
The E1a Adenoviral Gene Upregulates the Yamanaka Factors to Induce Partial Cellular Reprogramming

 , , , , ,
, , , , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
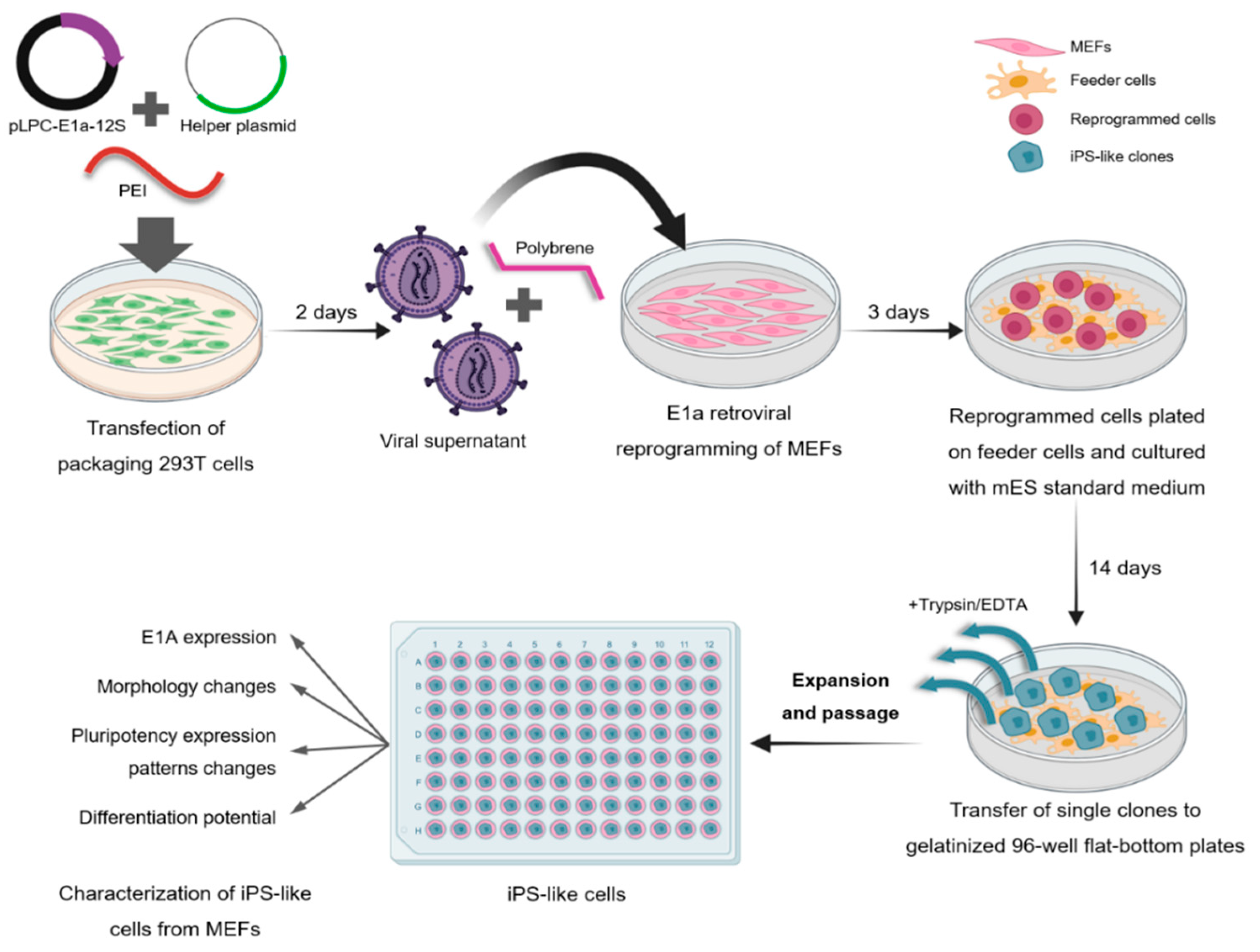
2. Materials and Methods
2.1. Cell Culture
2.2. E1a Retroviral Reprogramming of MEF Oct4-GFP
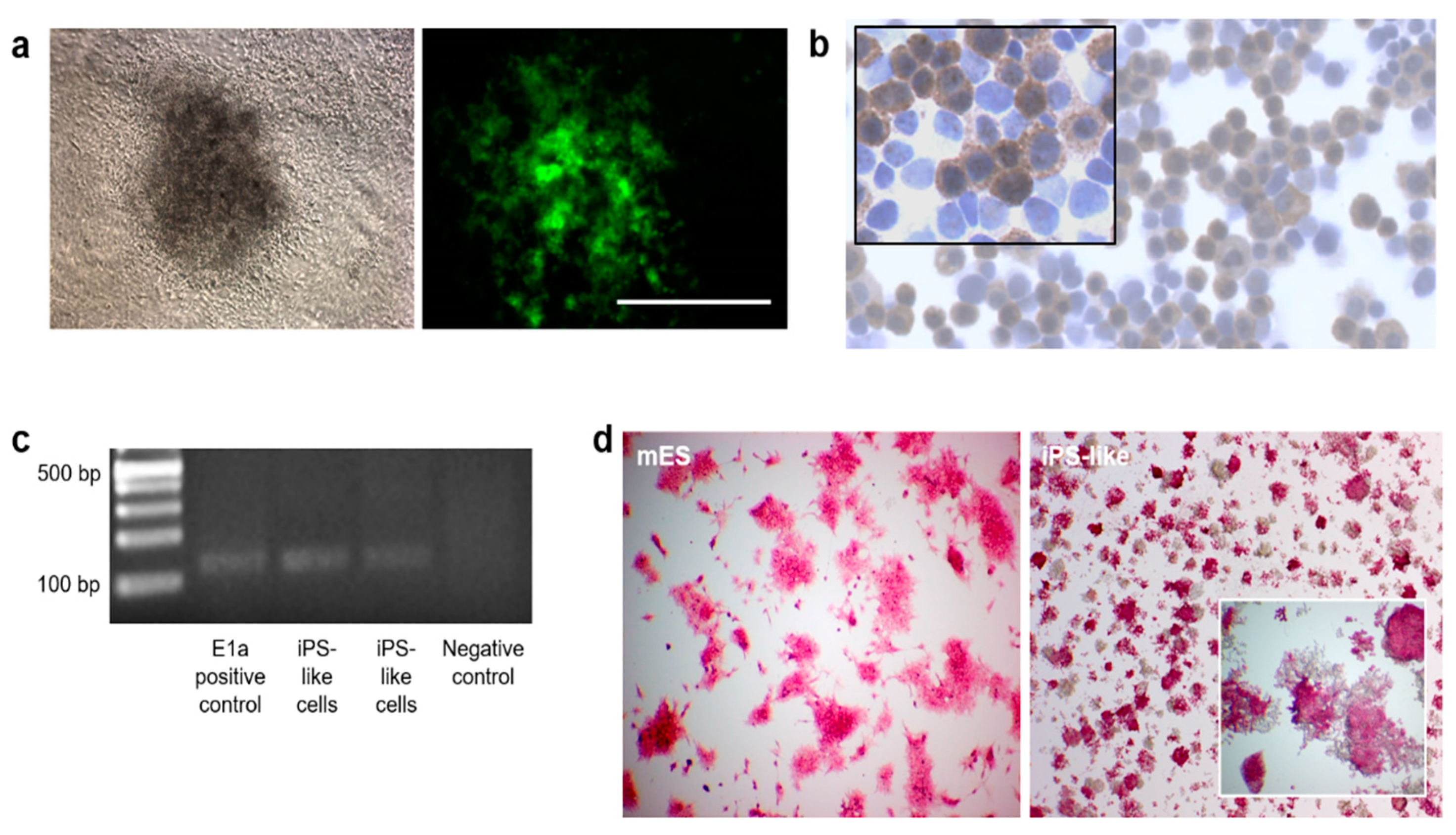
2.3. Alkaline Phosphatase Activity and E1a Expression
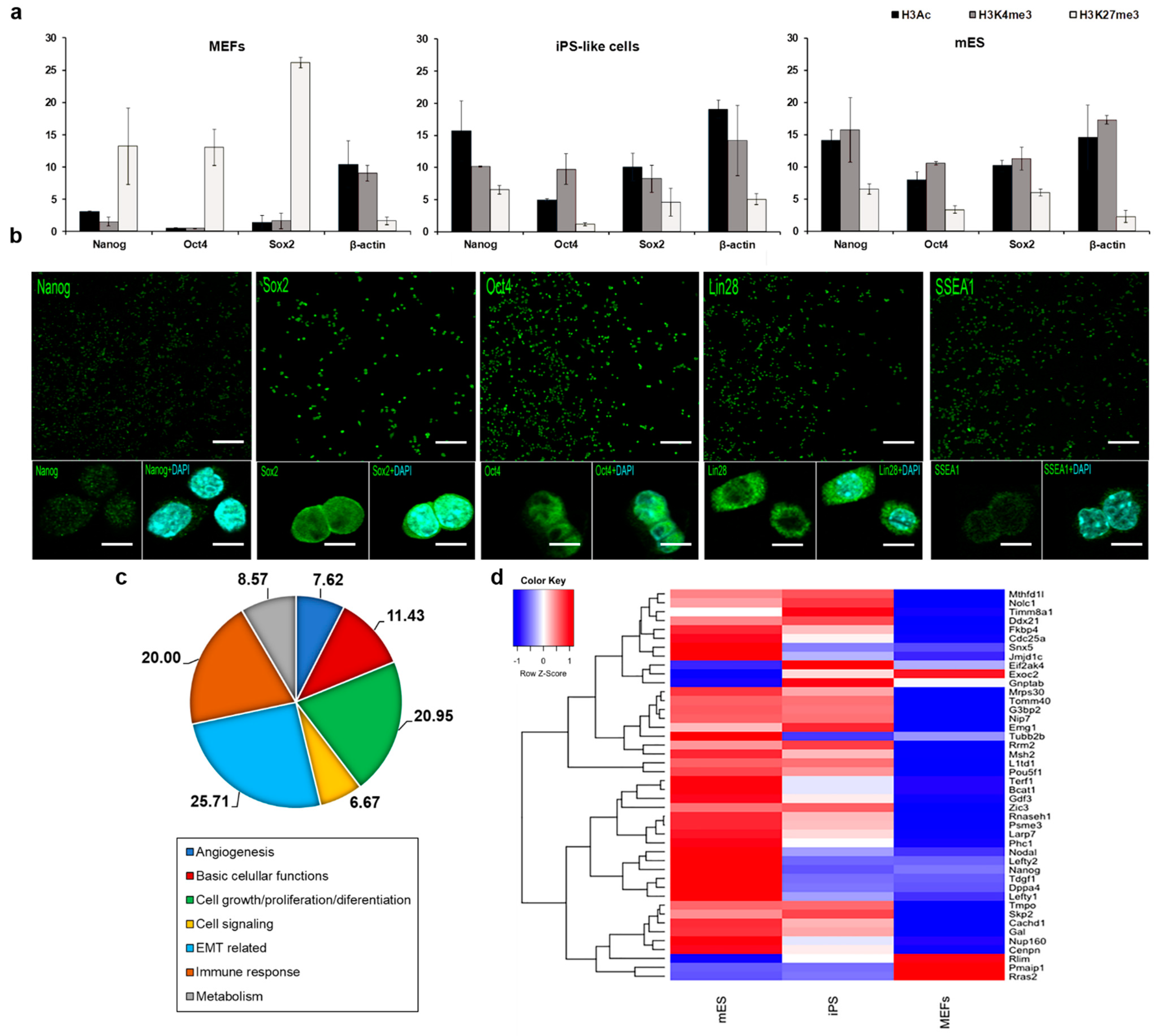
2.4. Immunocytochemistry
2.5. Embryoid Bodies Generation
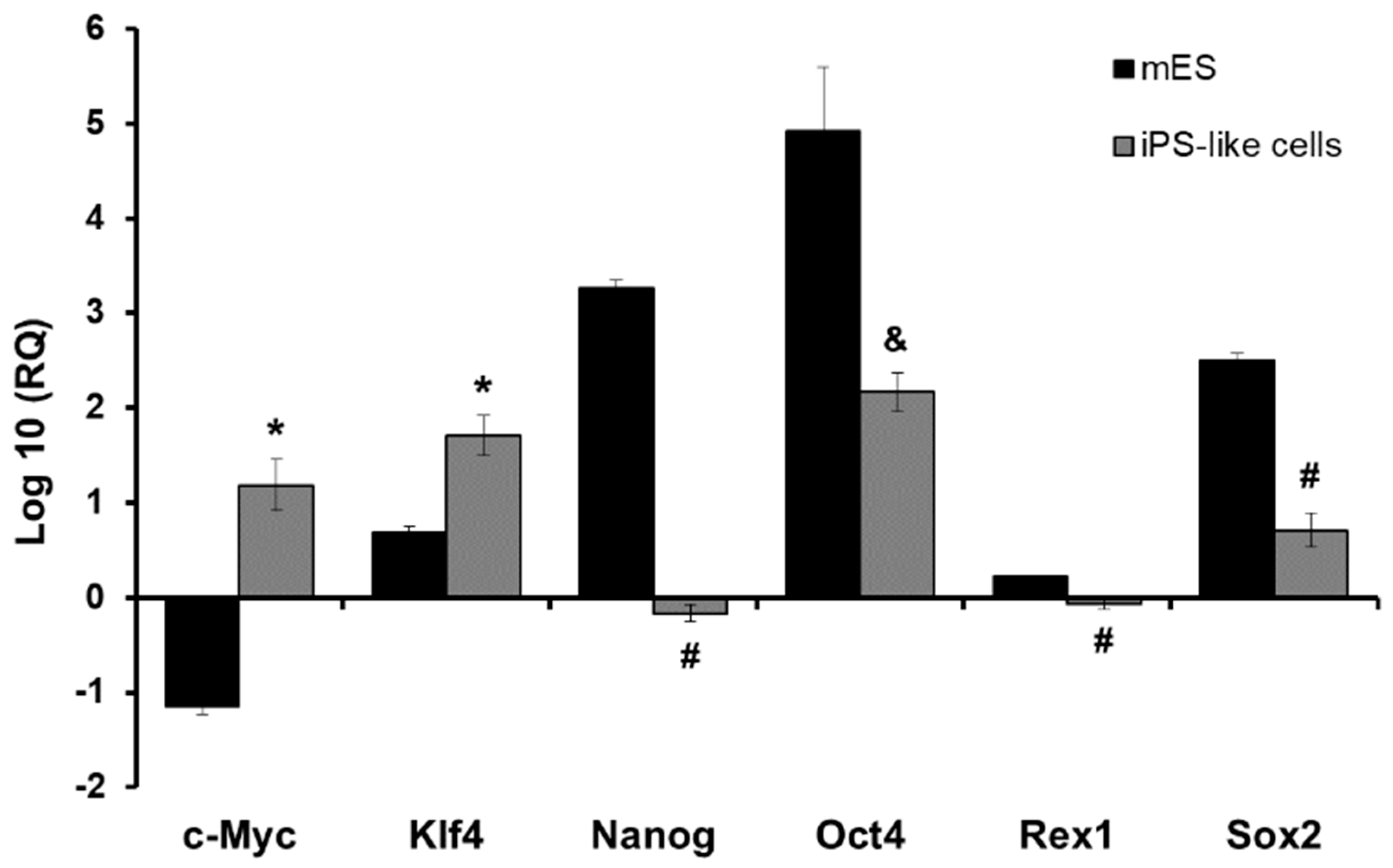
2.6. RNA Isolation and qPCR Analysis
2.7. Gene Expression Profiling
2.8. GeneChIP Assay
2.9. In Vivo Studies
2.9.1. Teratoma Formation
2.9.2. Chimera Production
2.10. Histological Studies
2.11. Statistical Analyses
3. Results
3.1. Generation and Characterization of iPS-like Cells from MEFs by E1a-12S Overexpression
3.2. E1a Derived iPS-like Cells Are Similar to mES Cells at the Molecular Level
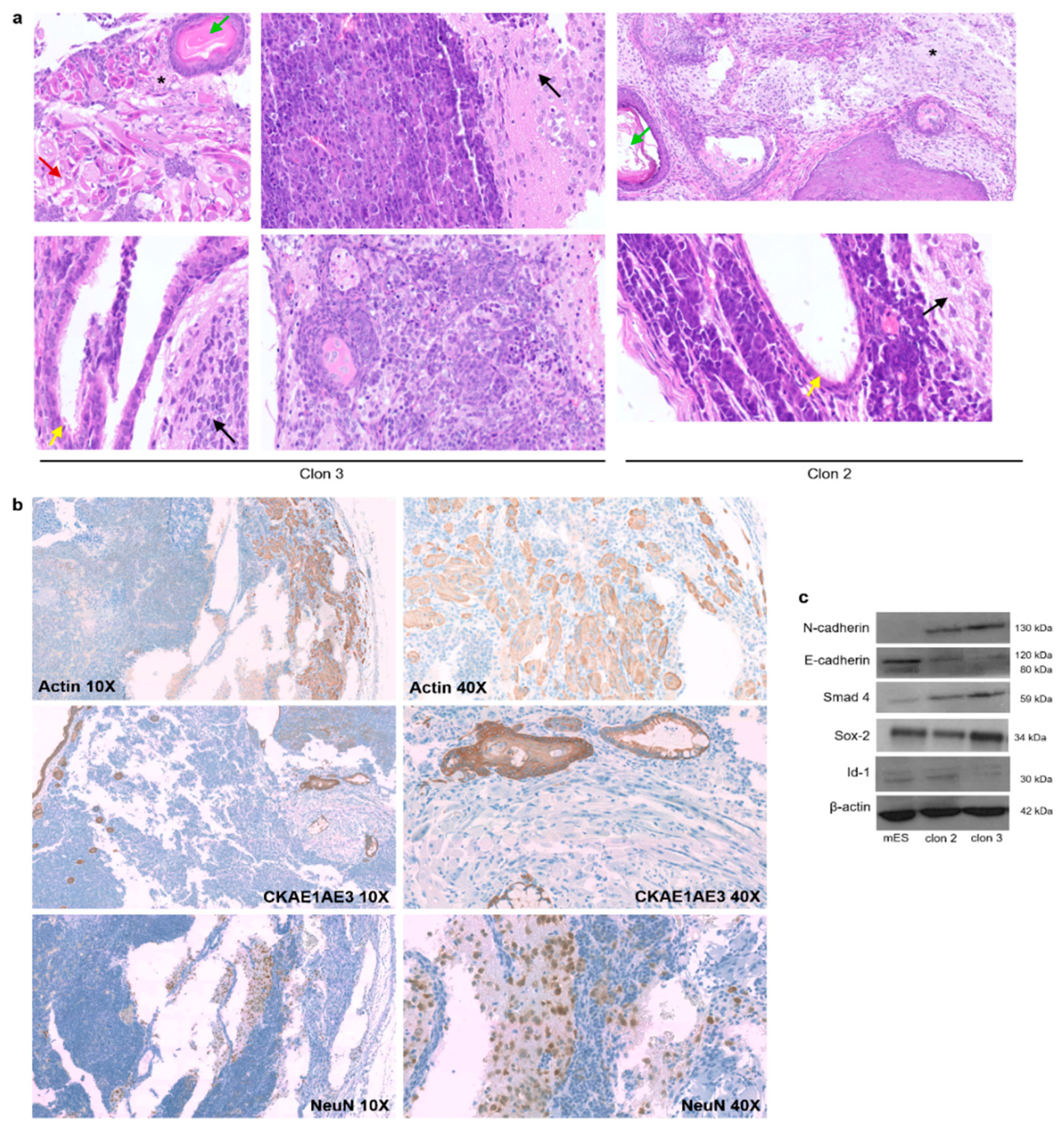
3.3. iPS-like Cells Can Be Differentiated into Three Germ Layers In Vitro
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Takahashi, K.; Yamanaka, S. Induction of Pluripotent Stem Cells from Mouse Embryonic and Adult Fibroblast Cultures by Defined Factors. Cell 2006, 126, 663–676. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Vodyanik, M.A.; Smuga-Otto, K.; Antosiewicz-Bourget, J.; Frane, J.L.; Tian, S.; Nie, J.; Jonsdottir, G.A.; Ruotti, V.; Stewart, R. Induced pluripotent stem cell lines derived from human somatic cells. Science 2007, 318, 1917–1920. [Google Scholar] [CrossRef] [PubMed]
- Yamanaka, S. A fresh look at iPS cells. Cell 2009, 137, 13–17. [Google Scholar] [CrossRef] [PubMed]
- Esteban, M.A.; Wang, T.; Qin, B.; Yang, J.; Qin, D.; Cai, J.; Li, W.; Weng, Z.; Chen, J.; Ni, S. Vitamin C enhances the generation of mouse and human induced pluripotent stem cells. Cell Stem Cell 2010, 6, 71–79. [Google Scholar] [CrossRef]
- Huangfu, D.; Osafune, K.; Maehr, R.; Guo, W.; Eijkelenboom, A.; Chen, S.; Muhlestein, W.; Melton, D. Induction of pluripotent stem cells from primary human fibroblasts with only Oct4 and Sox2. Nat. Biotechnol. 2008, 26, 1269–1275. [Google Scholar] [CrossRef] [PubMed]
