
Integrated Omic Analysis Delineates Pathways Modulating Toxic TDP-43 Protein Aggregates in Amyotrophic Lateral Sclerosis

 , , , ,
, , , ,  , ,
, ,
Abstract
:1. Introduction
2. Materials and Methods
3. Results
3.1. Metabolomic Analysis of TDP-43 Q331K Mutant Shows Deregulation of Pathways in the Yeast Model of TDP-43 Aggregation
3.2. Transcriptomic Analysis of the Yeast Model of TDP-43 Aggregation Expressing TDP-43 Q331K Shows Deregulated Metabolic and Signaling Pathways with Implications for Disease
3.3. Integrated Analysis of Transcriptomic and Metabolomic Datasets from the Yeast Model of TDP-43 Aggregation Show Significant Pathways with Potential Implications for Disease
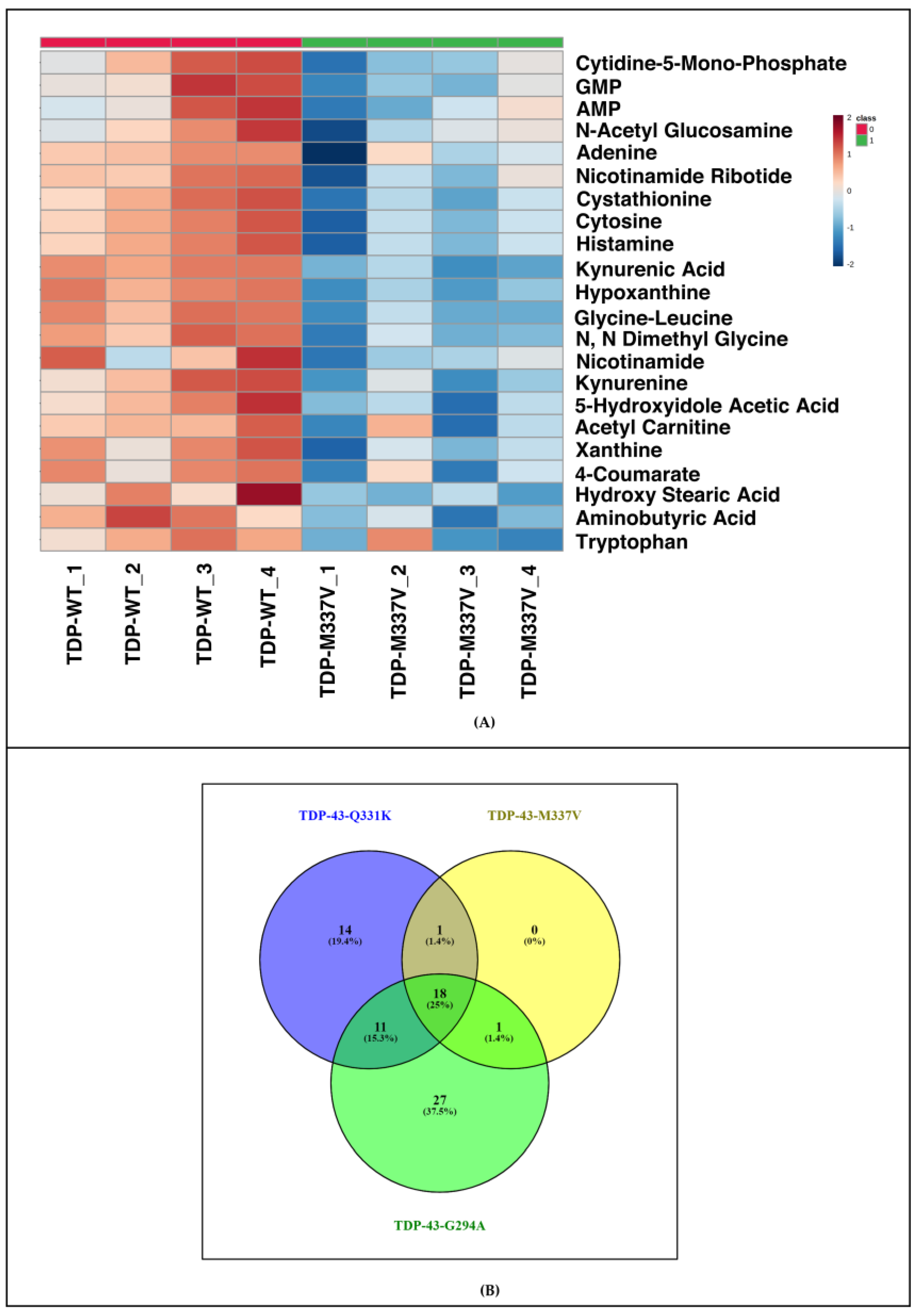
3.4. Metabolomic Analysis of Different TDP-43 Mutants Shows Deregulation of Similar Pathways in the Yeast Model of TDP-43 Aggregation
3.5. Analysis of Gene Expression Datasets from the Motor Neuron of the Mice Model of TDP-43 (A315T) and Post-Mortem Cortex of ALS Patients Shows Deregulation of Pathways with Potential Implications for Disease
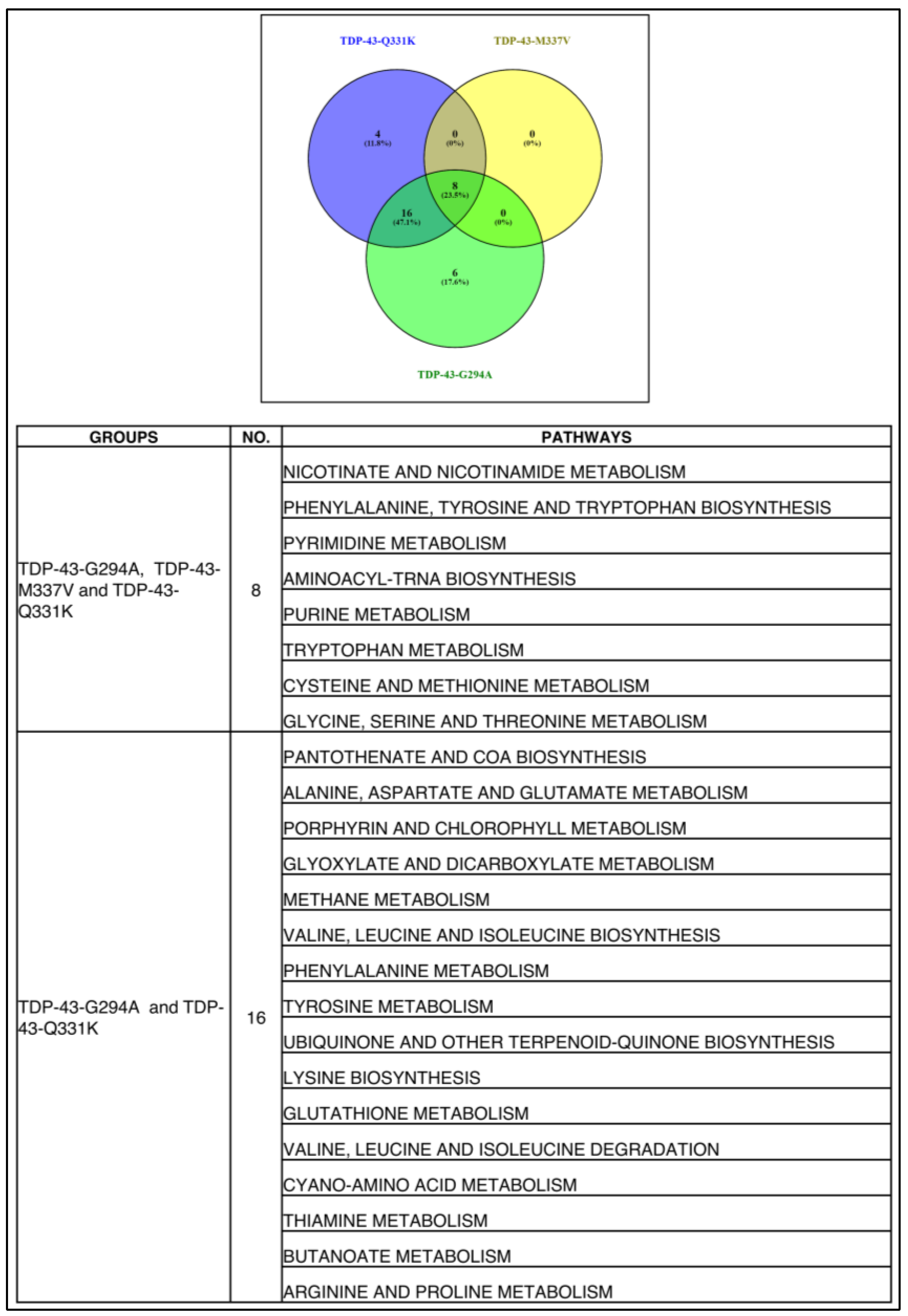
3.6. Commonality Analysis of Pathways from Yeast TDP-43 (Q331K), Mice TDP-43 (A315T) and Human ALS Shows Deregulated Pathways Conserved across Taxa, Study Setting, and Platforms
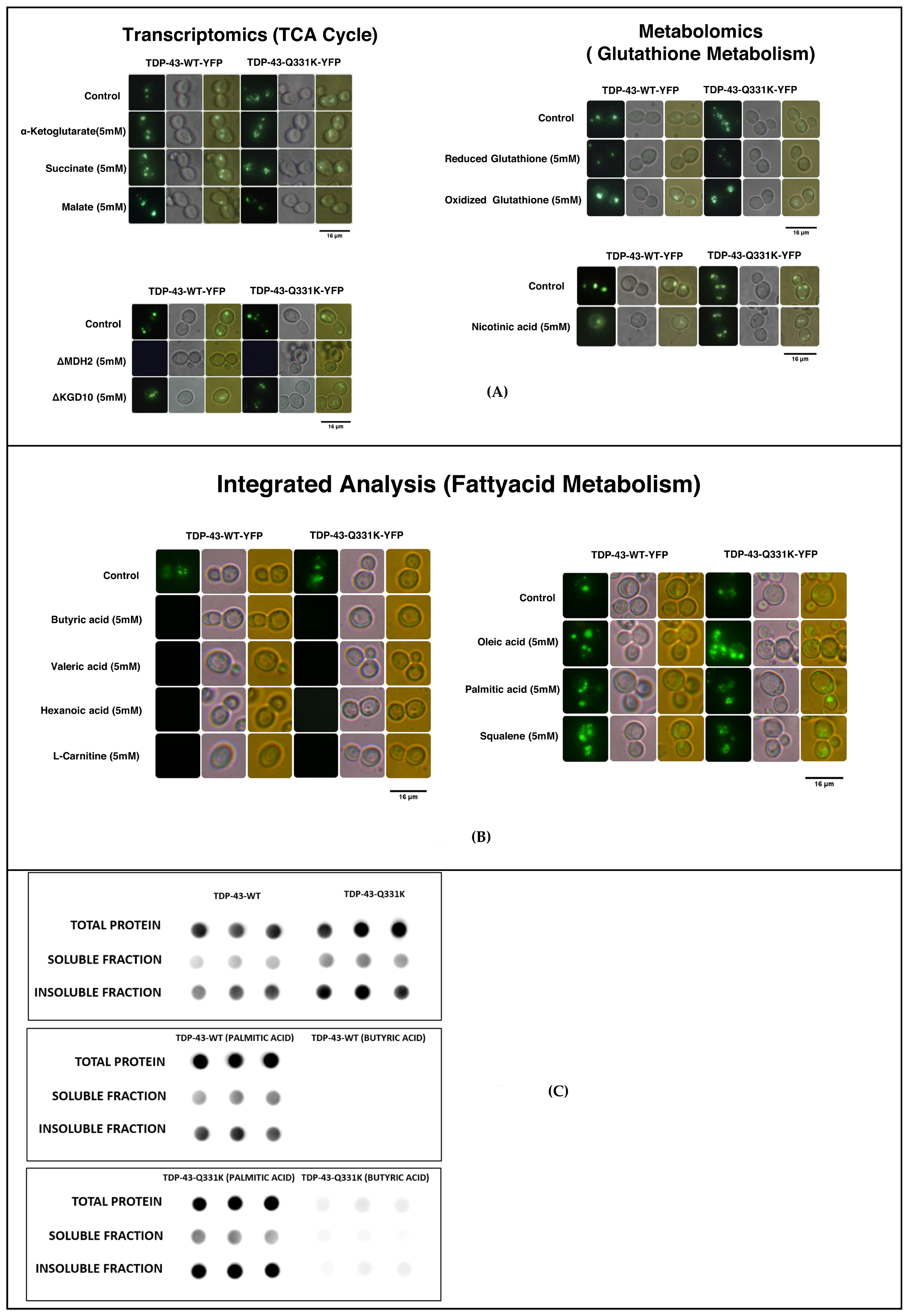
3.7. Metabolic Addition Experiments and Gene Knock-Out Experiments in the TDP-43 Yeast Model of TDP-43 Aggregation Reiterate a Role for Deregulated Pathways in the Disease Process
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Tandan, R.; Bradley, W.G. Amyotrophic lateral sclerosis: Part 1. Clinical features, pathology, and ethical issues in management. Ann. Neurol. 1985, 18, 271–280. [Google Scholar] [CrossRef] [PubMed]
- Koo, E.H.; Lansbury, P.T., Jr.; Kelly, J.W. Amyloid diseases: Abnormal protein aggregation in neurodegeneration. Proc. Natl. Acad. Sci. USA 1999, 96, 9989–9990. [Google Scholar] [CrossRef]
- Blokhuis, A.M.; Groen, E.J.N.; Koppers, M.; Berg, L.H.V.D.; Pasterkamp, R.J. Protein aggregation in amyotrophic lateral sclerosis. Acta Neuropathol. 2013, 125, 777–794. [Google Scholar] [CrossRef] [PubMed]
- Mathis, S.; Goizet, C.; Soulages, A.; Vallat, J.-M.; Le Masson, G. Genetics of amyotrophic lateral sclerosis: A review. J. Neurol. Sci. 2019, 399, 217–226. [Google Scholar] [CrossRef] [PubMed]
- McCann, E.P.; Henden, L.; Fifita, J.A.; Zhang, K.Y.; Grima, N.; Bauer, D.C.; Fat, S.C.M.; Twine, N.A.; Pamphlett, R.; Kiernan, M.C.; et al. Evidence for polygenic and oligogenic basis of Australian sporadic amyotrophic lateral sclerosis. J. Med. Genet. 2021, 58, 87–95. [Google Scholar] [CrossRef]
- Smith, E.F.; Shaw, P.J.; De Vos, K.J. The role of mitochondria in amyotrophic lateral sclerosis. Neurosci. Lett. 2019, 710, 132933. [Google Scholar] [CrossRef]
