
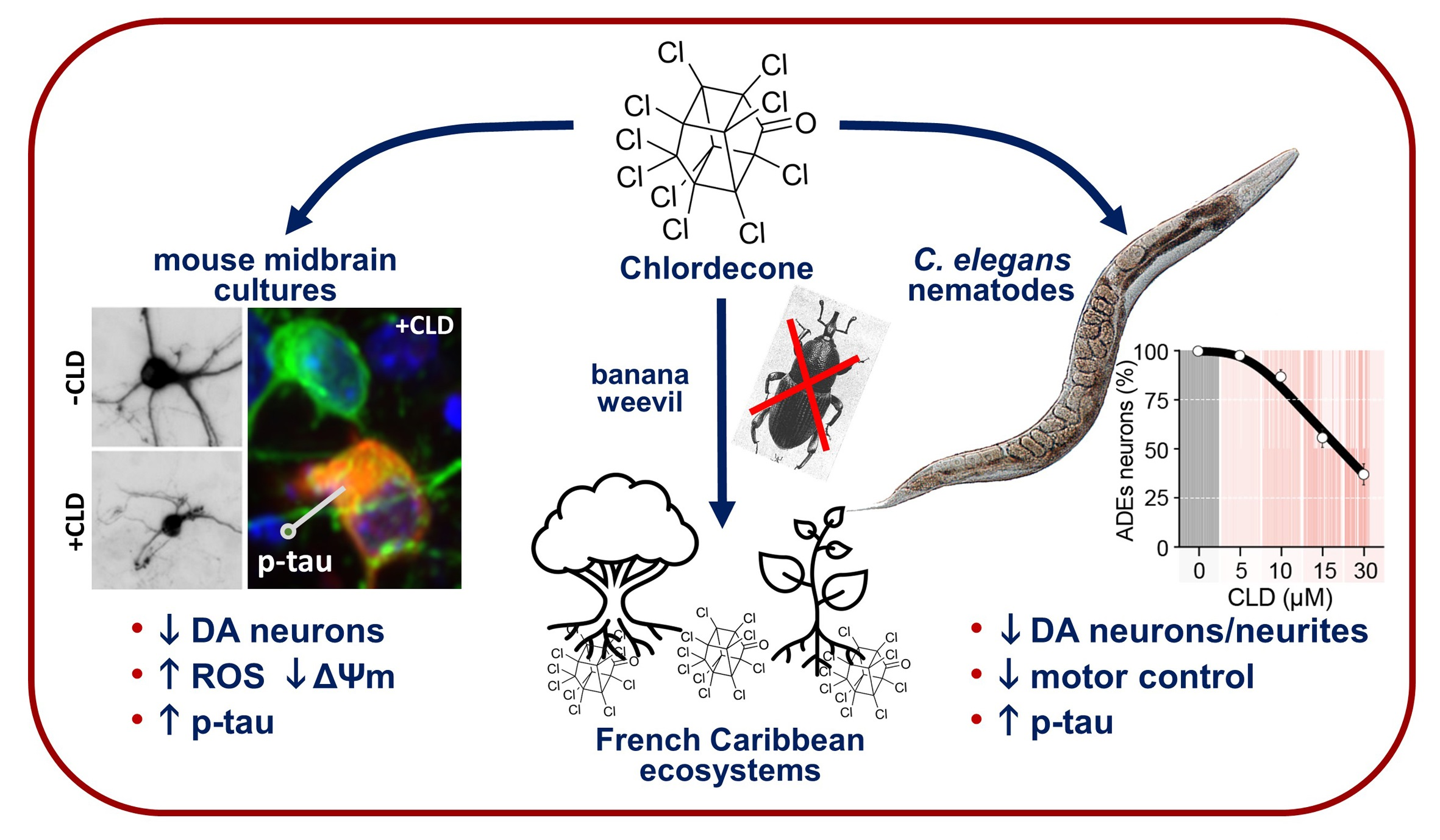
The Pesticide Chlordecone Promotes Parkinsonism-like Neurodegeneration with Tau Lesions in Midbrain Cultures and C. elegans Worms

 ,
,  and
and
Abstract
:
1. Introduction
2. Material and Methods
2.1. Chlordecone Treatments
2.2. In Vitro Studies
2.2.1. Pharmacological Reagents for Cell Culture Experiments
2.2.2. Animal Experimental Procedures
2.2.3. Neuronal and Glial Cell Cultures
2.2.4. TNF-α Measurement
2.2.5. Immunodetection Protocols for Cell Culture
2.2.6. Cell Counting in Cell Cultures
2.2.7. Tritiated-DA Uptake
2.2.8. Measurement of Reactive Oxygen Species (ROS) and Mitochondrial Membrane Potential (ΔΨm)
2.3. In Vivo Studies
2.3.1. C. elegans Lines
2.3.2. Growth and Maintenance of C. elegans Strains
2.3.3. C. elegans Liquid Culture and Treatments
2.3.4. Quantification of the Survival of C. elegans Neuronal Cell Populations
2.3.5. Immunostaining of C. elegans Samples
2.3.6. Quantification of Immunosignal Intensity
2.3.7. Behavioral Analysis
2.4. Statistical Analysis
3. Results
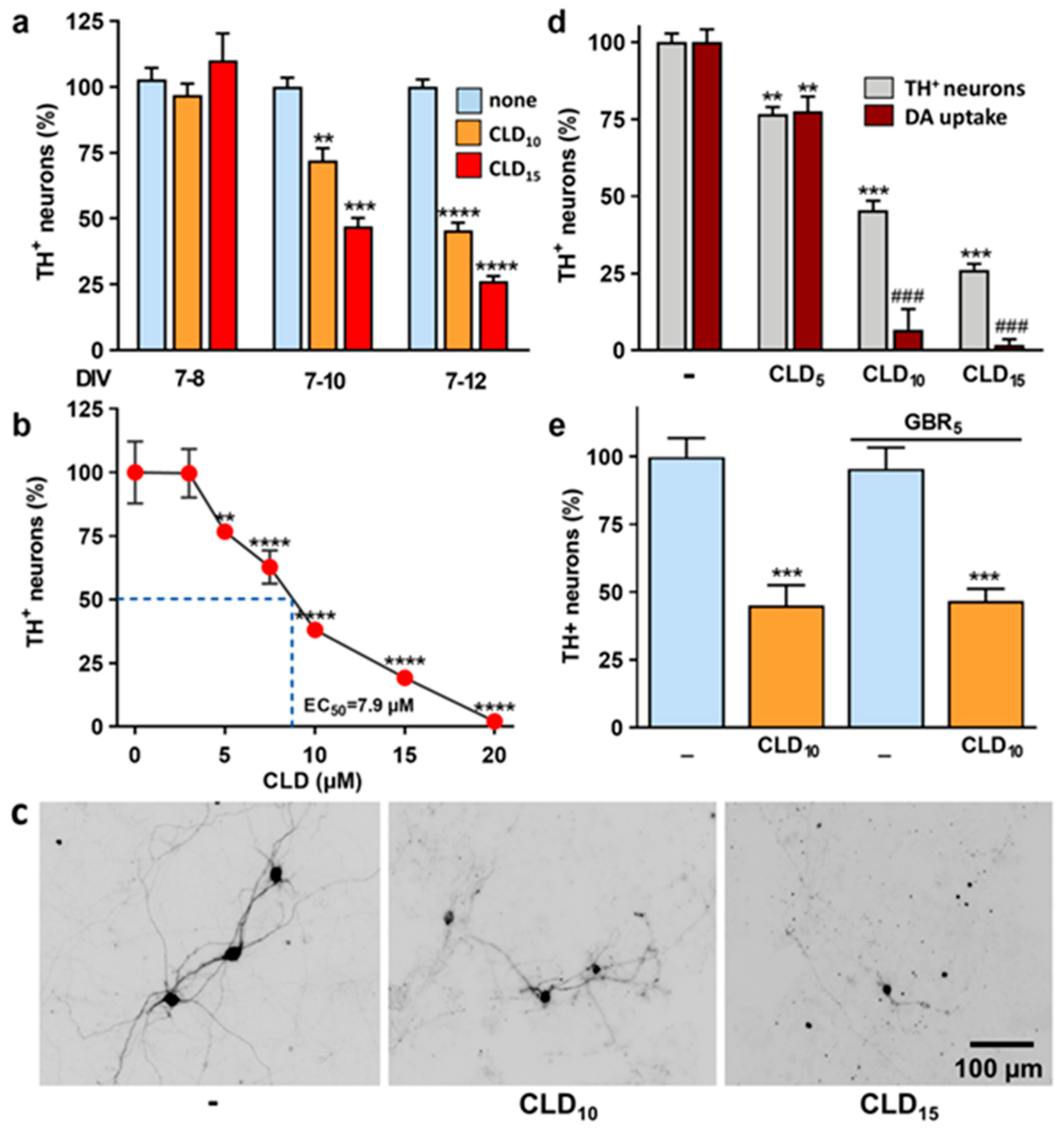
3.1. CLD Promotes Concentration- and Time-Dependent Dopaminergic Cell Loss in Mouse Midbrain Cultures
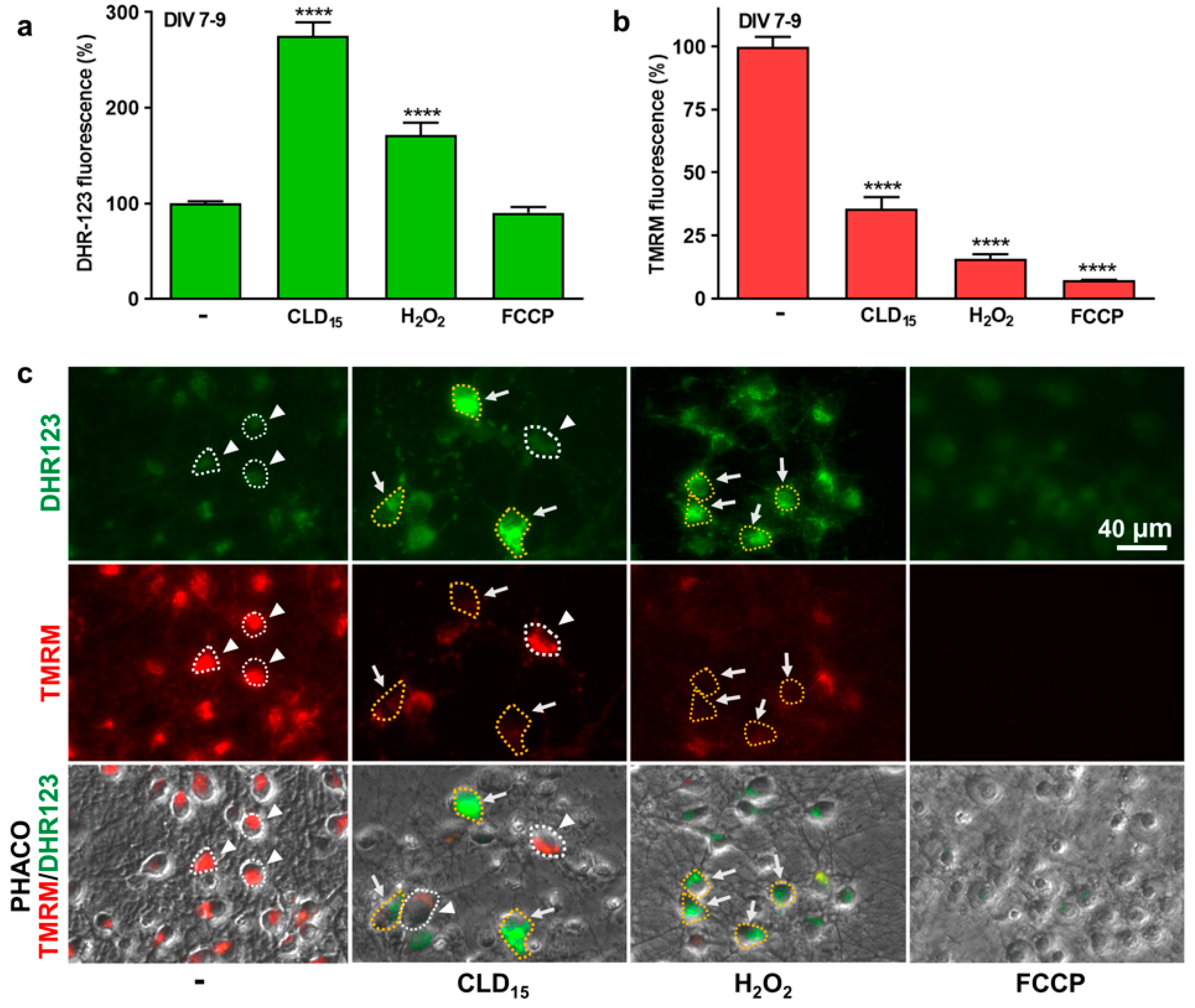
3.2. Neurodegeneration Induced by CLD Is Preceded by an Emission of ROS and a Drop in ΔΨm
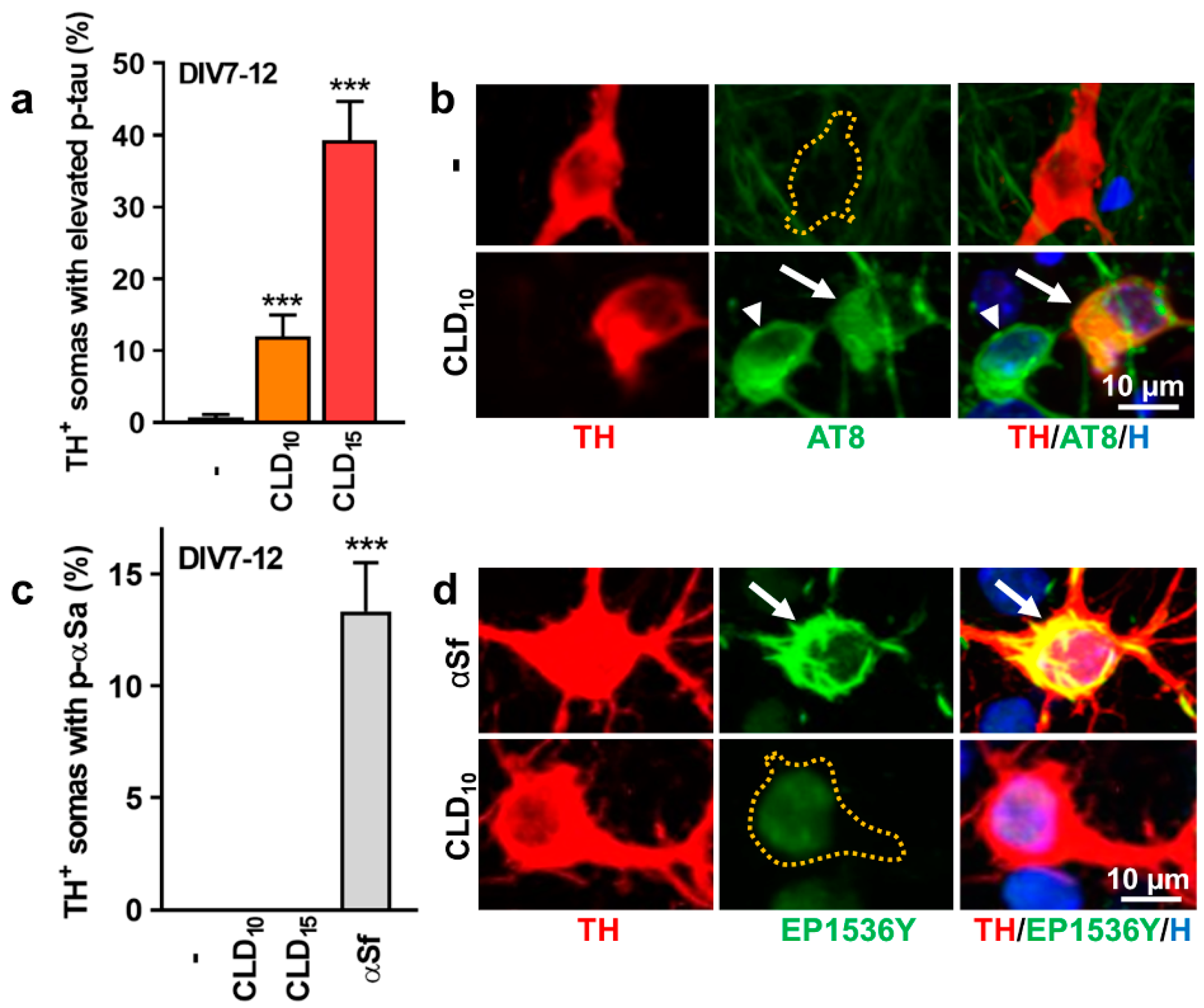
3.3. CLD Stimulates p-tau but Not p-αS Expression in DA Neurons
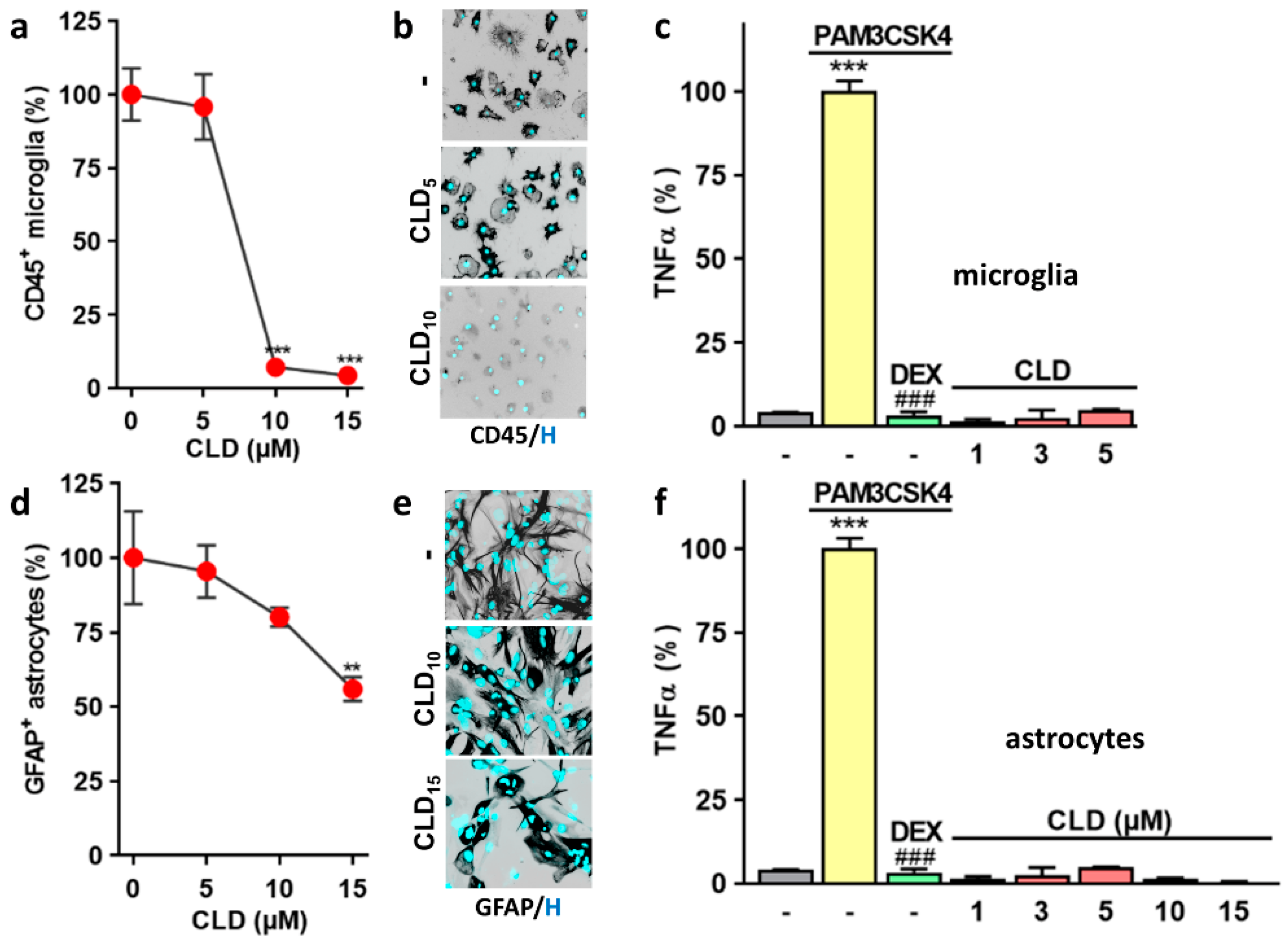
3.4. CLD Does Not Evoke an Inflammatory Response in Glial Cells
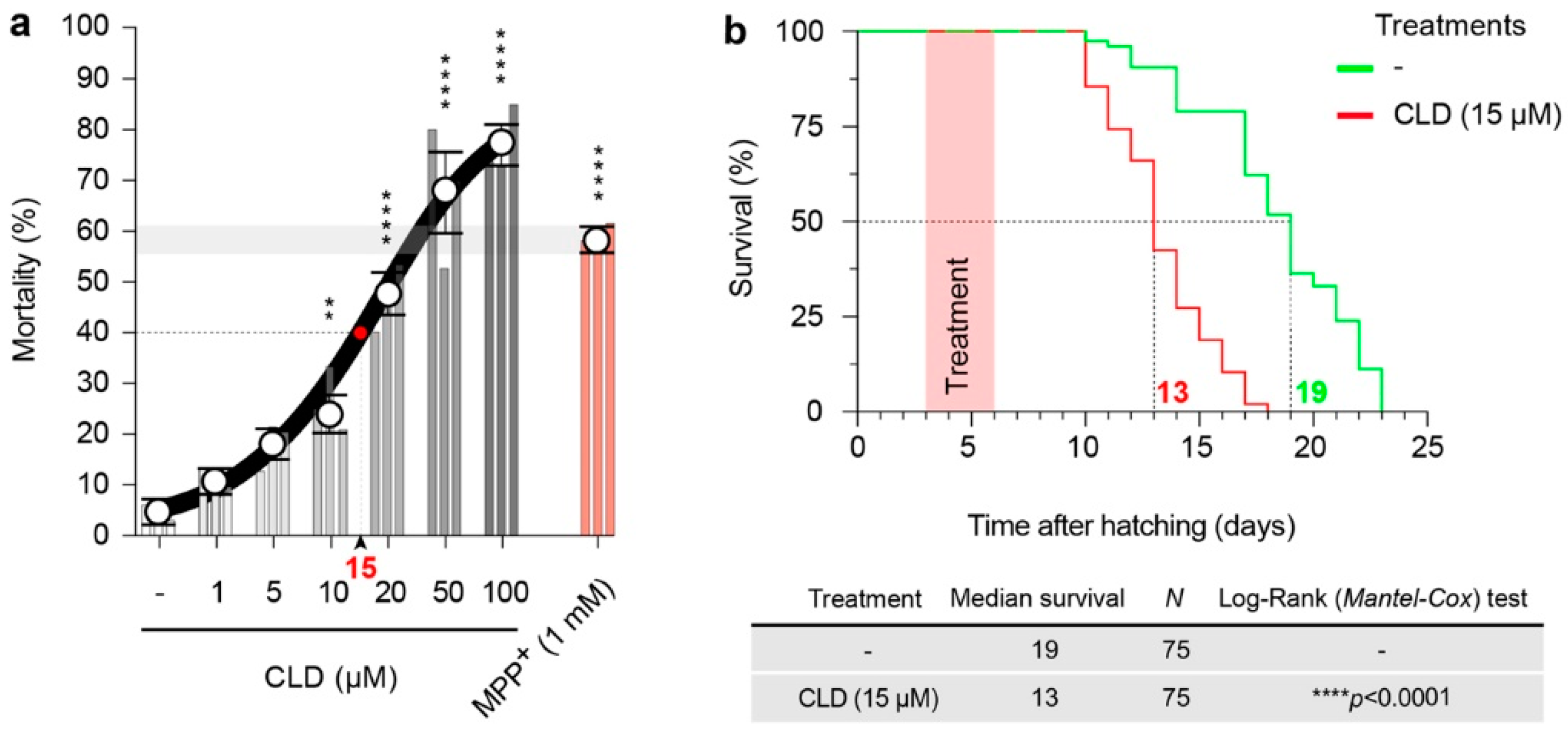
3.5. CLD Is Toxic and Alters the Lifespan of Live Nematodes
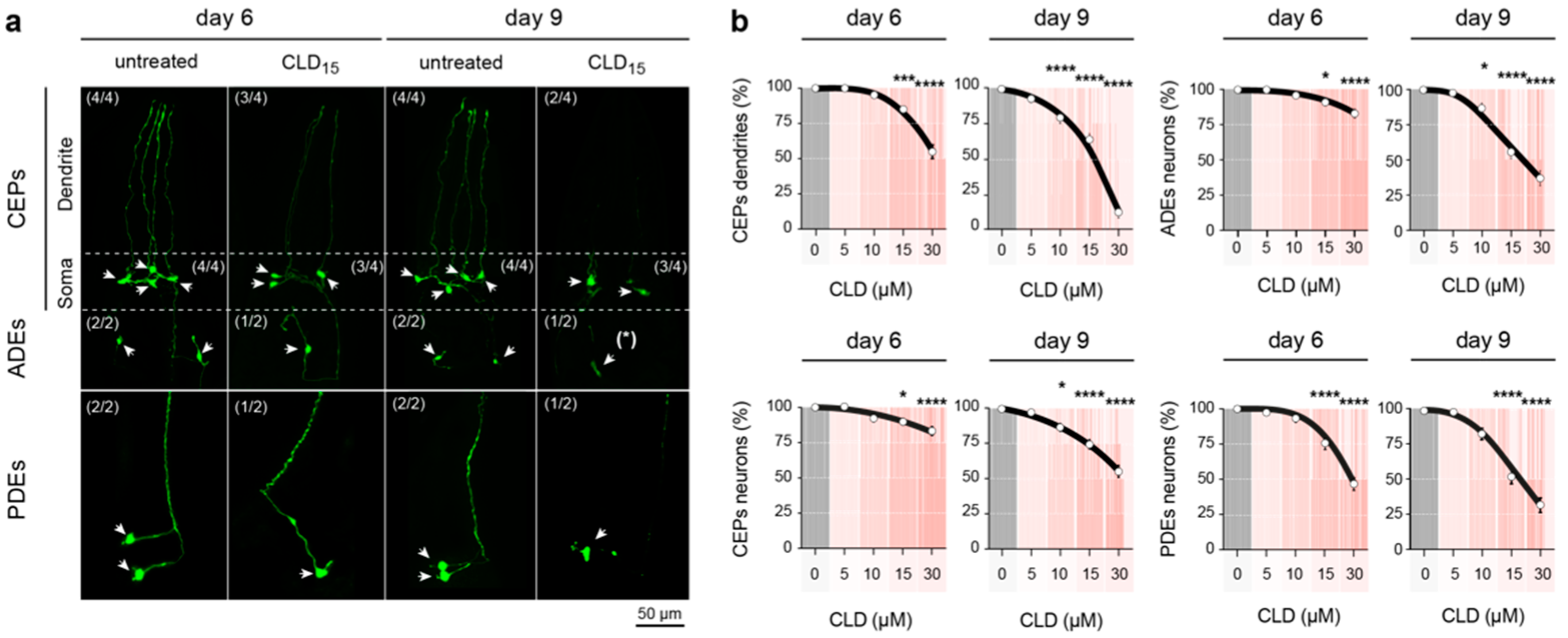
3.6. Chronic Exposure to CLD Promotes DA Cell Loss in C. elegans Worms
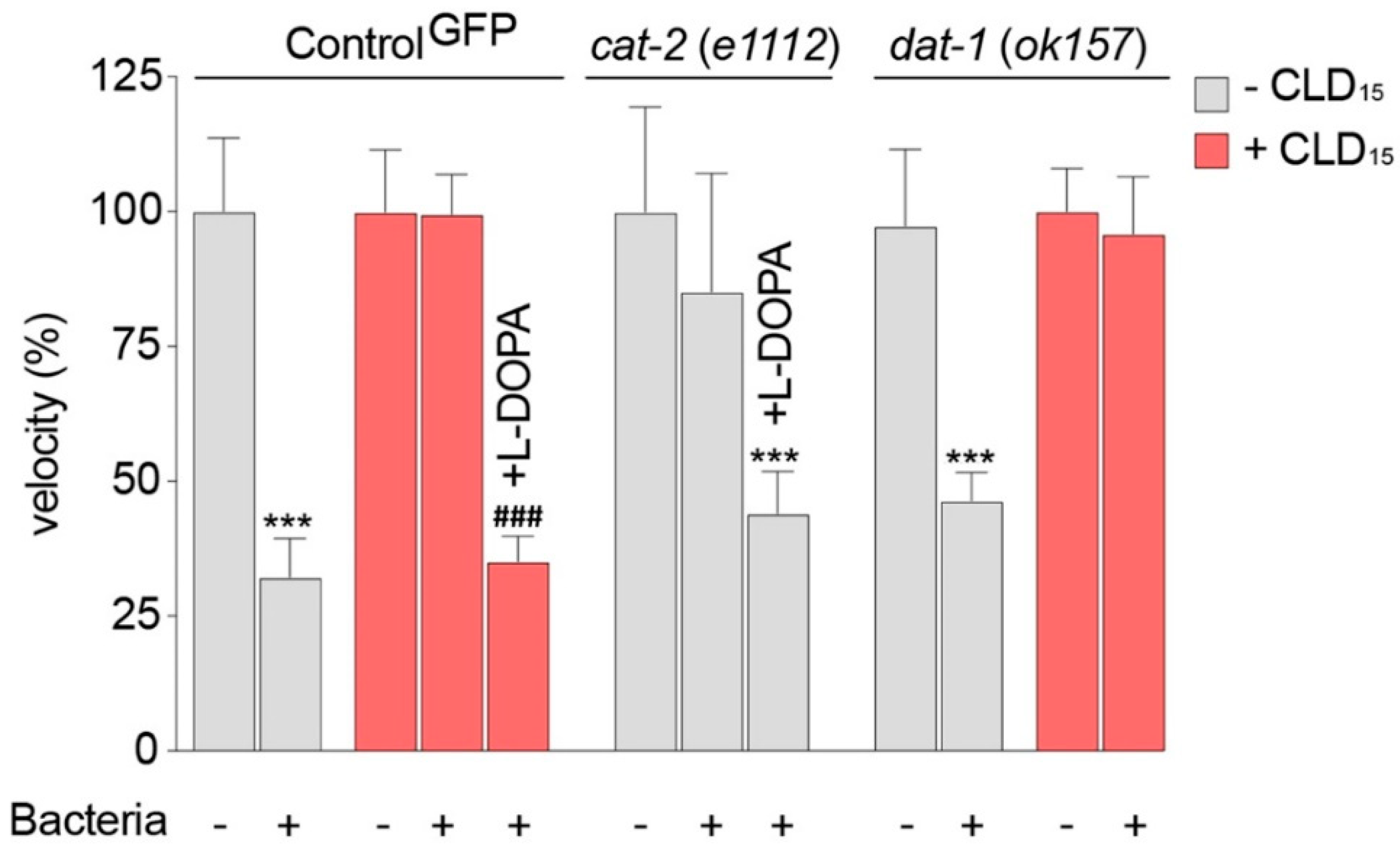
3.7. C. elegans Worms Exposed to CLD Exhibit Locomotor Behavior Deficits Due to Alterations in Food Perception
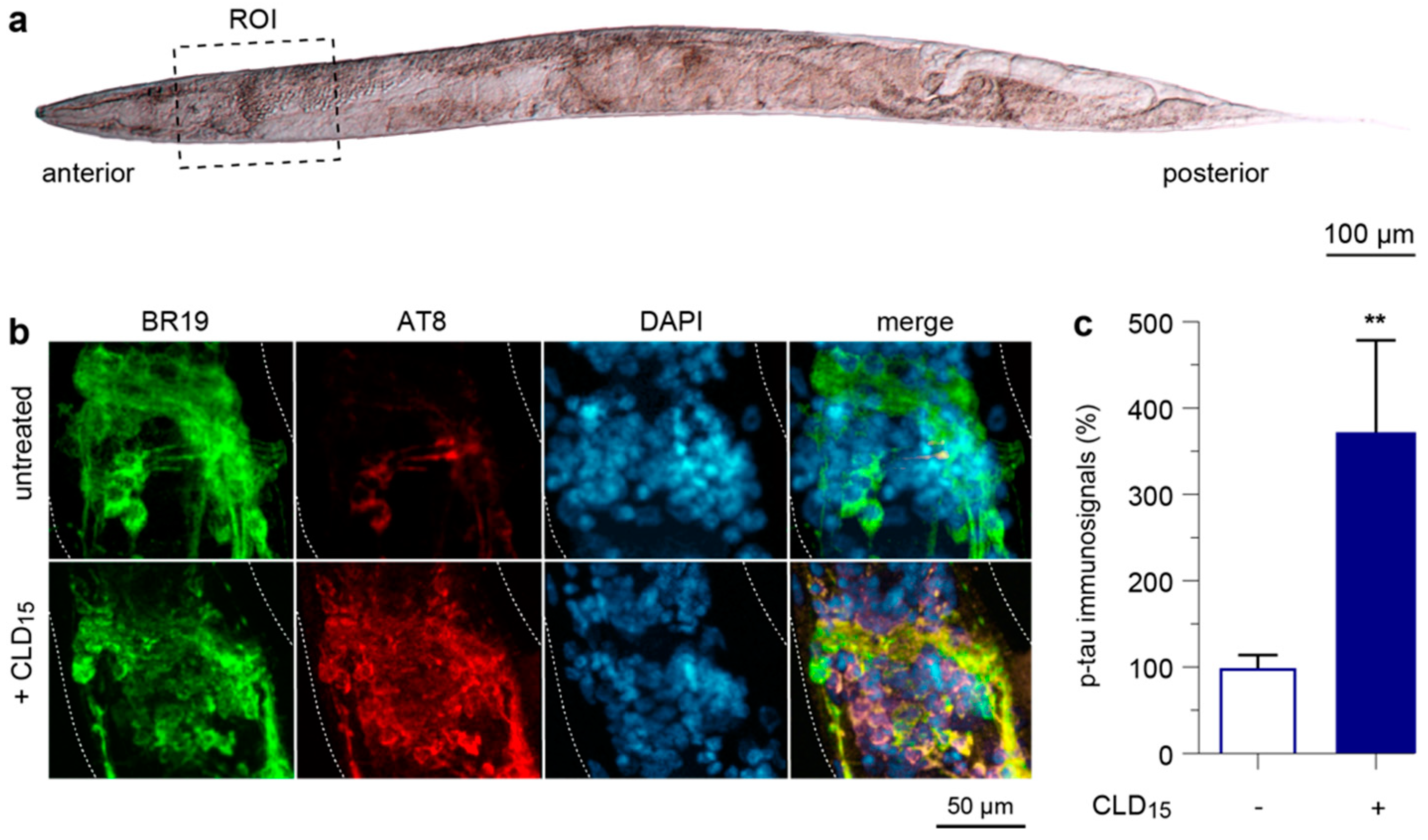
3.8. CLD Exposure Promotes Abnormal Tau Phosphorylation in a Transgenic C. elegans Line Expressing a Human 1N4R Tau Isoform
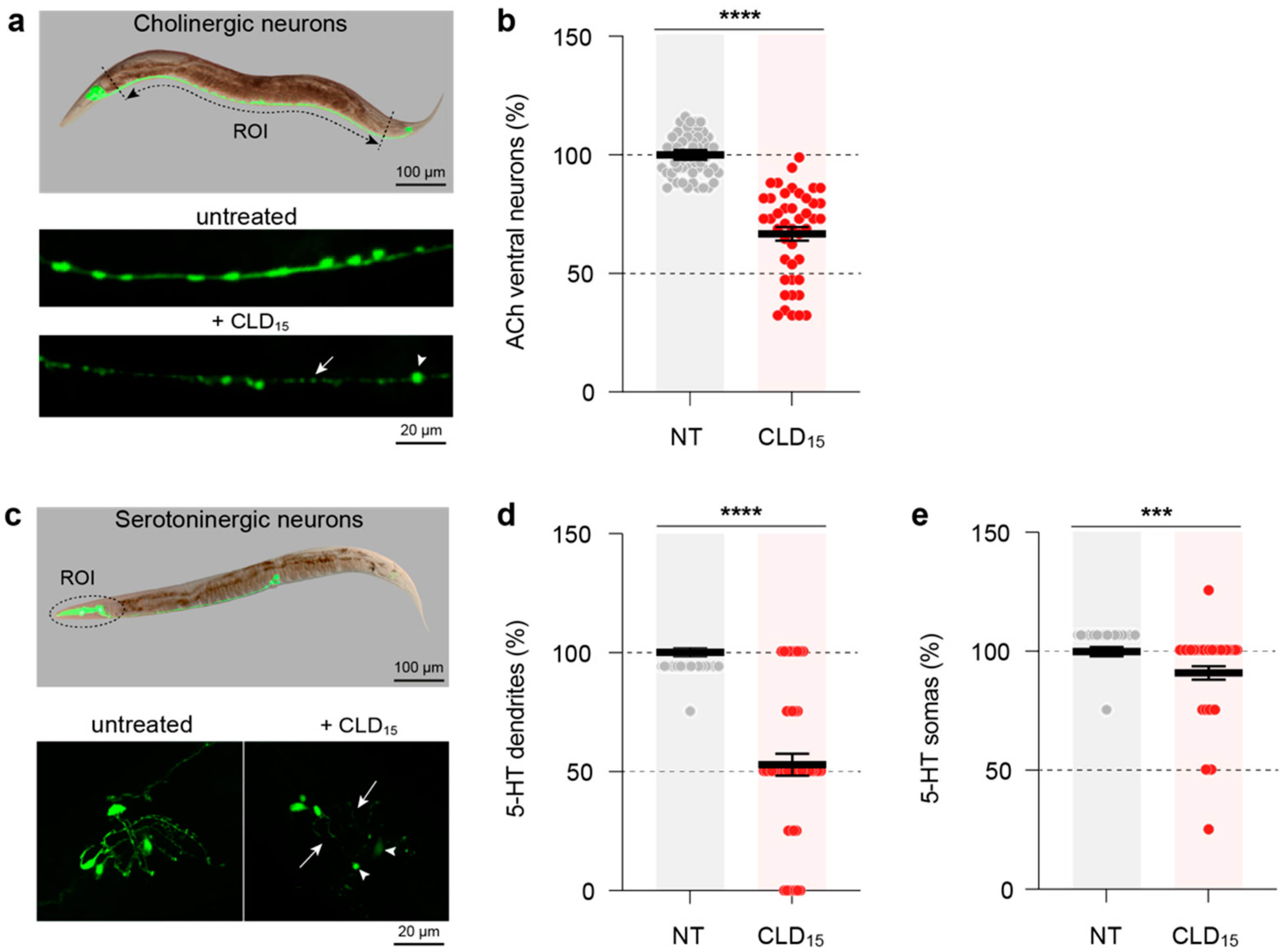
3.9. CLD Exposure Also Affects Cholinergic and Serotoninergic Neurotransmitter Systems in C. elegans Worms
4. Discussion
4.1. Neurotoxic Effects of CLD on Midbrain-Cultured DA Neurons
4.2. DA Neurons from the Nematode C. elegans Are Also Highly Vulnerable to CLD
4.3. The Neurotoxicity of CLD Is Not Restricted to DA Neurons
4.4. Impact of CLD on Brain Glial Cells
4.5. p-tau but Not p-αS Accumulates in Vulnerable DA Neurons
4.6. CLD Intoxication Results in Mitochondrial Depolarization and ROS Production
4.7. Potential Modulators of CLD-Mediated DA Cell Death
4.8. Circumstances under Which CLD Could Reach Neurotoxic Levels in the Human Brain
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Multigner, L.; Kadhel, P.; Rouget, F.; Blanchet, P.; Cordier, S. Chlordecone Exposure and Adverse Effects in French West Indies Populations. Environ. Sci. Pollut. Res. Int. 2016, 23, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Dereumeaux, C.; Saoudi, A.; Guldner, L.; Pecheux, M.; Chesneau, J.; Thomé, J.-P.; Ledrans, M.; Tertre, A.L.; Denys, S.; Fillol, C. Chlordecone and Organochlorine Compound Levels in the French West Indies Population in 2013–2014. Environ. Sci. Pollut. Res. Int. 2020, 27, 41033–41045. [Google Scholar] [CrossRef] [PubMed]