- Ying, Q.; Wray, J.; Nichols, J.; Batlle-Morera, L.; Doble, B.; Woodgett, J.; Cohen, P.; Smith, A. The ground state of embryonic stem cell self-renewal. Nature 2008, 453, 519. [Google Scholar] [CrossRef]
- Hammachi, F.; Morrison, G.M.; Sharov, A.A.; Livigni, A.; Narayan, S.; Papapetrou, E.P.; O’Malley, J.; Kaji, K.; Ko, M.S.; Ptashne, M. Transcriptional activation by Oct4 is sufficient for the maintenance and induction of pluripotency. Cell Rep. 2012, 1, 99–109. [Google Scholar] [CrossRef]
- Kim, J.B.; Greber, B.; Araúzo-Bravo, M.J.; Meyer, J.; Park, K.I.; Zaehres, H.; Schöler, H.R. Direct reprogramming of human neural stem cells by OCT4. Nature 2009, 461, 649–653. [Google Scholar] [CrossRef]
- Scognamiglio, R.; Cabezas-Wallscheid, N.; Thier, M.C.; Altamura, S.; Reyes, A.; Prendergast, Á.M.; Baumgärtner, D.; Carnevalli, L.S.; Atzberger, A.; Haas, S. Myc depletion induces a pluripotent dormant state mimicking diapause. Cell 2016, 164, 668–680. [Google Scholar] [CrossRef]
- Lang, S.E.; Hearing, P. The adenovirus E1A oncoprotein recruits the cellular TRRAP/GCN5 histone acetyltransferase complex. Oncogene 2003, 22, 2836–2841. [Google Scholar] [CrossRef]
- Pelka, P.; Ablack, J.N.; Fonseca, G.J.; Yousef, A.F.; Mymryk, J.S. Intrinsic structural disorder in adenovirus E1A: A viral molecular hub linking multiple diverse processes. J. Virol. 2008, 82, 7252–7263. [Google Scholar] [CrossRef]
- Chakraborty, A.A.; Tansey, W.P. Adenoviral E1A function through Myc. Cancer Res. 2009, 69, 6–9. [Google Scholar] [CrossRef] [PubMed]
- Singhal, G.; Leo, E.; Setty, S.K.G.; Pommier, Y.; Thimmapaya, B. Adenovirus E1A oncogene induces rereplication of cellular DNA and alters DNA replication dynamics. J. Virol. 2013, 87, 8767–8778. [Google Scholar] [CrossRef] [PubMed]
- Schöler, H.R.; Ciesiolka, T.; Gruss, P. A nexus between Oct-4 and E1 A: Implications for gene regulation in embryonic stem cells. Cell 1991, 66, 291–304. [Google Scholar] [CrossRef] [PubMed]
- Cartwright, P.; McLean, C.; Sheppard, A.; Rivett, D.; Jones, K.; Dalton, S. LIF/STAT3 controls ES cell self-renewal and pluripotency by a Myc-dependent mechanism. Development 2005, 132, 885–896. [Google Scholar] [CrossRef]
