
Retinal Cell Damage in Diabetic Retinopathy


{kind=link}
Abstract
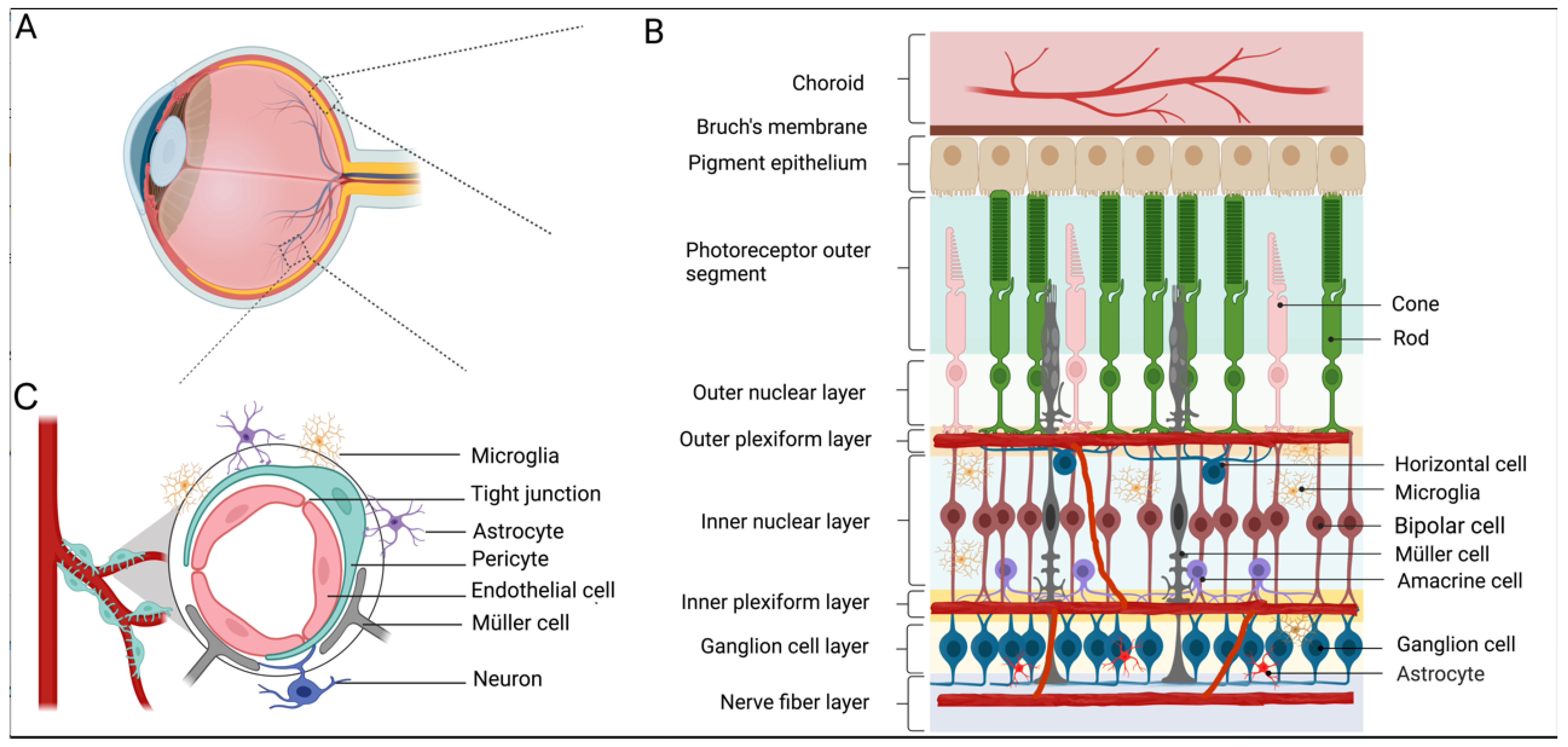
:1. An Overview of Diabetic Retinopathy
2. Diabetic Retinopathy and Retinal Vasculature
3. Neurodegeneration in Diabetic Retinopathy
3.1. RGCs and DR
3.2. Müller Cells and DR
3.3. Photoreceptors and DR
4. Molecular Mechanisms Underlying Neurodegeneration in DR
5. Future Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Cheung, N.; Mitchell, P.; Wong, T.Y. Diabetic retinopathy. Lancet 2010, 376, 124–136. [Google Scholar] [CrossRef] [PubMed]
- Cho, N.H.; Shaw, J.E.; Karuranga, S.; Huang, Y.; da Rocha Fernandes, J.D.; Ohlrogge, A.W.; Malanda, B. IDF Diabetes Atlas: Global estimates of diabetes prevalence for 2017 and projections for 2045. Diabetes Res. Clin. Pract. 2018, 138, 271–281. [Google Scholar] [CrossRef] [PubMed]
- Thomas, R.; Halim, S.; Gurudas, S.; Sivaprasad, S.; Owens, D. IDF Diabetes Atlas: A review of studies utilising retinal photography on the global prevalence of diabetes related retinopathy between 2015 and 2018. Diabetes Res. Clin. Pr. 2019, 157, 107840. [Google Scholar] [CrossRef] [PubMed]
- Kawasaki, R.; Tanaka, S.; Tanaka, S.; Abe, S.; Sone, H.; Yokote, K.; Ishibashi, S.; Katayama, S.; Ohashi, Y.; Akanuma, Y.; et al. Risk of cardiovascular diseases is increased even with mild diabetic retinopathy: The Japan Diabetes Complications Study. Ophthalmology 2013, 120, 574–582. [Google Scholar] [CrossRef] [PubMed]
- Tarr, J.M.; Kaul, K.; Chopra, M.; Kohner, E.M.; Chibber, R. Pathophysiology of Diabetic Retinopathy. ISRN Ophthalmol. 2013, 2013, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Fong, D.S.; Aiello, L.; Gardner, T.W.; King, G.L.; Blankenship, G.; Cavallerano, J.D.; Ferris, F.L.; Klein, R. Diabetic retinopathy. Diabetes Care 2004, 27, 2540–2553. [Google Scholar] [CrossRef] [PubMed]
- Hartnett, M.E.; Baehr, W.; Le, Y.Z. Diabetic retinopathy, an overview. Vision Res. 2017, 139, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Stitt, A.W.; Curtis, T.M.; Chen, M.; Medina, R.J.; McKay, G.J.; Jenkins, A.; Gardiner, T.A.; Lyons, T.J.; Hammes, H.-P.; Simó, R.; et al. The progress in understanding and treatment of diabetic retinopathy. Prog. Retin. Eye Res. 2016, 51, 156–186. [Google Scholar] [CrossRef] [PubMed]
- Klein, R.; Moss, S.E.; Klein, B.E.; Dams, M.D.; DeMets, D.L. The Wisconsin Epidemiologic Study of Diabetic Retinopathy. Ophthalmology 1989, 96, 1501–1510. [Google Scholar] [CrossRef]
- Yau, J.W.Y.; Rogers, S.L.; Kawasaki, R.; Lamoureux, E.L.; Kowalski, J.W.; Bek, T.; Chen, S.-J.; Dekker, J.M.; Fletcher, A.; Grauslund, J.; et al. Global Prevalence and Major Risk Factors of Diabetic Retinopathy. Diabetes Care 2012, 35, 556–564. [Google Scholar] [CrossRef] [PubMed]
- Stewart, M.W.; Browning, D.J.; Lee, C. Diabetic macular edema: Evidence-based management. Indian J. Ophthalmol. 2018, 66, 1736–1750. [Google Scholar] [CrossRef]
- Lai, A.K.W.; Lo, A.C.Y. Animal Models of Diabetic Retinopathy: Summary and Comparison. J. Diabetes Res. 2013, 2013, 1–29. [Google Scholar] [CrossRef]
- Kumar, S.; Singh, R.; Vasudeva, N.; Sharma, S. Acute and chronic animal models for the evaluation of anti-diabetic agents. Cardiovasc. Diabetol. 2012, 11, 9. [Google Scholar] [CrossRef]
- Rakieten, N.; Rakieten, M.L.; Nadkarni, M.V. Studies on the diabetogenic action of streptozotocin (NSC-37917). Cancer Chemother. Rep. 1963, 29, 91–98. [Google Scholar]