- Boucher, O.; Simard, M.-N.; Muckle, G.; Rouget, F.; Kadhel, P.; Bataille, H.; Chajès, V.; Dallaire, R.; Monfort, C.; Thomé, J.-P.; et al. Exposure to an Organochlorine Pesticide (Chlordecone) and Development of 18-Month-Old Infants. Neurotoxicology 2013, 35, 162–168. [Google Scholar] [CrossRef] [PubMed]
- Dallaire, R.; Muckle, G.; Rouget, F.; Kadhel, P.; Bataille, H.; Guldner, L.; Seurin, S.; Chajès, V.; Monfort, C.; Boucher, O.; et al. Cognitive, Visual, and Motor Development of 7-Month-Old Guadeloupean Infants Exposed to Chlordecone. Environ. Res. 2012, 118, 79–85. [Google Scholar] [CrossRef]
- Taylor, J.R. Neurological Manifestations in Humans Exposed to Chlordecone and Follow-up Results. Neurotoxicology 1982, 3, 9–16. [Google Scholar]
- Taylor, J.R.; Selhorst, J.B.; Houff, S.A.; Martinez, A.J. Chlordecone Intoxication in Man. I. Clinical Observations. Neurology 1978, 28, 626–630. [Google Scholar] [CrossRef]
- Benet, H.; Fujimori, K.; Ho, I.K. The Basal Ganglia in Chlordecone-Induced Neurotoxicity in the Mouse. Neurotoxicology 1985, 6, 151–158. [Google Scholar]
- Fujimori, K.; Benet, H.; Mehendale, H.M.; Ho, I.K. Comparison of Brain Discrete Area Distributions of Chlordecone and Mirex in the Mouse. Neurotoxicology 1982, 3, 125–129. [Google Scholar]
- Dardiotis, E.; Aloizou, A.-M.; Sakalakis, E.; Siokas, V.; Koureas, M.; Xiromerisiou, G.; Petinaki, E.; Wilks, M.; Tsatsakis, A.; Hadjichristodoulou, C.; et al. Organochlorine Pesticide Levels in Greek Patients with Parkinson’s Disease. Toxicol. Rep. 2020, 7, 596–601. [Google Scholar] [CrossRef]
- Elbaz, A.; Clavel, J.; Rathouz, P.J.; Moisan, F.; Galanaud, J.-P.; Delemotte, B.; Alpérovitch, A.; Tzourio, C. Professional Exposure to Pesticides and Parkinson Disease. Ann. Neurol. 2009, 66, 494–504. [Google Scholar] [CrossRef]
- Jayaraj, R.; Megha, P.; Sreedev, P. Review Article. Organochlorine Pesticides, Their Toxic Effects on Living Organisms and Their Fate in the Environment. Interdiscip. Toxicol. 2016, 9, 90–100. [Google Scholar] [CrossRef]
- Moisan, F.; Spinosi, J.; Delabre, L.; Gourlet, V.; Mazurie, J.-L.; Bénatru, I.; Goldberg, M.; Weisskopf, M.G.; Imbernon, E.; Tzourio, C.; et al. Association of Parkinson’s Disease and Its Subtypes with Agricultural Pesticide Exposures in Men: A Case-Control Study in France. Environ. Health Perspect. 2015, 123, 1123–1129. [Google Scholar] [CrossRef] [PubMed]
- Caparros-Lefebvre, D.; Elbaz, A. Possible Relation of Atypical Parkinsonism in the French West Indies with Consumption of Tropical Plants: A Case-Control Study. Lancet 1999, 354, 281–286. [Google Scholar] [CrossRef] [PubMed]
- Lannuzel, A.; Edragas, R.; Lackmy, A.; Tressières, B.; Pelonde, V.; Kaptué, M.E.N.; Mécharles, S.; Demas, A.; François, B.; McGovern, E.; et al. Further Evidence for a Distinctive Atypical Degenerative Parkinsonism in the Caribbean: A New Cluster in the French West Indian Island of Martinique. J. Neurol. Sci. 2018, 388, 214–219. [Google Scholar] [CrossRef] [PubMed]
- Lannuzel, A.; Höglinger, G.U.; Verhaeghe, S.; Gire, L.; Belson, S.; Escobar-Khondiker, M.; Poullain, P.; Oertel, W.H.; Hirsch, E.C.; Dubois, B.; et al. Atypical Parkinsonism in Guadeloupe: A Common Risk Factor for Two Closely Related Phenotypes? Brain 2007, 130, 816–827. [Google Scholar] [CrossRef]