- Nowling, T.K.; Johnson, L.R.; Wiebe, M.S.; Rizzino, A. Identification of the transactivation domain of the transcription factor Sox-2 and an associated co-activator. J. Biol. Chem. 2000, 275, 3810–3818. [Google Scholar] [CrossRef]
- Kadeppagari, R.-K.; Sankar, N.; Thimmapaya, B. Adenovirus transforming protein E1A induces c-Myc in quiescent cells by a novel mechanism. J. Virol. 2009, 83, 4810–4822. [Google Scholar] [CrossRef]
- Ferrari, R.; Pellegrini, M.; Horwitz, G.A.; Xie, W.; Berk, A.J.; Kurdistani, S.K. Epigenetic reprogramming by adenovirus e1a. Science 2008, 321, 1086–1088. [Google Scholar] [CrossRef]
- Horwitz, G.A.; Zhang, K.; McBrian, M.A.; Grunstein, M.; Kurdistani, S.K.; Berk, A.J. Adenovirus small e1a alters global patterns of histone modification. Science 2008, 321, 1084–1085. [Google Scholar] [CrossRef]
- Ying, Q.-L.; Nichols, J.; Evans, E.P.; Smith, A.G. Changing potency by spontaneous fusion. Nature 2002, 416, 545–548. [Google Scholar] [CrossRef]
- Samuelson, A.V.; Lowe, S.W. Selective induction of p53 and chemosensitivity in RB-deficient cells by E1A mutants unable to bind the RB-related proteins. Proc. Natl. Acad. Sci. USA 1997, 94, 12094–12099. [Google Scholar] [CrossRef] [PubMed]
- Gustafsson, B.; Huang, W.; Bogdanovic, G.; Gauffin, F.; Nordgren, A.; Talekar, G.; Ornelles, D.; Gooding, L. Adenovirus DNA is detected at increased frequency in Guthrie cards from children who develop acute lymphoblastic leukaemia. Br. J. Cancer 2007, 97, 992–994. [Google Scholar] [CrossRef] [PubMed]
- Honkaniemi, E.; Talekar, G.; Huang, W.; Bogdanovic, G.; Forestier, E.; Von Doblen, U.; Engvall, M.; Ornelles, D.; Gooding, L.; Gustafsson, B. Adenovirus DNA in Guthrie cards from children who develop acute lymphoblastic leukaemia (ALL). Br. J. Cancer 2010, 102, 796–798. [Google Scholar] [CrossRef] [PubMed]
- Vasconcelos, G.; Kang, M.; Pombo-de-Oliveira, M.; Schiffman, J.; Lorey, F.; Buffler, P.; Wiemels, J. Adenovirus detection in Guthrie cards from paediatric leukaemia cases and controls. Br. J. Cancer 2008, 99, 1668–1672. [Google Scholar] [CrossRef]