- Van Damme, P.; Robberecht, W.; Van Den Bosch, L. Modelling amyotrophic lateral sclerosis: Progress and possibilities. Dis. Model. Mech. 2017, 10, 537–549. [Google Scholar] [CrossRef]
- Suk, T.; Rousseaux, M.W.C. The role of TDP-43 mislocalization in amyotrophic lateral sclerosis. Mol. Neurodegener. 2020, 15, 1–16. [Google Scholar] [CrossRef]
- Sreedharan, J.; Blair, I.P.; Tripathi, V.B.; Hu, X.; Vance, C.; Rogelj, B.; Ackerley, S.; Durnall, J.C.; Williams, K.L.; Buratti, E.; et al. TDP-43 Mutations in Familial and Sporadic Amyotrophic Lateral Sclerosis. Science 2008, 319, 1668–1672. [Google Scholar] [CrossRef]
- Lattante, S.; Rouleau, G.A.; Kabashi, E. TARDBP and FUS mutations associated with amyotrophic lateral sclerosis: Summary and update. Hum. Mutat. 2013, 34, 812–826. [Google Scholar] [CrossRef]
- Budini, M.; Romano, V.; Quadri, Z.; Buratti, E.; Baralle, F.E. TDP-43 loss of cellular function through aggregation requires additional structural determinants beyond its C-terminal Q/N prion-like domain. Hum. Mol. Genet. 2014, 24, 9–20. [Google Scholar] [CrossRef] [PubMed]
- Johnson, B.S.; McCaffery, J.M.; Lindquist, S.; Gitler, A.D. A yeast TDP-43 proteinopathy model: Exploring the molecular determinants of TDP-43 aggregation and cellular toxicity. Proc. Natl. Acad. Sci. USA 2008, 105, 6439–6444. [Google Scholar] [CrossRef]
- Chen-Plotkin, A.S.; Lee, V.M.-Y.; Trojanowski, J.Q. TAR DNA-binding protein 43 in neurodegenerative disease. Nat. Rev. Neurol. 2010, 6, 211–220. [Google Scholar] [CrossRef]
- Neelagandan, N.; Gonnella, G.; Dang, S.; Janiesch, P.C.; Miller, K.; Küchler, K.; Marques, R.F.; Indenbirken, D.; Alawi, M.; Grundhoff, A.; et al. TDP-43 enhances translation of specific mRNAs linked to neurodegenerative disease. Nucleic Acids Res. 2018, 47, 341–361. [Google Scholar] [CrossRef]
- Veyrat-Durebex, C.; Corcia, P.; Piver, E.; Devos, D.; Dangoumau, A.; Gouel, F.; Vourc’h, P.; Emond, P.; Laumonnier, F.; Nadal-Desbarats, L.; et al. Disruption of TCA Cycle and Glutamate Metabolism Identified by Metabolomics in an In Vitro Model of Amyotrophic Lateral Sclerosis. Mol. Neurobiol. 2016, 53, 6910–6924. [Google Scholar] [CrossRef] [PubMed]
- Dogan, I.; Eickhoff, C.R.; Fox, P.T.; Laird, A.R.; Schulz, J.B.; Eickhoff, S.B.; Reetz, K. Functional connectivity modeling of consistent cortico-striatal degeneration in Huntington’s disease. NeuroImage Clin. 2015, 7, 640–652. [Google Scholar] [CrossRef]
- Wang, W.; Li, L.; Lin, W.-L.; Dickson, D.W.; Petrucelli, L.; Zhang, T.; Wang, X. The ALS disease-associated mutant TDP-43 impairs mitochondrial dynamics and function in motor neurons. Hum. Mol. Genet. 2013, 22, 4706–4719. [Google Scholar] [CrossRef]