- Cleret de Langavant, L.; Roze, E.; Petit, A.; Tressières, B.; Gharbi-Meliani, A.; Chaumont, H.; Michel, P.P.; Bachoud-Lévi, A.-C.; Remy, P.; Edragas, R.; et al. Annonaceae Consumption Worsens Disease Severity and Cognitive Deficits in Degenerative Parkinsonism. Mov. Disord. 2022, 37, 2355–2366. [Google Scholar] [CrossRef]
- Caparros-Lefebvre, D.; Sergeant, N.; Lees, A.; Camuzat, A.; Daniel, S.; Lannuzel, A.; Brice, A.; Tolosa, E.; Delacourte, A.; Duyckaerts, C. Guadeloupean Parkinsonism: A Cluster of Progressive Supranuclear Palsy-like Tauopathy. Brain 2002, 125, 801–811. [Google Scholar] [CrossRef] [PubMed]
- Lannuzel, A.; Michel, P.P.; Höglinger, G.U.; Champy, P.; Jousset, A.; Medja, F.; Lombès, A.; Darios, F.; Gleye, C.; Laurens, A.; et al. The Mitochondrial Complex I Inhibitor Annonacin Is Toxic to Mesencephalic Dopaminergic Neurons by Impairment of Energy Metabolism. Neuroscience 2003, 121, 287–296. [Google Scholar] [CrossRef]
- Lannuzel, A.; Ruberg, M.; Michel, P.P. Atypical Parkinsonism in the Caribbean Island of Guadeloupe: Etiological Role of the Mitochondrial Complex I Inhibitor Annonacin. Mov. Disord. 2008, 23, 2122–2128. [Google Scholar] [CrossRef]
- German Federal Institute for Risk Assessment (BfR); Department of Food Safety, Berlin, Germany; Raclariu-Manolica, A.C.; Bakhiya, N.; Hirsch-Ernst, K.I. Risk Assessment Regarding the Use of Annona Muricata in Food Supplements. EFSA J. 2020, 18, e181112. [Google Scholar] [CrossRef]
- González-Rodríguez, P.; Zampese, E.; Stout, K.A.; Guzman, J.N.; Ilijic, E.; Yang, B.; Tkatch, T.; Stavarache, M.A.; Wokosin, D.L.; Gao, L.; et al. Disruption of Mitochondrial Complex I Induces Progressive Parkinsonism. Nature 2021, 599, 650–656. [Google Scholar] [CrossRef] [PubMed]
- Rousseau, E.; Michel, P.P.; Hirsch, E.C. The Iron-Binding Protein Lactoferrin Protects Vulnerable Dopamine Neurons from Degeneration by Preserving Mitochondrial Calcium Homeostasis. Mol. Pharmacol. 2013, 84, 888–898. [Google Scholar] [CrossRef] [PubMed]
- Tourville, A.; Akbar, D.; Corti, O.; Prehn, J.H.M.; Melki, R.; Hunot, S.; Michel, P.P. Modelling α-Synuclein Aggregation and Neurodegeneration with Fibril Seeds in Primary Cultures of Mouse Dopaminergic Neurons. Cells 2022, 11, 1640. [Google Scholar] [CrossRef] [PubMed]
- Poehler, A.-M.; Xiang, W.; Spitzer, P.; May, V.E.L.; Meixner, H.; Rockenstein, E.; Chutna, O.; Outeiro, T.F.; Winkler, J.; Masliah, E.; et al. Autophagy Modulates SNCA/α-Synuclein Release, Thereby Generating a Hostile Microenvironment. Autophagy 2014, 10, 2171–2192. [Google Scholar] [CrossRef]
- Bizat, N.; Parrales, V.; Laoues, S.; Normant, S.; Levavasseur, E.; Roussel, J.; Privat, N.; Gougerot, A.; Ravassard, P.; Beaudry, P.; et al. An in Vivo Caenorhabditis elegans Model for Therapeutic Research in Human Prion Diseases. Brain 2021, 144, 2745–2758. [Google Scholar] [CrossRef]
- Bizat, N.; Peyrin, J.M.; Haik, S.; Cochois, V.; Beaudry, P.; Laplanche, J.L.; Neri, C. Neuron Dysfunction Is Induced by Prion Protein with an Insertional Mutation via a Fyn Kinase and Reversed by Sirtuin Activation in Caenorhabditis Elegans. J. Neurosci. 2010, 30, 5394–5403. [Google Scholar] [CrossRef]