- Marthaler, A.G.; Adachi, K.; Tiemann, U.; Wu, G.; Sabour, D.; Velychko, S.; Kleiter, I.; Schöler, H.R.; Tapia, N. Enhanced OCT4 transcriptional activity substitutes for exogenous SOX2 in cellular reprogramming. Sci. Rep. 2016, 6, 19415. [Google Scholar] [CrossRef]
- Zhang, R.; Zhang, L.-h.; Xie, X. iPSCs and small molecules: A reciprocal effort towards better approaches for drug discovery. Acta Pharmacol. Sin. 2013, 34, 765–776. [Google Scholar] [CrossRef]
- Haeussler, D.J.; Evangelista, A.M.; Burgoyne, J.R.; Cohen, R.A.; Bachschmid, M.M.; Pimental, D.R. Checkpoints in adenoviral production: Cross-contamination and E1A. PLoS ONE 2011, 6, e23160. [Google Scholar] [CrossRef]
- Kuwano, K.; Nomoto, Y.; Kunitake, R.; Hagimoto, N.; Matsuba, T.; Nakanishi, Y.; Hara, N. Detection of adenovirus E1A DNA in pulmonary fibrosis using nested polymerase chain reaction. Eur. Respir. J. 1997, 10, 1445–1449. [Google Scholar] [CrossRef]
- Lavine, J.A.; Raess, P.W.; Davis, D.B.; Rabaglia, M.E.; Presley, B.K.; Keller, M.P.; Beinfeld, M.C.; Kopin, A.S.; Newgard, C.B.; Attie, A.D. Contamination with E1A-positive wild-type adenovirus accounts for species-specific stimulation of islet cell proliferation by CCK: A cautionary note. Mol. Endocrinol. 2010, 24, 464–467. [Google Scholar] [CrossRef]
- Chambers, I.; Colby, D.; Robertson, M.; Nichols, J.; Lee, S.; Tweedie, S.; Smith, A. Functional expression cloning of Nanog, a pluripotency sustaining factor in embryonic stem cells. Cell 2003, 113, 643–655. [Google Scholar] [CrossRef]
- Golan-Mashiach, M.; Dazard, J.-E.; Gerecht-Nir, S.; Amariglio, N.; Fisher, T.; Jacob-Hirsch, J.; Bielorai, B.; Osenberg, S.; Barad, O.; Getz, G. Design principle of gene expression used by human stem cells: Implication for pluripotency. FASEB J. 2005, 19, 147–149. [Google Scholar] [CrossRef] [PubMed]
- Mitsui, K.; Tokuzawa, Y.; Itoh, H.; Segawa, K.; Murakami, M.; Takahashi, K.; Maruyama, M.; Maeda, M.; Yamanaka, S. The homeoprotein Nanog is required for maintenance of pluripotency in mouse epiblast and ES cells. Cell 2003, 113, 631–642. [Google Scholar] [CrossRef]
