. Med. 2022, 50, 90. [Google Scholar] [CrossRef]
- Loughery, J.; Cox, M.; Smith, L.M.; Meek, D.W. Critical role for p53-serine 15 phosphorylation in stimulating transactivation at p53-responsive promoters. Nucleic Acids Res. 2014, 42, 7666–7680. [Google Scholar] [CrossRef] [PubMed]
- Meek, D.W. Tumour suppression by p53: A role for the DNA damage response? Nat. Rev. Cancer 2009, 9, 714–723. [Google Scholar] [CrossRef] [PubMed]
- Mirzayans, R.; Andrais, B.; Scott, A.; Murray, D. New insights into p53 signaling and cancer cell response to DNA damage: Implications for cancer therapy. J. Biomed. Biotechnol. 2012, 2012, 170325. [Google Scholar] [CrossRef] [PubMed]
- Rothkamm, K.; Kruger, I.; Thompson, L.H.; Lobrich, M. Pathways of DNA double-strand break repair during the mammalian cell cycle. Mol. Cell. Biol. 2003, 23, 5706–5715. [Google Scholar] [CrossRef] [PubMed]
- Schmitt, C.A. Cellular senescence and cancer treatment. Biochim. Biophys. Acta 2007, 1775, 5–20. [Google Scholar] [CrossRef]
- Ojha, R.; Ishaq, M.; Singh, S.K. Caspase-mediated crosstalk between autophagy and apoptosis: Mutual adjustment or matter of dominance. J. Cancer Res. Ther. 2015, 11, 514–524. [Google Scholar] [CrossRef]
- Marcotte, R.; Lacelle, C.; Wang, E. Senescent fibroblasts resist apoptosis by downregulating caspase-3. Mech. Ageing Dev. 2004, 125, 777–783. [Google Scholar] [CrossRef]
- Aliper, A.M.; Bozdaganyan, M.E.; Orekhov, P.S.; Zhavoronkov, A.; Osipov, A.N. Replicative and radiation-induced aging: A comparison of gene expression profiles. Aging 2019, 11, 2378–2387. [Google Scholar] [CrossRef]
- Kwon, Y.; Kim, J.W.; Jeoung, J.A.; Kim, M.S.; Kang, C. Autophagy Is Pro-Senescence When Seen in Close-Up, but Anti-Senescence in Long-Shot. Mol. Cells 2017, 40, 607–612. [Google Scholar] [CrossRef]
- Young, A.R.; Narita, M.; Ferreira, M.; Kirschner, K.; Sadaie, M.; Darot, J.F.; Tavare, S.; Arakawa, S.; Shimizu, S.; Watt, F.M.; et al. Autophagy mediates the mitotic senescence transition. Genes Dev. 2009, 23, 798–803. [Google Scholar] [CrossRef]
- Campisi, J. Senescent cells, tumor suppression, and organismal aging: Good citizens, bad neighbors. Cell 2005, 120, 513–522. [Google Scholar] [CrossRef] [PubMed]
- Carafa, V.; Altucci, L. Deregulation of Cell Death in Cancer: Recent Highlights. Cancers 2020, 12, 3517. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; You, L.; Xue, J.; Lu, Y. Ionizing Radiation-Induced Cellular Senescence in Normal, Non-transformed Cells and the Involved DNA Damage Response: A Mini Review. Front. Pharmacol. 2018, 9, 522. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Liu, M.; Hong, D.; Zeng, M.; Zhang, X. The Paradoxical Role of Cellular Senescence in Cancer. Front. Cell Dev. Biol. 2021, 9, 722205. [Google Scholar] [CrossRef] [PubMed]












Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Osipov, A.; Chigasova, A.; Yashkina, E.; Ignatov, M.; Fedotov, Y.; Molodtsova, D.; Vorobyeva, N.; Osipov, A.N. Residual Foci of DNA Damage Response Proteins in Relation to Cellular Senescence and Autophagy in X-Ray Irradiated Fibroblasts. Cells 2023, 12, 1209. https://doi.org/10.3390/cells12081209
Osipov A, Chigasova A, Yashkina E, Ignatov M, Fedotov Y, Molodtsova D, Vorobyeva N, Osipov AN. Residual Foci of DNA Damage Response Proteins in Relation to Cellular Senescence and Autophagy in X-Ray Irradiated Fibroblasts. Cells. 2023; 12(8):1209. https://doi.org/10.3390/cells12081209
Chicago/Turabian StyleOsipov, Andrey, Anna Chigasova, Elizaveta Yashkina, Maxim Ignatov, Yuriy Fedotov, Daria Molodtsova, Natalia Vorobyeva, and Andreyan N. Osipov. 2023. "Residual Foci of DNA Damage Response Proteins in Relation to Cellular Senescence and Autophagy in X-Ray Irradiated Fibroblasts" Cells 12, no. 8: 1209. https://doi.org/10.3390/cells12081209
APA StyleOsipov, A., Chigasova, A., Yashkina, E., Ignatov, M., Fedotov, Y., Molodtsova, D., Vorobyeva, N., & Osipov, A. N. (2023). Residual Foci of DNA Damage Response Proteins in Relation to Cellular Senescence and Autophagy in X-Ray Irradiated Fibroblasts. Cells, 12(8), 1209. https://doi.org/10.3390/cells12081209