- Dominguez-Meijide, A.; Parrales, V.; Vasili, E.; González-Lizárraga, F.; König, A.; Lázaro, D.F.; Lannuzel, A.; Haik, S.; Del Bel, E.; Chehín, R.; et al. Doxycycline Inhibits α-Synuclein-Associated Pathologies in Vitro and in Vivo. Neurobiol. Dis. 2021, 151, 105256. [Google Scholar] [CrossRef]
- Dexter, P.M.; Caldwell, K.A.; Caldwell, G.A. A Predictable Worm: Application of Caenorhabditis Elegans for Mechanistic Investigation of Movement Disorders. Neurotherapeutics 2012, 9, 393–404. [Google Scholar] [CrossRef]
- Gaeta, A.L.; Caldwell, K.A.; Caldwell, G.A. Found in Translation: The Utility of C. Elegans Alpha-Synuclein Models of Parkinson’s Disease. Brain Sci. 2019, 9, 73. [Google Scholar] [CrossRef]
- Hannan, S.B.; Dräger, N.M.; Rasse, T.M.; Voigt, A.; Jahn, T.R. Cellular and Molecular Modifier Pathways in Tauopathies: The Big Picture from Screening Invertebrate Models. J. Neurochem. 2016, 137, 12–25. [Google Scholar] [CrossRef]
- Sepulveda-Diaz, J.E.; Ouidja, M.O.; Socias, S.B.; Hamadat, S.; Guerreiro, S.; Raisman-Vozari, R.; Michel, P.P. A Simplified Approach for Efficient Isolation of Functional Microglial Cells: Application for Modeling Neuroinflammatory Responses in Vitro. Glia 2016, 64, 1912–1924. [Google Scholar] [CrossRef] [PubMed]
- Traver, S.; Marien, M.; Martin, E.; Hirsch, E.C.; Michel, P.P. The Phenotypic Differentiation of Locus Ceruleus Noradrenergic Neurons Mediated by Brain-Derived Neurotrophic Factor Is Enhanced by Corticotropin Releasing Factor through the Activation of a CAMP-Dependent Signaling Pathway. Mol. Pharmacol. 2006, 70, 30–40. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, E.; Almeida, Z.L.; Cruz, P.F.; Silva E Sousa, M.; Veríssimo, P.; Brito, R.M.M. Searching for the Best Transthyretin Aggregation Protocol to Study Amyloid Fibril Disruption. Int. J. Mol. Sci. 2021, 23, 391. [Google Scholar] [CrossRef] [PubMed]
- Dos-Santos-Pereira, M.; Acuña, L.; Hamadat, S.; Rocca, J.; González-Lizárraga, F.; Chehín, R.; Sepulveda-Diaz, J.; Del-Bel, E.; Raisman-Vozari, R.; Michel, P.P. Microglial Glutamate Release Evoked by α-Synuclein Aggregates Is Prevented by Dopamine. Glia 2018, 66, 2353–2365. [Google Scholar] [CrossRef]
- Joshi, K.K.; Matlack, T.L.; Rongo, C. Dopamine Signaling Promotes the Xenobiotic Stress Response and Protein Homeostasis. EMBO J. 2016, 35, 1885–1901. [Google Scholar] [CrossRef]
- Tourville, A.; Viguier, S.; González-Lizárraga, F.; Tomas-Grau, R.H.; Ramirez, P.; Brunel, J.-M.; Dos Santos Pereira, M.; Del-Bel, E.; Chehin, R.; Ferrié, L.; et al. Rescue of Dopamine Neurons from Iron-Dependent Ferroptosis by Doxycycline and Demeclocycline and Their Non-Antibiotic Derivatives. Antioxidants 2023, 12, 575. [Google Scholar] [CrossRef]
- Schindelin, J.; Arganda-Carreras, I.; Frise, E.; Kaynig, V.; Longair, M.; Pietzsch, T.; Preibisch, S.; Rueden, C.; Saalfeld, S.; Schmid, B.; et al. Fiji: An Open-Source Platform for Biological-Image Analysis. Nat. Methods 2012, 9, 676–682. [Google Scholar] [CrossRef]
- Lints, R.; Emmons, S.W. Patterning of Dopaminergic Neurotransmitter Identity among Caenorhabditis Elegans Ray Sensory Neurons by a TGFbeta Family Signaling Pathway and a Hox Gene. Development 1999, 126, 5819–5831. [Google Scholar] [CrossRef]
- Nass, R.; Hall, D.H.; Miller, D.M.; Blakely, R.D. Neurotoxin-Induced Degeneration of Dopamine Neurons in Caenorhabditis Elegans. Proc. Natl. Acad. Sci. USA 2002, 99, 3264–3269. [Google Scholar] [CrossRef]