- Picanço-Castro, V.; Russo-Carbolante, E.; Covas, D.T. Forced expression of Nanog in human bone marrow-derived endothelial cells activates other six pluripotent genes. Cell. Reprogramming (Former. Cloning Stem Cells) 2012, 14, 187–192. [Google Scholar] [CrossRef]
- O’Malley, J.; Skylaki, S.; Iwabuchi, K.A.; Chantzoura, E.; Ruetz, T.; Johnsson, A.; Tomlinson, S.R.; Linnarsson, S.; Kaji, K. High-resolution analysis with novel cell-surface markers identifies routes to iPS cells. Nature 2013, 499, 88–91. [Google Scholar] [CrossRef] [PubMed]
- Mikkelsen, T.S.; Hanna, J.; Zhang, X.; Ku, M.; Wernig, M.; Schorderet, P.; Bernstein, B.E.; Jaenisch, R.; Lander, E.S.; Meissner, A. Dissecting direct reprogramming through integrative genomic analysis. Nature 2008, 454, 49–55. [Google Scholar] [CrossRef] [PubMed]
- Plath, K.; Lowry, W.E. Progress in understanding reprogramming to the induced pluripotent state. Nat. Rev. Genet. 2011, 12, 253–265. [Google Scholar] [CrossRef]
- Silva, J.; Barmndon, O.; Nichols, J.; Kawagnchi, l.; Theunisscn, T.; Smith, A. Promotion of reprogramming to ground state pluripotency by signal inhibition. PLoS Biol. 2008, 6, e253. [Google Scholar] [CrossRef]
- Dejosez, M.; Zwaka, T.P. Pluripotency and nuclear reprogramming. Annu. Rev. Biochem. 2012, 81, 737–765. [Google Scholar] [CrossRef]
- Brons, I.G.M.; Smithers, L.E.; Trotter, M.W.; Rugg-Gunn, P.; Sun, B.; Chuva de Sousa Lopes, S.M.; Howlett, S.K.; Clarkson, A.; Ahrlund-Richter, L.; Pedersen, R.A. Derivation of pluripotent epiblast stem cells from mammalian embryos. Nature 2007, 448, 191–195. [Google Scholar] [CrossRef]
- Mallon, B.S.; Chenoweth, J.G.; Johnson, K.R.; Hamilton, R.S.; Tesar, P.J.; Yavatkar, A.S.; Tyson, L.J.; Park, K.; Chen, K.G.; Fann, Y.C. StemCellDB: The human pluripotent stem cell database at the National Institutes of Health. Stem Cell Res. 2013, 10, 57–66. [Google Scholar] [CrossRef]
- Zhou, H.; Li, W.; Zhu, S.; Joo, J.Y.; Do, J.T.; Xiong, W.; Kim, J.B.; Zhang, K.; Schöler, H.R.; Ding, S. Conversion of mouse epiblast stem cells to an earlier pluripotency state by small molecules. J. Biol. Chem. 2010, 285, 29676–29680. [Google Scholar] [CrossRef] [PubMed]