- Grossniklaus, H.E.; Kang, S.J.; Berglin, L. Animal models of choroidal and retinal neovascularization. Prog. Retin. Eye Res. 2010, 29, 500–519. [Google Scholar] [CrossRef]
- Sakano, D.; Inoue, A.; Enomoto, T.; Imasaka, M.; Okada, S.; Yokota, M.; Koike, M.; Araki, K.; Kume, S. Insulin2Q104del (Kuma) mutant mice develop diabetes with dominant inheritance. Sci. Rep. 2020, 10, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Nickla, D.L.; Wallman, J. The multifunctional choroid. Prog. Retin. Eye Res. 2010, 29, 144–168. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Smith, L.E.H. Retinal Vasculature in Development and Diseases. Annu. Rev. Vis. Sci. 2018, 4, 101–122. [Google Scholar] [CrossRef] [PubMed]
- Cunha-Vaz, J.G.; Shakib, M.; Ashton, N. Studies on the permeability of the blood-retinal barrier. I. On the existence, development, and site of a blood-retinal barrier. Br. J. Ophthalmol. 1966, 50, 441–453. [Google Scholar] [CrossRef] [PubMed]
- Yu, D.-Y.; Cringle, S.J.; Yu, P.K.; Balaratnasingam, C.; Mehnert, A.; Sarunic, M.V.; An, D.; Su, E.-N. Retinal capillary perfusion: Spatial and temporal heterogeneity. Prog. Retin. Eye Res. 2019, 70, 23–54. [Google Scholar] [CrossRef]
- Usui, Y.; Westenskow, P.D.; Kurihara, T.; Aguilar, E.; Sakimoto, S.; Paris, L.P.; Wittgrove, C.; Feitelberg, D.; Friedlander, M.S.; Moreno, S.K.; et al. Neurovascular crosstalk between interneurons and capillaries is required for vision. J. Clin. Investig. 2015, 125, 2335–2346. [Google Scholar] [CrossRef] [PubMed]
- Metea, M.R.; Newman, E.A. Signalling within the neurovascular unit in the mammalian retina. Exp. Physiol. 2007, 92, 635–640. [Google Scholar] [CrossRef]
- Hawkins, B.; Davis, T. The Blood-Brain Barrier/Neurovascular Unit in Health and Disease. Pharmacol. Rev. 2005, 57, 173–185. [Google Scholar] [CrossRef]
- Kur, J.; Newman, E.A.; Chan-Ling, T. Cellular and physiological mechanisms underlying blood flow regulation in the retina and choroid in health and disease. Prog. Retin. Eye Res. 2012, 31, 377–406. [Google Scholar] [CrossRef] [PubMed]
- Abbott, N.J.; Rönnbäck, L.; Hansson, E. Astrocyte-endothelial interactions at the blood-brain barrier. Nat. Rev. Neurosci. 2006, 7, 41–53. [Google Scholar] [CrossRef] [PubMed]
- Leal, E.C.; Martins, J.; Voabil, P.; Liberal, J.; Chiavaroli, C.; Bauer, J.; Cunha-Vaz, J.; Ambrósio, A.F. Calcium Dobesilate Inhibits the Alterations in Tight Junction Proteins and Leukocyte Adhesion to Retinal Endothelial Cells Induced by Diabetes. Diabetes 2010, 59, 2637–2645. [Google Scholar] [CrossRef] [PubMed]
- Cunha-Vaz, J.; de Abreu, J.R.F.; Campos, A.J. Early breakdown of the blood-retinal barrier in diabetes. Br. J. Ophthalmol. 1975, 59, 649–656. [Google Scholar] [CrossRef]
- Navaratna, D.; McGuire, P.G.; Menicucci, G.; Das, A. Proteolytic Degradation of VE-Cadherin Alters the Blood-Retinal Barrier in Diabetes. Diabetes 2007, 56, 2380–2387. [Google Scholar] [CrossRef]
- Fresta, C.G.; Fidilio, A.; Caruso, G.; Caraci, F.; Giblin, F.J.; Marco Leggio, G.; Salomone, S.; Drago, F.; Bucolo, C. A New Human Blood-Retinal Barrier Model Based on Endothelial Cells, Pericytes, and Astrocytes. Int. J. Mol. Sci. 2020, 21, 1636. [Google Scholar] [CrossRef]