- Bennett, S.A.; Tanaz, R.; Cobos, S.N.; Torrente, M.P. Epigenetics in amyotrophic lateral sclerosis: A role for histone post-translational modifications in neurodegenerative disease. Transl. Res. 2018, 204, 19–30. [Google Scholar] [CrossRef]
- Amlie-Wolf, A.; Ryvkin, P.; Tong, R.; Dragomir, I.; Suh, E.; Xu, Y.; Van Deerlin, V.M.; Gregory, B.D.; Kwong, L.K.; Trojanowski, J.Q.; et al. Transcriptomic Changes Due to Cytoplasmic TDP-43 Expression Reveal Dysregulation of Histone Transcripts and Nuclear Chromatin. PLoS ONE 2015, 10, e0141836. [Google Scholar] [CrossRef] [PubMed]
- Prasad, A.; Bharathi, V.; Sivalingam, V.; Girdhar, A.; Patel, B.K. Molecular Mechanisms of TDP-43 Misfolding and Pathology in Amyotrophic Lateral Sclerosis. Front. Mol. Neurosci. 2019, 12, 25. [Google Scholar] [CrossRef]
- Caballero-Hernandez, D.; Toscano, M.G.; Cejudo-Guillen, M.; Garcia-Martin, M.L.; Lopez, S.; Franco, J.M.; Quintana, F.J.; Roodveldt, C.; Pozo, D. The ‘Omics’ of Amyotrophic Lateral Sclerosis. Trends Mol. Med. 2016, 22, 53–67. [Google Scholar] [CrossRef]
- Love, R. Knockout mice help to pin down the cause of neurodegeneration. Lancet Neurol. 2003, 2, 521. [Google Scholar] [CrossRef]
- Giorgini, F.; Muchowski, P.J. Screening for Genetic Modifiers of Amyloid Toxicity in Yeast. Methods Enzymol. 2006, 412, 201–222. [Google Scholar] [CrossRef]
- Pradhan, S.S.; Thota, S.M.; Rajaratnam, S.; Bhagavatham, S.K.; Pulukool, S.K.; Rathnakumar, S.; Phalguna, K.S.; Dandamudi, R.B.; Pargaonkar, A.; Joseph, P.; et al. Integrated multi-omic analysis of Huntington disease and yeast model delineates pathways modulating protein aggregation. Dis. Model. Mech. 2022, 15, dmm049492. [Google Scholar] [CrossRef]
- Chernoff, Y.O.; Grizel, A.V.; Rubel, A.A.; Zelinsky, A.A.; Chandramowlishwaran, P.; Chernova, T.A. Application of Yeast to Studying Amyloid and Prion Diseases; Elsevier Inc.: Amsterdam, The Netherlands, 2020; Volume 105, pp. 293–380. [Google Scholar] [CrossRef]
- Swaroop, R.S.; Akhil, P.S.; Sanwid, P.S.; Bandana, P.; Raksha, R.K.; Meghana, M.; Bibha, C.; Sivaramakrishnan, V. Integrated multi-omic data analysis and validation with yeast model show oxidative phosphorylation modulates protein aggregation in amyotrophic lateral sclerosis. J. Biomol. Struct. Dyn. 2022, 41, 1–20. [Google Scholar] [CrossRef]
- Swaroop, R.S.; Pradhan, S.S.; Darshan, V.M.D.; Phalguna, K.S.; Sivaramakrishnan, V. Integrated network pharmacology approach shows a potential role of Ginseng catechins and ginsenosides in modulating protein aggregation in Amyotrophic Lateral Sclerosis. 3 Biotech 2022, 12, 333. [Google Scholar] [CrossRef]