- Song, L.; Chen, J.; Peng, G.; Tang, K.; Jing, N. Dynamic Heterogeneity of Brachyury in Mouse Epiblast Stem Cells Mediates Distinct Response to Extrinsic Bone Morphogenetic Protein (BMP) Signaling. J. Biol. Chem. 2016, 291, 15212–15225. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, R.; Plath, K. The roles of the reprogramming factors Oct4, Sox2 and Klf4 in resetting the somatic cell epigenome during induced pluripotent stem cell generation. Genome Biol. 2012, 13, 251. [Google Scholar] [CrossRef]
- Teshigawara, R.; Cho, J.; Kameda, M.; Tada, T. Mechanism of human somatic reprogramming to iPS cell. Lab. Investig. 2017, 97, 1152–1157. [Google Scholar] [CrossRef]
- Higuchi, A.; Ling, Q.-D.; Kumar, S.S.; Munusamy, M.A.; Alarfaj, A.A.; Chang, Y.; Kao, S.-H.; Lin, K.-C.; Wang, H.-C.; Umezawa, A. Generation of pluripotent stem cells without the use of genetic material. Lab. Investig. 2015, 95, 26–42. [Google Scholar] [CrossRef] [PubMed]
- Lin, T.; Wu, S. Reprogramming with small molecules instead of exogenous transcription factors. Stem Cells Int. 2015, 2015, 794632. [Google Scholar] [CrossRef]
- Costa, R.; Akkerman, N.; Graves, D.; Crisostomo, L.; Bachus, S.; Pelka, P. Characterization of adenovirus 5 E1A exon 1 deletion mutants in the viral replicative cycle. Viruses 2020, 12, 213. [Google Scholar] [CrossRef]
- Zemke, N.R.; Gou, D.; Berk, A.J. Dedifferentiation by adenovirus E1A due to inactivation of Hippo pathway effectors YAP and TAZ. Genes Dev. 2019, 33, 828–843. [Google Scholar] [CrossRef]
- Hu, K. All roads lead to induced pluripotent stem cells: The technologies of iPSC generation. Stem Cells Dev. 2014, 23, 1285–1300. [Google Scholar] [CrossRef]
- Kelaini, S.; Cochrane, A.; Margariti, A. Direct reprogramming of adult cells: Avoiding the pluripotent state. Stem Cells Cloning Adv. Appl. 2014, 7, 19. [Google Scholar]
- Pelka, P.; Ablack, J.N.; Torchia, J.; Turnell, A.S.; Grand, R.J.; Mymryk, J.S. Transcriptional control by adenovirus E1A conserved region 3 via p300/CBP. Nucleic Acids Res. 2009, 37, 1095–1106. [Google Scholar] [CrossRef] [PubMed]