- Heng, L.Z.; Comyn, O.; Peto, T.; Tadros, C.; Ng, E.; Sivaprasad, S.; Hykin, P.G. Diabetic retinopathy: Pathogenesis, clinical grading, management and future developments. Diabet. Med. 2013, 30, 640–650. [Google Scholar] [CrossRef]
- Wolter, J.R. Diabetic retinopathy. Am. J. Ophthalmol. 1961, 51, 1123–1141. [Google Scholar] [CrossRef] [PubMed]
- Sima, A.A.F.; Zhang, W.-X.; Cherian, P.V.; Chakrabarti, S. Impaired visual evoked potential and primary axonopathy of the optic nerve in the diabetic BB/W-rat. Diabetologia 1992, 35, 602–607. [Google Scholar] [CrossRef] [PubMed]
- Kamijo, M.; Cherian, P.V.; Sima, A.A.F. The preventive effect of aldose reductase inhibition on diabetic optic neuropathy in the BB/W-rat. Diabetologia 1993, 36, 893–898. [Google Scholar] [CrossRef] [PubMed]
- Aizu, Y.; Oyanagi, K.; Hu, J.; Nakagawa, H. Degeneration of retinal neuronal processes and pigment epithelium in the early stage of the streptozotocin-diabetic rats. Neuropathology 2002, 22, 161–170. [Google Scholar] [CrossRef]
- Barber, A.; Lieth, E.; Khin, S.A.; Antonetti, D.; Buchanan, A.G.; Gardner, T. Neural apoptosis in the retina during experimental and human diabetes. Early onset and effect of insulin. J. Clin. Investig. 1998, 102, 783–791. [Google Scholar] [CrossRef]
- Qin, Y.; Xu, G.; Wang, W. Dendritic Abnormalities in Retinal Ganglion Cells of Three-Month Diabetic Rats. Curr. Eye Res. 2006, 31, 967–974. [Google Scholar] [CrossRef]
- Fernandez, D.C.; Pasquini, L.A.; Dorfman, D.; Marcos, H.J.A.; Rosenstein, R.E. Early Distal Axonopathy of the Visual Pathway in Experimental Diabetes. Am. J. Pathol. 2012, 180, 303–313. [Google Scholar] [CrossRef]
- Howell, S.J.; Mekhail, M.N.; Azem, R.; Ward, N.L.; Kern, T.S. Degeneration of retinal ganglion cells in diabetic dogs and mice: Relationship to glycemic control and retinal capillary degeneration. Mol. Vis. 2013, 19, 1413–1421. [Google Scholar]
- Simo, R.; Stitt, A.W.; Gardner, T.W. Neurodegeneration in diabetic retinopathy: Does it really matter? Diabetologia 2018, 61, 1902–1912. [Google Scholar] [CrossRef]
- Frydkjaer-Olsen, U.; Hansen, R.S.; Peto, T.; Grauslund, J. Structural neurodegeneration correlates with early diabetic retinopathy. Int. Ophthalmol. 2017, 38, 1621–1626. [Google Scholar] [CrossRef]
- Yang, Q.; Xu, Y.; Xie, P.; Cheng, H.; Song, Q.; Su, T.; Yuan, S.; Liu, Q. Retinal Neurodegeneration in db/db Mice at the Early Period of Diabetes. J. Ophthalmol. 2015, 2015, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Barber, A.J.; Antonetti, D.A.; Kern, T.S.; Reiter, C.E.N.; Soans, R.S.; Krady, J.K.; Levison, S.W.; Gardner, T.W.; Bronson, S.K. The Ins2AkitaMouse as a Model of Early Retinal Complications in Diabetes. Investig. Opthalmology Vis. Sci. 2005, 46, 2210–2218. [Google Scholar] [CrossRef] [PubMed]
- Gastinger, M.J.; Kunselman, A.R.; Conboy, E.E.; Bronson, S.K.; Barber, A.J. Dendrite Remodeling and Other Abnormalities in the Retinal Ganglion Cells of Ins2Akita Diabetic Mice. Investig. Opthalmology Vis. Sci. 2008, 49, 2635–2642. [Google Scholar] [CrossRef]
- Gastinger, M.J.; Singh, R.S.J.; Barber, A.J. Loss of Cholinergic and Dopaminergic Amacrine Cells in Streptozotocin-Diabetic Rat and Ins2Akita-Diabetic Mouse Retinas. Investig. Opthalmology Vis. Sci. 2006, 47, 3143–3150. [Google Scholar] [CrossRef] [PubMed]