- Narayanan, A.; Khanchandani, P.; Borkar, R.M.; Ambati, C.R.; Roy, A.; Han, X.; Bhoskar, R.N.; Ragampeta, S.; Gannon, F.; Mysorekar, V.; et al. Avascular Necrosis of Femoral Head: A Metabolomic, Biophysical, Biochemical, Electron Microscopic and Histopathological Characterization. Sci. Rep. 2017, 7, 10721. [Google Scholar] [CrossRef]
- Pulukool, S.K.; Bhagavatham, S.K.S.; Kannan, V.; Sukumar, P.; Dandamudi, R.B.; Ghaisas, S.; Kunchala, H.; Saieesh, D.; Naik, A.A.; Pargaonkar, A.; et al. Elevated dimethylarginine, ATP, cytokines, metabolic remodeling involving tryptophan metabolism and potential microglial inflammation characterize primary open angle glaucoma. Sci. Rep. 2021, 11, 9766. [Google Scholar] [CrossRef] [PubMed]
- Naik, A.A.; Narayanan, A.; Khanchandani, P.; Sridharan, D.; Sukumar, P.; Bhagavatam, S.K.S.; Seshagiri, P.B.; Sivaramakrishnan, V. Systems analysis of avascular necrosis of femoral head using integrative data analysis and literature mining delineates pathways associated with disease. Sci. Rep. 2020, 10, 18099. [Google Scholar] [CrossRef]
- Bhagavatham, S.K.S.; Khanchandani, P.; Kannan, V.; Potikuri, D.; Sridharan, D.; Pulukool, S.K.; Naik, A.A.; Dandamudi, R.B.; Divi, S.M.; Pargaonkar, A.; et al. Adenosine deaminase modulates metabolic remodeling and orchestrates joint destruction in rheumatoid arthritis. Sci. Rep. 2021, 11, 15129. [Google Scholar] [CrossRef]
- Cordeiro, R.D.A.; Aguiar, A.L.R.; Pereira, V.S.; Pereira, L.M.G.; Portela, F.V.M.; Brilhante, R.S.N.; de Camargo, Z.P.; Sidrim, J.J.C.; Castelo-Branco, D.D.S.C.M.; Rocha, M.F.G. Sodium butyrate inhibits planktonic cells and biofilms of Trichosporon spp. Microb. Pathog. 2019, 130, 219–225. [Google Scholar] [CrossRef]
- Schindelin, J.; Rueden, C.T.; Hiner, M.C.; Eliceiri, K.W. The ImageJ ecosystem: An open platform for biomedical image analysis. Mol. Reprod. Dev. 2015, 82, 518–529. [Google Scholar] [CrossRef]
- Pang, Z.; Chong, J.; Zhou, G.; de Lima Morais, D.A.; Chang, L.; Barrette, M.; Gauthier, C.; Jacques, P.-É.; Li, S.; Xia, J. MetaboAnalyst 5.0: Narrowing the gap between raw spectra and functional insights. Nucleic Acids Res. 2021, 49, W388–W396. [Google Scholar] [CrossRef]
- Wingett, S.W.; Andrews, S. FastQ Screen: A tool for multi-genome mapping and quality control. F1000Research 2018, 7, 1338. [Google Scholar] [CrossRef]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef]
- Ramirez-Gonzalez, R.H.; Bonnal, R.; Caccamo, M.; MacLean, D. Bio-samtools: Ruby bindings for SAMtools, a library for accessing BAM files containing high-throughput sequence alignments. Source Code Biol. Med. 2012, 7, 1–6. [Google Scholar] [CrossRef]
- Love, M.; Anders, S.; Huber, W. Differential analysis of count data—The DESeq2 package. Genome Biol. 2016, 15, 10–1186. [Google Scholar]