- Dai, X.; Liu, P.; Lau, A.W.; Liu, Y.; Inuzuka, H. Acetylation-dependent regulation of essential iPS-inducing factors: A regulatory crossroad for pluripotency and tumorigenesis. Cancer Med. 2014, 3, 1211–1224. [Google Scholar] [CrossRef] [PubMed]
- Nowling, T.; Bernadt, C.; Johnson, L.; Desler, M.; Rizzino, A. The Co-activator p300 Associates Physically with and Can Mediate the Action of the Distal Enhancer of the FGF-4 gene. J. Biol. Chem. 2003, 278, 13696–13705. [Google Scholar] [CrossRef] [PubMed]
- Berk, A.J. Recent lessons in gene expression, cell cycle control, and cell biology from adenovirus. Oncogene 2005, 24, 7673–7685. [Google Scholar] [CrossRef]
- Lin, T.; Lin, Y. p53 switches off pluripotency on differentiation. Stem Cell Res. Ther. 2017, 8, 44. [Google Scholar] [CrossRef] [PubMed]
- Volman, Y.; Hefetz, R.; Galun, E.; Rachmilewitz, J. DNA damage alters EGFR signaling and reprograms cellular response via Mre-11. Sci. Rep. 2022, 12, 5760. [Google Scholar] [CrossRef]
- Missero, C.; Serra, C.; Stenn, K.; Dotto, G.P. Skin-specific expression of a truncated E1a oncoprotein binding to p105-Rb leads to abnormal hair follicle maturation without increased epidermal proliferation. J. Cell Biol. 1993, 121, 1109–1120. [Google Scholar] [CrossRef]
- Walboomers, J.M.; Jacobs, M.V.; Manos, M.M.; Bosch, F.X.; Kummer, J.A.; Shah, K.V.; Snijders, P.J.; Peto, J.; Meijer, C.J.; Muñoz, N. Human papillomavirus is a necessary cause of invasive cervical cancer worldwide. J. Pathol. 1999, 189, 12–19. [Google Scholar] [CrossRef]
- Brechot, C.; Pourcel, C.; Louise, A.; Rain, B.; Tiollais, P. Presence of integrated hepatitis B virus DNA sequences in cellular DNA of human hepatocellular carcinoma. Nature 1980, 286, 533–535. [Google Scholar] [CrossRef]
- Boulakia, C.A.; Chen, G.; Ng, F.W.; Teodoro, J.G.; Branton, P.E.; Nicholson, D.W.; Poirier, G.G.; Shore, G.C. Bcl-2 and adenovirus E1B 19 kDA protein prevent E1A-induced processing of CPP32 and cleavage of poly(ADP-ribose) polymerase. Oncogene 1996, 12, 529–535. [Google Scholar]