- Pitale, P.M.; Saltykova, I.V.; Adu-Agyeiwaah, Y.; Calzi, S.L.; Satoh, T.; Akira, S.; Gorbatyuk, O.; Boulton, M.E.; Pardue, M.T.; Garvey, W.T.; et al. Tribbles Homolog 3 Mediates the Development and Progression of Diabetic Retinopathy. Diabetes 2021, 70, 1738–1753. [Google Scholar] [CrossRef] [PubMed]
- Sohn, E.H.; van Dijk, H.W.; Jiao, C.; Kok, P.H.B.; Jeong, W.; Demirkaya, N.; Garmager, A.; Wit, F.; Kucukevcilioglu, M.; van Velthoven, M.E.J.; et al. Retinal neurodegeneration may precede microvascular changes characteristic of diabetic retinopathy in diabetes mellitus. Proc. Natl. Acad. Sci. USA 2016, 113, E2655–E2664. [Google Scholar] [CrossRef]
- Sergeys, J.; Etienne, I.; Van Hove, I.; Lefevere, E.; Stalmans, I.; Feyen, J.H.M.; Moons, L.; Van Bergen, T. Longitudinal In Vivo Characterization of the Streptozotocin-Induced Diabetic Mouse Model: Focus on Early Inner Retinal Responses. Investig. Opthalmology Vis. Sci. 2019, 60, 807–822. [Google Scholar] [CrossRef]
- Cui, R.-Z.; Wang, L.; Qiao, S.-N.; Wang, Y.-C.; Wang, X.; Yuan, F.; Weng, S.-J.; Yang, X.-L.; Zhong, Y.-M. ON-Type Retinal Ganglion Cells are Preferentially Affected in STZ-Induced Diabetic Mice. Investig. Opthalmology Vis. Sci. 2019, 60, 1644–1656. [Google Scholar] [CrossRef]
- Amato, R.; Lazzara, F.; Chou, T.-H.; Romano, G.L.; Cammalleri, M.; Monte, M.D.; Casini, G.; Porciatti, V. Diabetes Exacerbates the Intraocular Pressure-Independent Retinal Ganglion Cells Degeneration in the DBA/2J Model of Glaucoma. Investig. Opthalmology Vis. Sci. 2021, 62, 9. [Google Scholar] [CrossRef]
- Amato, R.; Catalani, E.; Monte, M.D.; Cammalleri, M.; Cervia, D.; Casini, G. Morpho-functional analysis of the early changes induced in retinal ganglion cells by the onset of diabetic retinopathy: The effects of a neuroprotective strategy. Pharmacol. Res. 2022, 185, 106516. [Google Scholar] [CrossRef]
- Newman, E.; Reichenbach, A. The Müller cell: A functional element of the retina. Trends Neurosci. 1996, 19, 307–312. [Google Scholar] [CrossRef]
- Shen, W.; Fruttiger, M.; Zhu, L.; Chung, S.H.; Barnett, N.L.; Kirk, J.K.; Lee, S.; Coorey, N.J.; Killingsworth, M.; Sherman, L.S.; et al. Conditional Müllercell ablation causes independent neuronal and vascular pathologies in a novel transgenic model. J. Neurosci. 2012, 32, 15715–15727. [Google Scholar] [CrossRef]
- Amaducci, L.; Forno, K.I.; Eng, L.F. Glial fibrillary acidic protein in cryogenic lesions of the rat brain. Neurosci. Lett. 1981, 21, 27–32. [Google Scholar] [CrossRef]
- Rungger-Brändle, E.; Dosso, A.A.; Leuenberger, P.M. Glial reactivity, an early feature of diabetic retinopathy. Investig. Opthalmology Vis. Sci. 2000, 41, 1971–1980. [Google Scholar]
- Barber, A.; Antonetti, D.; Gardner, T. Altered expression of retinal occludin and glial fibrillary acidic protein in experimental diabetes. The Penn State Retina Research Group. Investig. Opthalmology Vis. Sci. 2000, 41, 3561–3568. [Google Scholar]
- Lieth, E.; Barber, A.J.; Xu, B.; Dice, C.; Ratz, M.J.; Tanase, D.; Strother, J.M. Glial reactivity and impaired glutamate metabolism in short-term experimental diabetic retinopathy. Penn State Retina Research Group. Diabetes 1998, 47, 815–820. [Google Scholar] [CrossRef] [PubMed]
- Zhang, R.; Huang, C.; Chen, Y.; Li, T.; Pang, L. Single-cell transcriptomic analysis revealing changes in retinal cell subpopulation levels and the pathways involved in diabetic retinopathy. Ann. Transl. Med. 2022, 10, 562. [Google Scholar] [CrossRef] [PubMed]