- Kuleshov, M.V.; Jones, M.R.; Rouillard, A.D.; Fernandez, N.F.; Duan, Q.; Wang, Z.; Koplev, S.; Jenkins, S.L.; Jagodnik, K.M.; Lachmann, A.; et al. Enrichr: A comprehensive gene set enrichment analysis web server 2016 update. Nucleic Acids Res. 2016, 44, W90–W97. [Google Scholar] [CrossRef]
- Chen, E.Y.; Tan, C.M.; Kou, Y.; Duan, Q.; Wang, Z.; Meirelles, G.V.; Clark, N.R.; Ma’Ayan, A. Enrichr: Interactive and collaborative HTML5 gene list enrichment analysis tool. BMC Bioinform. 2013, 14, 128. [Google Scholar] [CrossRef]
- Xie, Z.; Bailey, A.; Kuleshov, M.V.; Clarke, D.J.B.; Evangelista, J.E.; Jenkins, S.L.; Lachmann, A.; Wojciechowicz, M.L.; Kropiwnicki, E.; Jagodnik, K.M.; et al. Gene Set Knowledge Discovery with Enrichr. Curr. Protoc. 2021, 1, e90. [Google Scholar] [CrossRef]
- Zhou, G.; Soufan, O.; Ewald, J.; Hancock, R.E.W.; Basu, N.; Xia, J. NetworkAnalyst 3.0: A visual analytics platform for comprehensive gene expression profiling and meta-analysis. Nucleic Acids Res. 2019, 47, W234–W241. [Google Scholar] [CrossRef]
- Chong, J.; Soufan, O.; Li, C.; Caraus, I.; Li, S.; Bourque, G.; Wishart, D.S.; Xia, J. MetaboAnalyst 4.0: Towards more transparent and integrative metabolomics analysis. Nucleic Acids Res. 2018, 46, W486–W494. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M. Toward understanding the origin and evolution of cellular organisms. Protein Sci. 2019, 28, 1947–1951. [Google Scholar] [CrossRef] [PubMed]
- Ogata, H.; Goto, S.; Sato, K.; Fujibuchi, W.; Bono, H.; Kanehisa, M. KEGG: Kyoto Encyclopedia of Genes and Genomes. Nucleic Acids Res. 2000, 28, 29–34. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M.; Furumichi, M.; Sato, Y.; Ishiguro-Watanabe, M.; Tanabe, M. KEGG: Integrating viruses and cellular organisms. Nucleic Acids Res. 2021, 49, D545–D551. [Google Scholar] [CrossRef]
- Tam, O.H. Postmortem Cortex Samples Identify Distinct Molecular Subtypes of ALS: Retrotransposon Activation, Oxidative Stress, and Activated Glia. HHS Public Access 2019, 29, 1164–1177. [Google Scholar] [CrossRef]
- Wuolikainen, A.; Moritz, T.; Marklund, S.L.; Antti, H.; Andersen, P.M. Disease-Related Changes in the Cerebrospinal Fluid Metabolome in Amyotrophic Lateral Sclerosis Detected by GC/TOFMS. PLoS ONE 2011, 6, e17947. [Google Scholar] [CrossRef]
- Gautam, M.; Gunay, A.; Chandel, N.S.; Ozdinler, P.H. Mitochondrial dysregulation occurs early in ALS motor cortex with TDP-43 pathology and suggests maintaining NAD+ balance as a therapeutic strategy. Sci. Rep. 2022, 12, 4287. [Google Scholar] [CrossRef]
- Alsabeeh, N.; Chausse, B.; Kakimoto, P.A.; Kowaltowski, A.J.; Shirihai, O.; Angeles, L. Cell culture models of fatty acid overload: Problems and solutions. HHS Public Access 2019, 1863, 143–151. [Google Scholar] [CrossRef]