- Chiou, S.-K.; Tseng, C.-C.; Rao, L.; White, E. Functional complementation of the adenovirus E1B 19-kilodalton protein with Bcl-2 in the inhibition of apoptosis in infected cells. J. Virol. 1994, 68, 6553–6566. [Google Scholar] [CrossRef] [PubMed]
- Slack, R.S.; El-Bizri, H.; Wong, J.; Belliveau, D.J.; Miller, F.D. A critical temporal requirement for the retinoblastoma protein family during neuronal determination. J. Cell Biol. 1998, 140, 1497–1509. [Google Scholar] [CrossRef]
- Goding, C.R.; Pei, D.; Lu, X. Cancer: Pathological nuclear reprogramming? Nat. Rev. Cancer 2014, 14, 568–573. [Google Scholar] [CrossRef] [PubMed]
- Trosko, J.E. Induction of iPS cells and of cancer stem cells: The stem cell or reprogramming hypothesis of cancer? Anat. Rec. 2014, 297, 161–173. [Google Scholar] [CrossRef] [PubMed]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mendoza, G.; González-Pastor, R.; Sánchez, J.M.; Arce-Cerezo, A.; Quintanilla, M.; Moreno-Bueno, G.; Pujol, A.; Belmar-López, C.; de Martino, A.; Riu, E.; et al. The E1a Adenoviral Gene Upregulates the Yamanaka Factors to Induce Partial Cellular Reprogramming. Cells 2023, 12, 1338. https://doi.org/10.3390/cells12091338
Mendoza G, González-Pastor R, Sánchez JM, Arce-Cerezo A, Quintanilla M, Moreno-Bueno G, Pujol A, Belmar-López C, de Martino A, Riu E, et al. The E1a Adenoviral Gene Upregulates the Yamanaka Factors to Induce Partial Cellular Reprogramming. Cells. 2023; 12(9):1338. https://doi.org/10.3390/cells12091338
Chicago/Turabian StyleMendoza, Gracia, Rebeca González-Pastor, Juan Miguel Sánchez, Altamira Arce-Cerezo, Miguel Quintanilla, Gema Moreno-Bueno, Anna Pujol, Carolina Belmar-López, Alba de Martino, Efrén Riu, and et al. 2023. "The E1a Adenoviral Gene Upregulates the Yamanaka Factors to Induce Partial Cellular Reprogramming" Cells 12, no. 9: 1338. https://doi.org/10.3390/cells12091338
APA StyleMendoza, G., González-Pastor, R., Sánchez, J. M., Arce-Cerezo, A., Quintanilla, M., Moreno-Bueno, G., Pujol, A., Belmar-López, C., de Martino, A., Riu, E., Rodriguez, T. A., & Martin-Duque, P. (2023). The E1a Adenoviral Gene Upregulates the Yamanaka Factors to Induce Partial Cellular Reprogramming. Cells, 12(9), 1338. https://doi.org/10.3390/cells12091338