- Miller, W.P.; Toro, A.L.; Sunilkumar, S.; Stevens, S.A.; VanCleave, A.M.; Williamson, D.L.; Barber, A.J.; Dennis, M.D. Müller Glial Expression of REDD1 Is Required for Retinal Neurodegeneration and Visual Dysfunction in Diabetic Mice. Diabetes 2022, 71, 1051–1062. [Google Scholar] [CrossRef]
- Okawa, H.; Sampath, A.P.; Laughlin, S.B.; Fain, G.L. ATP Consumption by Mammalian Rod Photoreceptors in Darkness and in Light. Curr. Biol. 2008, 18, 1917–1921. [Google Scholar] [CrossRef]
- Meng, E.C.; Bourne, H.R. Receptor activation: What does the rhodopsin structure tell us? Trends Pharmacol. Sci. 2001, 22, 587–593. [Google Scholar] [CrossRef]
- Vanderkooi, J.M.; Erecinska, M.; Silver, I.A. Oxygen in mammalian tissue: Methods of measurement and affinities of various reactions. Am. J. Physiol. Physiol. 1991, 260, C1131–C1150. [Google Scholar] [CrossRef] [PubMed]
- Ames, A. Energy requirements of CNS cells as related to their function and to their vulnerability to ischemia: A commentary based on studies on retina. Can. J. Physiol. Pharmacol. 1992, 70, S158–S164. [Google Scholar] [CrossRef] [PubMed]
- Arden, G.B.; Sidman, R.L.; Arap, W.; Schlingemann, R.O. Spare the rod and spoil the eye. Br. J. Ophthalmol. 2005, 89, 764–769. [Google Scholar] [CrossRef] [PubMed]
- Havelius, U.; Berglund, S.; Falke, P.; Hindfelt, B.; Krakau, T. Impaired dark adaptation in polycythemia. Improvement after treatment. Acta Ophthalmol. Scand. 2000, 78, 53–57. [Google Scholar] [CrossRef] [PubMed]
- Havelius, U.; Bergqvist, D.; Falke, P.; Hindfelt, B.; Krakau, T.I. Impaired dark adaptation in symptomatic carotid artery disease. Neurology 1997, 49, 1353–1359. [Google Scholar] [CrossRef] [PubMed]
- Havelius, U.; Bergqvist, D.; Hindfelt, B.; Krakau, T. II. Improved dark adaptation after carotid endarterectomy. Neurology 1997, 49, 1360–1364. [Google Scholar] [CrossRef] [PubMed]
- Kinnear, P.R.; Aspinall, P.; Lakowski, R. The diabetic eye and colour vision. Trans. Ophthalmol. Soc. UK 1972, 92, 69–78. [Google Scholar] [PubMed]
- Lakowski, R.; Aspinall, P.A.; Kinnear, P.R. Association between Colour Vision Losses and Diabetes Mellitus. Ophthalmic Res. 1972, 4, 145–159. [Google Scholar] [CrossRef]
- Daley, M.L.; Watzke, R.C.; Riddle, M.C. Early Loss of Blue-Sensitive Color Vision in Patients with Type I Diabetes. Diabetes Care 1987, 10, 777–781. [Google Scholar] [CrossRef]
- Roy, M.S.; Gunkel, R.D.; Podgor, M.J. Color Vision Defects in Early Diabetic Retinopathy. Arch. Ophthalmol. 1986, 104, 225–228. [Google Scholar] [CrossRef]
- Trick, G.L.; Burde, R.M.; Cordon, M.O.; Santiago, J.V.; Kilo, C. The Relationship between Hue Discrimination and Contrast Sensitivity Deficits in Patients with Diabetes Mellitus. Ophthalmology 1988, 95, 693–698. [Google Scholar] [CrossRef] [PubMed]
- Sokol, S.; Moskowitz, A.; Skarf, B.; Evans, R.; Molitch, M.; Senior, B. Contrast Sensitivity in Diabetics with and without Background Retinopathy. Arch. Ophthalmol. 1985, 103, 51–54. [Google Scholar] [CrossRef] [PubMed]
- Park, S.-J.; Park, J.-W.; Kim, K.-Y.; Chung, J.-W.; Chun, M.-H.; Oh, S.-J. Apoptotic death of photoreceptors in the streptozotocin-induced diabetic rat retina. Diabetologia 2003, 46, 1260–1268. [Google Scholar] [CrossRef] [PubMed]