- De Chaumont, F.; Dallongeville, S.; Chenouard, N.; Hervé, N.; Pop, S.; Provoost, T.; Meas-Yedid, V.; Pankajakshan, P.; LeComte, T.; Le Montagner, Y.; et al. Icy: An open bioimage informatics platform for extended reproducible research. Nat. Methods 2012, 9, 690–696. [Google Scholar] [CrossRef]
- Wirths, O. Extraction of Soluble and Insoluble Protein Fractions from Mouse Brains and Spinal Cords. Bio-Protocol 2017, 7, e2422. [Google Scholar] [CrossRef] [PubMed]
- Cohen, A.; Ross, L.; Nachman, I.; Bar-Nun, S. Aggregation of PolyQ Proteins Is Increased upon Yeast Aging and Affected by Sir2 and Hsf1: Novel Quantitative Biochemical and Microscopic Assays. PLoS ONE 2012, 7, e44785. [Google Scholar] [CrossRef] [PubMed]
- Bogie, J.F.; Haidar, M.; Kooij, G.; Hendriks, J.J. Fatty acid metabolism in the progression and resolution of CNS disorders. Adv. Drug Deliv. Rev. 2020, 159, 198–213. [Google Scholar] [CrossRef] [PubMed]
- Lissouba, A.; Liao, M.; Kabashi, E.; Drapeau, P. Transcriptomic Analysis of Zebrafish TDP-43 Transgenic Lines. Front. Mol. Neurosci. 2018, 11, 463. [Google Scholar] [CrossRef]
- Yang, C.; Wang, H.; Qiao, T.; Yang, B.; Aliaga, L.; Qiu, L.; Tan, W.; Salameh, J.; McKenna-Yasek, D.M.; Smith, T.; et al. Partial loss of TDP-43 function causes phenotypes of amyotrophic lateral sclerosis. Proc. Natl. Acad. Sci. USA 2014, 111, E1121–E1129. [Google Scholar] [CrossRef]
- Wang, P.; Deng, J.; Dong, J.; Liu, J.; Bigio, E.H.; Mesulam, M.; Wang, T.; Sun, L.; Wang, L.; Lee, A.Y.-L.; et al. TDP-43 induces mitochondrial damage and activates the mitochondrial unfolded protein response. PLoS Genet. 2019, 15, e1007947. [Google Scholar] [CrossRef]
- Perera, N.D.; Turner, B.J. AMPK Signalling and Defective Energy Metabolism in Amyotrophic Lateral Sclerosis. Neurochem. Res. 2016, 41, 544–553. [Google Scholar] [CrossRef]
- Guo, C.; Sun, L.; Chen, X.; Zhang, D. Oxidative stress, mitochondrial damage and neurodegenerative diseases. Neural Regen. Res. 2021, 8, 2003–2014. [Google Scholar] [CrossRef]
- Bharathi, V.; Bajpai, V.B.A.; Parappuram, I.T.; Patel, B.K. Elevated constitutive expression of Hsp40 chaperone Sis1 reduces TDP-43 aggregation-induced oxidative stress in Ire1 pathway dependent-manner in yeast TDP-43 proteinopathy model of amyotrophic lateral sclerosis. Biochem. Biophys. Res. Commun. 2022, 595, 28–34. [Google Scholar] [CrossRef]
- Gupta, A.; Puri, A.; Singh, P.; Sonam, S.; Pandey, R.; Sharma, D. The yeast stress inducible Ssa Hsp70 reduces α-synuclein toxicity by promoting its degradation through autophagy. PLoS Genet. 2018, 14, e1007751. [Google Scholar] [CrossRef]