- Kraemer, B.C.; Zhang, B.; Leverenz, J.B.; Thomas, J.H.; Trojanowski, J.Q.; Schellenberg, G.D. Neurodegeneration and Defective Neurotransmission in a Caenorhabditis Elegans Model of Tauopathy. Proc. Natl. Acad. Sci. USA 2003, 100, 9980–9985. [Google Scholar] [CrossRef] [PubMed]
- Brenner, S. The Genetics of Caenorhabditis Elegans. Genetics 1974, 77, 71–94. [Google Scholar] [CrossRef] [PubMed]
- Stiernagle, T. Maintenance of C. Elegans; WormBook: Columbia, NY, USA, 2006; pp. 1–11. [Google Scholar] [CrossRef]
- Couillault, C.; Ewbank, J.J. Diverse Bacteria Are Pathogens of Caenorhabditis Elegans. Infect. Immun. 2002, 70, 4705–4707. [Google Scholar] [CrossRef]
- Albertson, D.G. Localization of the Ribosomal Genes in Caenorhabditis Elegans Chromosomes by in Situ Hybridization Using Biotin-Labeled Probes. EMBO J. 1984, 3, 1227–1234. [Google Scholar] [CrossRef] [PubMed]
- Sawin, E.R.; Ranganathan, R.; Horvitz, H.R.C. Elegans Locomotory Rate Is Modulated by the Environment through a Dopaminergic Pathway and by Experience through a Serotonergic Pathway. Neuron 2000, 26, 619–631. [Google Scholar] [CrossRef] [PubMed]
- Lavaur, J.; Le Nogue, D.; Lemaire, M.; Pype, J.; Farjot, G.; Hirsch, E.C.; Michel, P.P. The Noble Gas Xenon Provides Protection and Trophic Stimulation to Midbrain Dopamine Neurons. J. Neurochem. 2017, 142, 14–28. [Google Scholar] [CrossRef]
- Michel, P.P.; Dandapani, B.K.; Knusel, B.; Sanchez-Ramos, J.; Hefti, F. Toxicity of 1-Methyl-4-Phenylpyridinium for Rat Dopaminergic Neurons in Culture: Selectivity and Irreversibility. J. Neurochem. 1990, 54, 1102–1109. [Google Scholar] [CrossRef]
- Gerencser, A.A.; Chinopoulos, C.; Birket, M.J.; Jastroch, M.; Vitelli, C.; Nicholls, D.G.; Brand, M.D. Quantitative Measurement of Mitochondrial Membrane Potential in Cultured Cells: Calcium-Induced de- and Hyperpolarization of Neuronal Mitochondria. J. Physiol. 2012, 590, 2845–2871. [Google Scholar] [CrossRef]
- Takeda, A.; Smith, M.A.; Avilá, J.; Nunomura, A.; Siedlak, S.L.; Zhu, X.; Perry, G.; Sayre, L.M. In Alzheimer’s Disease, Heme Oxygenase Is Coincident with Alz50, an Epitope of Tau Induced by 4-Hydroxy-2-Nonenal Modification. J. Neurochem. 2000, 75, 1234–1241. [Google Scholar] [CrossRef]
- Parrales-Macias, V.; Harfouche, A.; Ferrié, L.; Haïk, S.; Michel, P.P.; Raisman-Vozari, R.; Figadère, B.; Bizat, N.; Maciuk, A. Effects of a New Natural Catechol-O-Methyl Transferase Inhibitor on Two In Vivo Models of Parkinson’s Disease. ACS Chem. Neurosci. 2022, 13, 3303–3313. [Google Scholar] [CrossRef]
- Fujiwara, M.; Sengupta, P.; McIntire, S.L. Regulation of Body Size and Behavioral State of C. Elegans by Sensory Perception and the EGL-4 CGMP-Dependent Protein Kinase. Neuron 2002, 36, 1091–1102. [Google Scholar] [CrossRef]
- Rivard, L.; Srinivasan, J.; Stone, A.; Ochoa, S.; Sternberg, P.W.; Loer, C.M. A Comparison of Experience-Dependent Locomotory Behaviors and Biogenic Amine Neurons in Nematode Relatives of Caenorhabditis Elegans. BMC Neurosci. 2010, 11, 22. [Google Scholar] [CrossRef]
- Brion, J.P.; Hanger, D.P.; Bruce, M.T.; Couck, A.M.; Flament-Durand, J.; Anderton, B.H. Tau in Alzheimer Neurofibrillary Tangles. N- and C-Terminal Regions Are Differentially Associated with Paired Helical Filaments and the Location of a Putative Abnormal Phosphorylation Site. Biochem. J. 1991, 273 Pt 1, 127–133. [Google Scholar] [CrossRef]