- Tonade, D.; Kern, T.S. Photoreceptor cells and RPE contribute to the development of diabetic retinopathy. Prog. Retin. Eye Res. 2020, 83, 100919. [Google Scholar] [CrossRef] [PubMed]
- Du, Y.; Veenstra, A.; Palczewski, K.; Kern, T.S. Photoreceptor cells are major contributors to diabetes-induced oxidative stress and local inflammation in the retina. Proc. Natl. Acad. Sci. USA 2013, 110, 16586–16591. [Google Scholar] [CrossRef] [PubMed]
- Tonade, D.; Liu, H.; Palczewski, K.; Kern, T.S. Photoreceptor cells produce inflammatory products that contribute to retinal vascular permeability in a mouse model of diabetes. Diabetologia 2017, 60, 2111–2120. [Google Scholar] [CrossRef]
- Tonade, D.; Liu, H.; Kern, T.S. Photoreceptor Cells Produce Inflammatory Mediators That Contribute to Endothelial Cell Death in Diabetes. Investig. Opthalmology Vis. Sci. 2016, 57, 4264–4271. [Google Scholar] [CrossRef]
- de Gooyer, T.E.; Stevenson, K.A.; Humphries, P.; Simpson, D.A.; Gardiner, T.A.; Stitt, A.W. Retinopathy is reduced during experimental diabetes in a mouse model of outer retinal degeneration. Investig. Ophthalmol. Vis. Sci. 2006, 47, 5561–5568. [Google Scholar] [CrossRef]
- Zheng, L.; Du, Y.; Miller, C.; Gubitosi-Klug, R.A.; Kern, T.S.; Ball, S.; Berkowitz, B.A. Critical role of inducible nitric oxide synthase in degeneration of retinal capillaries in mice with streptozotocin-induced diabetes. Diabetologia 2007, 50, 1987–1996. [Google Scholar] [CrossRef]
- Joussen, A.M.; Poulaki, V.; Le, M.L.; Koizumi, K.; Esser, C.; Janicki, H.; Schraermeyer, U.; Kociok, N.; Fauser, S.; Kirchhof, B.; et al. A central role for inflammation in the pathogenesis of diabetic retinopathy. FASEB J. 2004, 18, 1450–1452. [Google Scholar] [CrossRef]
- Scuderi, S.; D’amico, A.G.; Federico, C.; Saccone, S.; Magro, G.; Bucolo, C.; Drago, F.; D’agata, V. Different Retinal Expression Patterns of IL-1α, IL-1β, and Their Receptors in a Rat Model of Type 1 STZ-Induced Diabetes. J. Mol. Neurosci. 2015, 56, 431–439. [Google Scholar] [CrossRef]
- Rochfort, K.D.; Collins, L.E.; McLoughlin, A.; Cummins, P.M. Tumour necrosis factor-α-mediated disruption of cerebrovascular endothelial barrier integrity in vitro involves the production of proinflammatory interleukin-6. J. Neurochem. 2015, 136, 564–572. [Google Scholar] [CrossRef] [PubMed]
- Powell, E.D.; Field, R.A. Diabetic retinopathy and rheumatoid arthritis. Lancet 1964, 2, 17–18. [Google Scholar] [CrossRef] [PubMed]
- Kern, T.S.; Engerman, R.L. Pharmacological inhibition of diabetic retinopathy: Aminoguanidine and aspirin. Diabetes 2001, 50, 1636–1642. [Google Scholar] [CrossRef] [PubMed]
- Joussen, A.M.; Huang, S.; Poulaki, V.; Camphausen, K.; Beecken, W.D.; Kirchhof, B.; Adamis, A.P. In vivo retinal gene expression in early diabetes. Investig. Opthalmology Vis. Sci. 2001, 42, 3047–3057. [Google Scholar]
- Brownlee, M.; Cerami, A.; Vlassara, H. Advanced Glycosylation End Products in Tissue and the Biochemical Basis of Diabetic Complications. N. Engl. J. Med. 1988, 318, 1315–1321. [Google Scholar] [CrossRef]
- Hammes, H.-P. Diabetic retinopathy: Hyperglycaemia, oxidative stress and beyond. Diabetologia 2018, 61, 29–38. [Google Scholar] [CrossRef]
- Lorenzi, M. The Polyol Pathway as a Mechanism for Diabetic Retinopathy: Attractive, Elusive, and Resilient. Exp. Diabetes Res. 2007, 2007, 1–10. [Google Scholar] [CrossRef]