- Caplliure-Llopis, J.; Peralta-Chamba, T.; Carrera-Juliá, S.; Cuerda-Ballester, M.; Drehmer-Rieger, E.; López-Rodriguez, M.M.; de la Rubia Ortí, J.E. Therapeutic alternative of the ketogenic Mediterranean diet to improve mitochondrial activity in Amyotrophic Lateral Sclerosis (ALS): A Comprehensive Review. Food Sci. Nutr. 2020, 8, 23–35. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, R.W.; On, N.H.; Del Bigio, M.R.; Miller, D.W.; Hatch, G.M. Fatty acid transport protein expression in human brain and potential role in fatty acid transport across human brain microvessel endothelial cells. J. Neurochem. 2011, 117, 735–746. [Google Scholar] [CrossRef]
- Tefera, T.W.; Borges, K. Metabolic Dysfunctions in Amyotrophic Lateral Sclerosis Pathogenesis and Potential Metabolic Treatments. Front. Neurosci. 2017, 10, 611. [Google Scholar] [CrossRef]
- Rosenfeld, J.; Ellis, A. Nutrition and Dietary Supplements in Motor Neuron Disease. Phys. Med. Rehabil. Clin. N. Am. 2008, 19, 573–589. [Google Scholar] [CrossRef]
- Díaz-García, C.M.; Mongeon, R.; Lahmann, C.; Koveal, D.; Zucker, H.; Yellen, G. Neuronal Stimulation Triggers Neuronal Glycolysis and Not Lactate Uptake. Cell Metab. 2017, 26, 361–374.e4. [Google Scholar] [CrossRef]
- Liu, D.; Pitta, M.; Mattson, M.P. Preventing NAD+ depletion protects neurons against excitotoxicity: Bioenergetic effects of mild mitochondrial uncoupling and caloric restriction. Ann. N. Y. Acad. Sci. 2008, 1147, 275. [Google Scholar] [CrossRef] [PubMed]
- Kim, E.J.; Yang, S.J. Nicotinamide Reduces Amyloid Precursor Protein and Presenilin 1 in Brain Tissues of Amyloid Beta-Tail Vein Injected Mice. Clin. Nutr. Res. 2017, 6, 130–135. [Google Scholar] [CrossRef]
- Pfister, J.A.; Ma, C.; Morrison, B.E.; D'Mello, S.R. Opposing Effects of Sirtuins on Neuronal Survival: SIRT1-Mediated Neuroprotection Is Independent of Its Deacetylase Activity. PLoS ONE 2008, 3, e4090. [Google Scholar] [CrossRef]
- Bharathi, V.; Girdhar, A.; Patel, B.K. Role of CNC1 gene in TDP-43 aggregation-induced oxidative stress-mediated cell death in S. cerevisiae model of ALS. Biochim. Et Biophys. Acta (BBA)-Mol. Cell Res. 2021, 1868, 118993. [Google Scholar] [CrossRef] [PubMed]
- Vogel, R.; Wiesinger, H.; Hamprecht, B.; Dringen, R. The regeneration of reduced glutathione in rat forebrain mitochondria identifies metabolic pathways providing the NADPH required. Neurosci. Lett. 1999, 275, 97–100. [Google Scholar] [CrossRef]
- Mali, Y.; Zisapel, N. A Novel Decoy That Interrupts G93A-Superoxide Dismutase Gain of Interaction with Malate Dehydrogenase Improves Survival in an Amyotrophic Lateral Sclerosis Cell Model. J. Med. Chem. 2009, 52, 5442–5448. [Google Scholar] [CrossRef] [PubMed]
- Balsa, E.; Perry, E.A.; Bennett, C.F.; Jedrychowski, M.; Gygi, S.P.; Doench, J.G.; Puigserver, P. Defective NADPH production in mitochondrial disease complex I causes inflammation and cell death. Nat. Commun. 2020, 11, 65. [Google Scholar] [CrossRef]