- Mercken, M.; Vandermeeren, M.; Lübke, U.; Six, J.; Boons, J.; Van de Voorde, A.; Martin, J.J.; Gheuens, J. Monoclonal Antibodies with Selective Specificity for Alzheimer Tau Are Directed against Phosphatase-Sensitive Epitopes. Acta Neuropathol. 1992, 84, 265–272. [Google Scholar] [CrossRef] [PubMed]
- Zlotkowski, K.; Eliasen, A.M.; Mitra, A.; Siegel, D. Small-Molecule Mechanism of Action Studies in Caenorhabditis Elegans. Chembiochem 2013, 14, 2338–2344. [Google Scholar] [CrossRef]
- Clark, S.G.; Chiu, C.C. Elegans ZAG-1, a Zn-Finger-Homeodomain Protein, Regulates Axonal Development and Neuronal Differentiation. Development 2003, 130, 3781–3794. [Google Scholar] [CrossRef]
- Inoue, K.; Nakazawa, K.; Obama, T.; Fujimori, K.; Takanaka, A. Chlordecone inhibits three types of ion channels in a neural cell line. Pharmacol. Toxicol. 1990, 67, 444–446. [Google Scholar] [CrossRef]
- Park, H.K.; Ilango, S.D.; Litvan, I. Environmental Risk Factors for Progressive Supranuclear Palsy. J. Mov. Disord. 2021, 14, 103–113. [Google Scholar] [CrossRef] [PubMed]
- Saint-Hilaire, M.; Fourcot, A.; Bousquet-Mélou, A.; Rychen, G.; Thomé, J.-P.; Parinet, J.; Feidt, C.; Fournier, A. Characterization and Quantification of Chlordecone Elimination in Ewes. Environ. Toxicol. Pharmacol. 2021, 87, 103698. [Google Scholar] [CrossRef] [PubMed]
- Emond, C.; Multigner, L. Chlordecone: Development of a Physiologically Based Pharmacokinetic Tool to Support Human Health Risks Assessments. Arch. Toxicol. 2022, 96, 1009–1019. [Google Scholar] [CrossRef]
- Omura, D.T.; Clark, D.A.; Samuel, A.D.T.; Horvitz, H.R. Dopamine Signaling Is Essential for Precise Rates of Locomotion by C. Elegans. PLoS ONE 2012, 7, e38649. [Google Scholar] [CrossRef]
- Salim, C.; Rajini, P.S. Glucose-Rich Diet Aggravates Monocrotophos-Induced Dopaminergic Neuronal Dysfunction in Caenorhabditis Elegans. J. Appl. Toxicol. 2017, 37, 772–780. [Google Scholar] [CrossRef] [PubMed]
- Richter, F.; Gabby, L.; McDowell, K.A.; Mulligan, C.K.; De La Rosa, K.; Sioshansi, P.C.; Mortazavi, F.; Cely, I.; Ackerson, L.C.; Tsan, L.; et al. Effects of Decreased Dopamine Transporter Levels on Nigrostriatal Neurons and Paraquat/Maneb Toxicity in Mice. Neurobiol. Aging 2017, 51, 54–66. [Google Scholar] [CrossRef] [PubMed]
- Felton, C.M.; Johnson, C.M. Dopamine Signaling in C. elegans Is Mediated in Part by HLH-17-Dependent Regulation of Extracellular Dopamine Levels. G3 (Bethesda) 2014, 4, 1081–1089. [Google Scholar] [CrossRef]
- Sevenich, L. Brain-Resident Microglia and Blood-Borne Macrophages Orchestrate Central Nervous System Inflammation in Neurodegenerative Disorders and Brain Cancer. Front. Immunol. 2018, 9, 697. [Google Scholar] [CrossRef] [PubMed]
- Sofroniew, M.V.; Vinters, H.V. Astrocytes: Biology and Pathology. Acta Neuropathol. 2010, 119, 7–35. [Google Scholar] [CrossRef]
- Zhao, T.; Hong, Y.; Yin, P.; Li, S.; Li, X.-J. Differential HspBP1 Expression Accounts for the Greater Vulnerability of Neurons than Astrocytes to Misfolded Proteins. Proc. Natl. Acad. Sci. USA 2017, 114, E7803–E7811. [Google Scholar] [CrossRef]