- Ishii, H.; Koya, D.; King, G.L. Protein kinase C activation and its role in the development of vascular complications in diabetes mellitus. J. Mol. Med. 1997, 76, 21–31. [Google Scholar] [CrossRef]
- Mohammad, G.; Kowluru, R.A. The role of Raf-1 kinase in diabetic retinopathy. Expert Opin. Ther. Targets 2011, 15, 357–364. [Google Scholar] [CrossRef]
- Cox, J.T.; Eliott, D.; Sobrin, L. Inflammatory Complications of Intravitreal Anti-VEGF Injections. J. Clin. Med. 2021, 10, 981. [Google Scholar] [CrossRef]
- El-Remessy, A.B.; Al-Shabrawey, M.; Khalifa, Y.; Tsai, N.-T.; Caldwell, R.B.; Liou, G.I. Neuroprotective and Blood-Retinal Barrier-Preserving Effects of Cannabidiol in Experimental Diabetes. Am. J. Pathol. 2006, 168, 235–244. [Google Scholar] [CrossRef] [PubMed]
- Kowluru, R.A.; Mishra, M. Epigenetic regulation of redox signaling in diabetic retinopathy: Role of Nrf2. Free Radic. Biol. Med. 2017, 103, 155–164. [Google Scholar] [CrossRef]
- Poprac, P.; Jomova, K.; Simunkova, M.; Kollar, V.; Rhodes, C.J.; Valko, M. Targeting Free Radicals in Oxidative Stress-Related Human Diseases. Trends Pharmacol. Sci. 2017, 38, 592–607. [Google Scholar] [CrossRef] [PubMed]
- Flaxel, C.J.; Adelman, R.A.; Bailey, S.T.; Fawzi, A.; Lim, J.I.; Vemulakonda, G.A.; Ying, G.-S. Diabetic Retinopathy Preferred Practice Pattern®. Ophthalmology 2019, 127, P66–P145. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.J.; Ehlers, J.P.; Sevgi, D.D.; Hach, J.; O’Connell, M.; Reese, J.L.; Srivastava, S.K.; Wykoff, C.C. Real-Time Photographic- and Fluorescein Angiographic-Guided Management of Diabetic Retinopathy: Randomized Prime Trial Outcomes. Am. J. Ophthalmol. 2021, 226, 126–136. [Google Scholar] [CrossRef] [PubMed]
- Lazzara, F.; Fidilio, A.; Platania, C.B.M.; Giurdanella, G.; Salomone, S.; Leggio, G.M.; Tarallo, V.; Cicatiello, V.; De Falco, S.; Eandi, C.M.; et al. Aflibercept regulates retinal inflammation elicited by high glucose via the PlGF/ERK pathway. Biochem. Pharmacol. 2019, 168, 341–351. [Google Scholar] [CrossRef]
- Maturi, R.K.; Glassman, A.R.; Josic, K.; Antoszyk, A.N.; Blodi, B.A.; Jampol, L.M.; Marcus, D.M.; Martin, D.F.; Melia, M.; Salehi-Had, H.; et al. Effect of Intravitreous Anti-Vascular Endothelial Growth Factor vs. Sham Treatment for Prevention of Vision-Threatening Complications of Diabetic Retinopathy: The Protocol W Randomized Clinical Trial. JAMA Ophthalmol. 2021, 139, 701–712. [Google Scholar] [CrossRef]
- LeBlanc, M.E.; Wang, W.; Chen, X.; Caberoy, N.B.; Guo, F.; Shen, C.; Ji, Y.; Tian, H.; Wang, H.; Chen, R.; et al. Secretogranin III as a disease-associated ligand for antiangiogenic therapy of diabetic retinopathy. J. Exp. Med. 2017, 214, 1029–1047. [Google Scholar] [CrossRef]
- Li, W.; Webster, K.A.; LeBlanc, M.E.; Tian, H. Secretogranin III: A diabetic retinopathy-selective angiogenic factor. Cell. Mol. Life Sci. 2017, 75, 635–647. [Google Scholar] [CrossRef]
- Gehlbach, P.; Demetriades, A.M.; Yamamoto, S.; Deering, T.; Xiao, W.H.; Duh, E.J.; Yang, H.S.; Lai, H.; Kovesdi, I.; Carrion, M.; et al. Periocular gene transfer of sFlt-1 suppresses ocular neovascularization and vascular endothelial growth factor-induced breakdown of the blood-retinal barrier. Hum. Gene Ther. 2003, 14, 129–141. [Google Scholar] [CrossRef] [PubMed]