- Holten, D.; Procsal, D.; Chang, H.L. Regulation of pentose phosphate pathway dehydrogenases by NADP+/NADPH ratios. Biochem. Biophys. Res. Commun. 1976, 68, 436–441. [Google Scholar] [CrossRef] [PubMed]
- Harlan, B.A.; Killoy, K.M.; Pehar, M.; Liu, L.; Auwerx, J.; Vargas, M.R. Evaluation of the NAD+ biosynthetic pathway in ALS patients and effect of modulating NAD+ levels in hSOD1-linked ALS mouse models. Exp. Neurol. 2020, 327, 113219. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Plasmid ID | Strain Features |
|---|---|
| 27447 | pRS416Gal TDP43 WT E-YFP |
| 27450 | pRS416 Gal Q331K E-YFP |
| 27449 | pRS416Gal-M337V-E-YFP |
| 27448 | pRS416-Gal G294A-E-YFP |
| Gene Names | Primer Sequences |
|---|---|
| ALG9 | Forward primer: CTTCTGCCGTTGCCATGTTG Reverse primer: GACCCAGTGGACAGATAGCG |
| CIT3 | Forward primer: TTTTGGGTGTTCAAGGGCCA Reverse primer:GCTTCCAGACCCTCCAAGTT |
| MIH1 | Forward primer: TGCAACGGCAAGATGGGAAA Reverse primer: CTGGATGACGCAGACGTGAA |
| FAA2 | Forward primer: CCGGTTACACCAAAGGCTCT Reverse primer: ATGGCAACCGCCTGTTTCTT |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rajaratnam, S.; Soman, A.P.; Phalguna, K.S.; Pradhan, S.S.; Manjunath, M.; Rao, R.K.; Dandamudi, R.B.; Bhagavatham, S.K.S.; Pulukool, S.K.; Rathnakumar, S.; et al. Integrated Omic Analysis Delineates Pathways Modulating Toxic TDP-43 Protein Aggregates in Amyotrophic Lateral Sclerosis. Cells 2023, 12, 1228. https://doi.org/10.3390/cells12091228
Rajaratnam S, Soman AP, Phalguna KS, Pradhan SS, Manjunath M, Rao RK, Dandamudi RB, Bhagavatham SKS, Pulukool SK, Rathnakumar S, et al. Integrated Omic Analysis Delineates Pathways Modulating Toxic TDP-43 Protein Aggregates in Amyotrophic Lateral Sclerosis. Cells. 2023; 12(9):1228. https://doi.org/10.3390/cells12091228
Chicago/Turabian StyleRajaratnam, Saiswaroop, Akhil P. Soman, Kanikaram Sai Phalguna, Sai Sanwid Pradhan, Meghana Manjunath, Raksha Kanthavara Rao, Rajesh Babu Dandamudi, Sai Krishna Srimadh Bhagavatham, Sujith Kumar Pulukool, Sriram Rathnakumar, and et al. 2023. "Integrated Omic Analysis Delineates Pathways Modulating Toxic TDP-43 Protein Aggregates in Amyotrophic Lateral Sclerosis" Cells 12, no. 9: 1228. https://doi.org/10.3390/cells12091228
APA StyleRajaratnam, S., Soman, A. P., Phalguna, K. S., Pradhan, S. S., Manjunath, M., Rao, R. K., Dandamudi, R. B., Bhagavatham, S. K. S., Pulukool, S. K., Rathnakumar, S., Kocherlakota, S., Pargaonkar, A., Veeranna, R. P., Arumugam, N., Almansour, A. I., Choudhary, B., & Sivaramakrishnan, V. (2023). Integrated Omic Analysis Delineates Pathways Modulating Toxic TDP-43 Protein Aggregates in Amyotrophic Lateral Sclerosis. Cells, 12(9), 1228. https://doi.org/10.3390/cells12091228