- Swanson, R.A.; Ying, W.; Kauppinen, T.M. Astrocyte Influences on Ischemic Neuronal Death. Curr. Mol. Med. 2004, 4, 193–205. [Google Scholar] [CrossRef] [PubMed]
- Janelsins, M.C.; Mastrangelo, M.A.; Park, K.M.; Sudol, K.L.; Narrow, W.C.; Oddo, S.; LaFerla, F.M.; Callahan, L.M.; Federoff, H.J.; Bowers, W.J. Chronic Neuron-Specific Tumor Necrosis Factor-Alpha Expression Enhances the Local Inflammatory Environment Ultimately Leading to Neuronal Death in 3xTg-AD Mice. Am. J. Pathol. 2008, 173, 1768–1782. [Google Scholar] [CrossRef]
- Acuña, L.; Hamadat, S.; Corbalán, N.S.; González-Lizárraga, F.; Dos-Santos-Pereira, M.; Rocca, J.; Díaz, J.S.; Del-Bel, E.; Papy-García, D.; Chehín, R.N.; et al. Rifampicin and Its Derivative Rifampicin Quinone Reduce Microglial Inflammatory Responses and Neurodegeneration Induced In Vitro by α-Synuclein Fibrillary Aggregates. Cells 2019, 8, 776. [Google Scholar] [CrossRef]
- Fischer, R.; Kontermann, R.E.; Pfizenmaier, K. Selective Targeting of TNF Receptors as a Novel Therapeutic Approach. Front. Cell. Dev. Biol. 2020, 8, 401. [Google Scholar] [CrossRef]
- Lassozé, S.; de Guilhem de Lataillade, A.; Oullier, T.; Neunslist, M.; Leclair-Visonneau, L.; Derkinderen, P.; Paillusson, S. Comparison of Commercially Available Antibodies for the Detection of Phosphorylated Alpha-Synuclein in Primary Culture of ENS. Neurogastroenterol. Motil. 2022, 34, e14354. [Google Scholar] [CrossRef] [PubMed]
- Iqbal, K.; Liu, F.; Gong, C.-X.; Alonso, A.D.C.; Grundke-Iqbal, I. Mechanisms of Tau-Induced Neurodegeneration. Acta Neuropathol. 2009, 118, 53–69. [Google Scholar] [CrossRef] [PubMed]
- Taylor, L.M.; McMillan, P.J.; Liachko, N.F.; Strovas, T.J.; Ghetti, B.; Bird, T.D.; Keene, C.D.; Kraemer, B.C. Pathological Phosphorylation of Tau and TDP-43 by TTBK1 and TTBK2 Drives Neurodegeneration. Mol. Neurodegener. 2018, 13, 7. [Google Scholar] [CrossRef] [PubMed]
- Gezer, A.O.; Kochmanski, J.; VanOeveren, S.E.; Cole-Strauss, A.; Kemp, C.J.; Patterson, J.R.; Miller, K.M.; Kuhn, N.C.; Herman, D.E.; McIntire, A.; et al. Developmental Exposure to the Organochlorine Pesticide Dieldrin Causes Male-Specific Exacerbation of α-Synuclein-Preformed Fibril-Induced Toxicity and Motor Deficits. Neurobiol. Dis. 2020, 141, 104947. [Google Scholar] [CrossRef] [PubMed]
- Douda, D.N.; Khan, M.A.; Grasemann, H.; Palaniyar, N. SK3 Channel and Mitochondrial ROS Mediate NADPH Oxidase-Independent NETosis Induced by Calcium Influx. Proc. Natl. Acad. Sci. USA 2015, 112, 2817–2822. [Google Scholar] [CrossRef]
- Creed, S.; McKenzie, M. Measurement of Mitochondrial Membrane Potential with the Fluorescent Dye Tetramethylrhodamine Methyl Ester (TMRM). Methods Mol. Biol. 2019, 1928, 69–76. [Google Scholar] [CrossRef]
- O’Connell, M.; McClure, N.; Lewis, S.E.M. The Effects of Cryopreservation on Sperm Morphology, Motility and Mitochondrial Function. Hum. Reprod. 2002, 17, 704–709. [Google Scholar] [CrossRef]
- Egaña, J.T.; Zambrano, C.; Nuñez, M.T.; Gonzalez-Billault, C.; Maccioni, R.B. Iron-Induced Oxidative Stress Modify Tau Phosphorylation Patterns in Hippocampal Cell Cultures. Biometals 2003, 16, 215–223. [Google Scholar] [CrossRef]
- Du, F.; Yu, Q.; Kanaan, N.M.; Yan, S.S. Mitochondrial Oxidative Stress Contributes to the Pathological Aggregation and Accumulation of Tau Oligomers in Alzheimer’s Disease. Hum. Mol. Genet. 2022, 31, 2498–2507. [Google Scholar] [CrossRef]