- Hernández, C.; Monte, M.D.; Simó, R.; Casini, G. Neuroprotection as a Therapeutic Target for Diabetic Retinopathy. J. Diabetes Res. 2016, 2016, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Pearsall, E.A.; Cheng, R.; Matsuzaki, S.; Zhou, K.; Ding, L.; Ahn, B.; Kinter, M.; Humphries, K.M.; Quiambao, A.B.; Farjo, R.A.; et al. Neuroprotective effects of PPARα in retinopathy of type 1 diabetes. PLoS ONE 2019, 14, e0208399. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.; Chen, Y.; Ding, L.; He, X.; Takahashi, Y.; Gao, Y.; Shen, W.; Cheng, R.; Chen, Q.; Qi, X.; et al. Pathogenic role of diabetes-induced PPAR-α down-regulation in microvascular dysfunction. Proc. Natl. Acad. Sci. USA 2013, 110, 15401–15406. [Google Scholar] [CrossRef]
- Bikbova, G.; Oshitari, T.; Baba, T.; Yamamoto, S. Neurotrophic factors for retinal ganglion cell neuropathy—With a special reference to diabetic neuropathy in the retina. Curr. Diabetes Rev. 2014, 10, 166–176. [Google Scholar] [CrossRef]
- Afarid, M.; Namvar, E.; Sanie-Jahromi, F. Diabetic Retinopathy and BDNF: A Review on Its Molecular Basis and Clinical Applications. J. Ophthalmol. 2020, 2020, 1–7. [Google Scholar] [CrossRef]
- McAnany, J.J.; Persidina, O.S.; Park, J.C. Clinical electroretinography in diabetic retinopathy: A review. Surv. Ophthalmol. 2021, 67, 712–722. [Google Scholar] [CrossRef]
- Wu, Z.; Asokan, A.; Samulski, R.J. Adeno-associated Virus Serotypes: Vector Toolkit for Human Gene Therapy. Mol. Ther. 2006, 14, 316–327. [Google Scholar] [CrossRef]
- Smalley, E. First AAV gene therapy poised for landmark approval. Nat. Biotechnol. 2017, 35, 998–999. [Google Scholar] [CrossRef]
- Morrison, C. Landmark gene therapy poised for US approval. Nat. Rev. Drug Discov. 2017, 16, 739–741. [Google Scholar] [CrossRef]
- Zhang, X.; Das, S.K.; Passi, S.F.; Uehara, H.; Bohner, A.; Chen, M.; Tiem, M.; Archer, B.; Ambati, B.K. AAV2 delivery of Flt23k intraceptors inhibits murine choroidal neovascularization. Mol. Ther. J. Am. Soc. Gene Ther. 2015, 23, 226–234. [Google Scholar] [CrossRef] [PubMed]
- Lai, Y.; Shen, W.; Brankov, M.; Lai, C.; Constable, I.; Rakoczy, P. Potential long-term inhibition of ocular neovascularisation by recombinant adeno-associated virus-mediated secretion gene therapy. Gene Ther. 2002, 9, 804–813. [Google Scholar] [CrossRef] [PubMed]
- Bainbridge, J.W.; Mistry, A.; De Alwis, M.; Paleolog, E.; Baker, A.; Thrasher, A.J.; Ali, R.R. Inhibition of retinal neovascularisation by gene transfer of soluble VEGF receptor sFlt-1. Gene Ther. 2002, 9, 320–326. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.; Zhang, L.; Gu, L.; Lu, L.; Gao, G.; Li, W.; Xu, G.; Wang, J.; Gao, F.; Xu, J.-Y.; et al. Subretinal Delivery of AAV2-Mediated Human Erythropoietin Gene Is Protective and Safe in Experimental Diabetic Retinopathy. Investig. Opthalmology Vis. Sci. 2014, 55, 1519–1530. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhou, J.; Chen, B. Retinal Cell Damage in Diabetic Retinopathy. Cells 2023, 12, 1342. https://doi.org/10.3390/cells12091342
Zhou J, Chen B. Retinal Cell Damage in Diabetic Retinopathy. Cells. 2023; 12(9):1342. https://doi.org/10.3390/cells12091342
Chicago/Turabian StyleZhou, Jing, and Bo Chen. 2023. "Retinal Cell Damage in Diabetic Retinopathy" Cells 12, no. 9: 1342. https://doi.org/10.3390/cells12091342
APA StyleZhou, J., & Chen, B. (2023). Retinal Cell Damage in Diabetic Retinopathy. Cells, 12(9), 1342. https://doi.org/10.3390/cells12091342