- Musgrove, R.E.; Helwig, M.; Bae, E.-J.; Aboutalebi, H.; Lee, S.-J.; Ulusoy, A.; Di Monte, D.A. Oxidative Stress in Vagal Neurons Promotes Parkinsonian Pathology and Intercellular α-Synuclein Transfer. J. Clin. Investig. 2019, 129, 3738–3753. [Google Scholar] [CrossRef]
- Brennan, L.A.; Kantorow, M. Mitochondrial Function and Redox Control in the Aging Eye: Role of MsrA and Other Repair Systems in Cataract and Macular Degenerations. Exp. Eye Res. 2009, 88, 195–203. [Google Scholar] [CrossRef] [PubMed]
- Cannon, S.B.; Veazey, J.M., Jr.; Jackson, R.S.; Burse, V.W.; Hayes, C.; Straub, W.E.; Landrigan, P.J.; Liddle, J.A. Epidemic kepone poisoning in chemical workers. Am. J. Epidemiol. 1978, 107, 529–537. [Google Scholar] [CrossRef]
- Wang, T.P.; Ho, I.K.; Mehendale, H.M. Correlation between neurotoxicity and chlordecone (Kepone) levels in brain and plasma in the mouse. Neurotoxicology 1981, 2, 373–381. [Google Scholar] [PubMed]
- Emeville, E.; Giusti, A.; Coumoul, X.; Thomé, J.P.; Blanchet, P.; Multigner, L. Associations of plasma concentrations of dichlorodiphenyldichloroethylene and polychlorinated biphenyls with prostate cancer: A case-control study in Guadeloupe (French West Indies). Environ. Health Perspect. 2015, 123, 317–323. [Google Scholar] [CrossRef] [PubMed]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Primary Antibodies | Host | Working Dilution | Source | Identifier |
|---|---|---|---|---|
| anti-TH [LNC1] | Mouse | 1:2500 | Immunostar | 22941 |
| anti-TH | Chicken | 1:1000 | Abcam | ab76442 |
| anti p-S202/pT205 Tau [AT8] | Mouse | 1:500 | Thermo Fisher Sci. | #MN1020 |
| anti-Tau [EP2456Y] * | Rabbit | 1:200 | Abcam | #ab76128 |
| anti-p-S129 αS [EP1536Y] | Rabbit | 1:2500 | Abcam | #ab51253 |
| BB515 anti-CD45 | Rat | 1:100 | BD BioSciences | #564590 |
| anti-GFAP | Chicken | 1:1000 | Sigma-Aldrich | #AB5541 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Parrales-Macias, V.; Michel, P.P.; Tourville, A.; Raisman-Vozari, R.; Haïk, S.; Hunot, S.; Bizat, N.; Lannuzel, A. The Pesticide Chlordecone Promotes Parkinsonism-like Neurodegeneration with Tau Lesions in Midbrain Cultures and C. elegans Worms. Cells 2023, 12, 1336. https://doi.org/10.3390/cells12091336
Parrales-Macias V, Michel PP, Tourville A, Raisman-Vozari R, Haïk S, Hunot S, Bizat N, Lannuzel A. The Pesticide Chlordecone Promotes Parkinsonism-like Neurodegeneration with Tau Lesions in Midbrain Cultures and C. elegans Worms. Cells. 2023; 12(9):1336. https://doi.org/10.3390/cells12091336
Chicago/Turabian StyleParrales-Macias, Valeria, Patrick P. Michel, Aurore Tourville, Rita Raisman-Vozari, Stéphane Haïk, Stéphane Hunot, Nicolas Bizat, and Annie Lannuzel. 2023. "The Pesticide Chlordecone Promotes Parkinsonism-like Neurodegeneration with Tau Lesions in Midbrain Cultures and C. elegans Worms" Cells 12, no. 9: 1336. https://doi.org/10.3390/cells12091336
APA StyleParrales-Macias, V., Michel, P. P., Tourville, A., Raisman-Vozari, R., Haïk, S., Hunot, S., Bizat, N., & Lannuzel, A. (2023). The Pesticide Chlordecone Promotes Parkinsonism-like Neurodegeneration with Tau Lesions in Midbrain Cultures and C. elegans Worms. Cells, 12(9), 1336. https://doi.org/10.3390/cells12091336