
Super-Resolution Analysis of the Origins of the Elementary Events of ER Calcium Release in Dorsal Root Ganglion Neurons


{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Microscope Setup
2.2. Isolation and Culture of DRG
2.3. Ca2+ Spark Imaging
2.4. dSTORM Imaging and Primary Processing
2.5. Correlative and Quantitative Image Analysis
2.5.1. Ca2+ Spark Detection
2.5.2. RyR3 and IP3R1 Puncta Localisation
2.5.3. Image Correlation and Alignment of Discretised Ca2+ Sparks and RyR3 Puncta
2.5.4. Local Sampling of RyR3 or IP3R1 Organisation Using Ca2+ Spark Footprint
2.6. Enhanced Expansion Microscopy
2.7. Statistics
3. Results
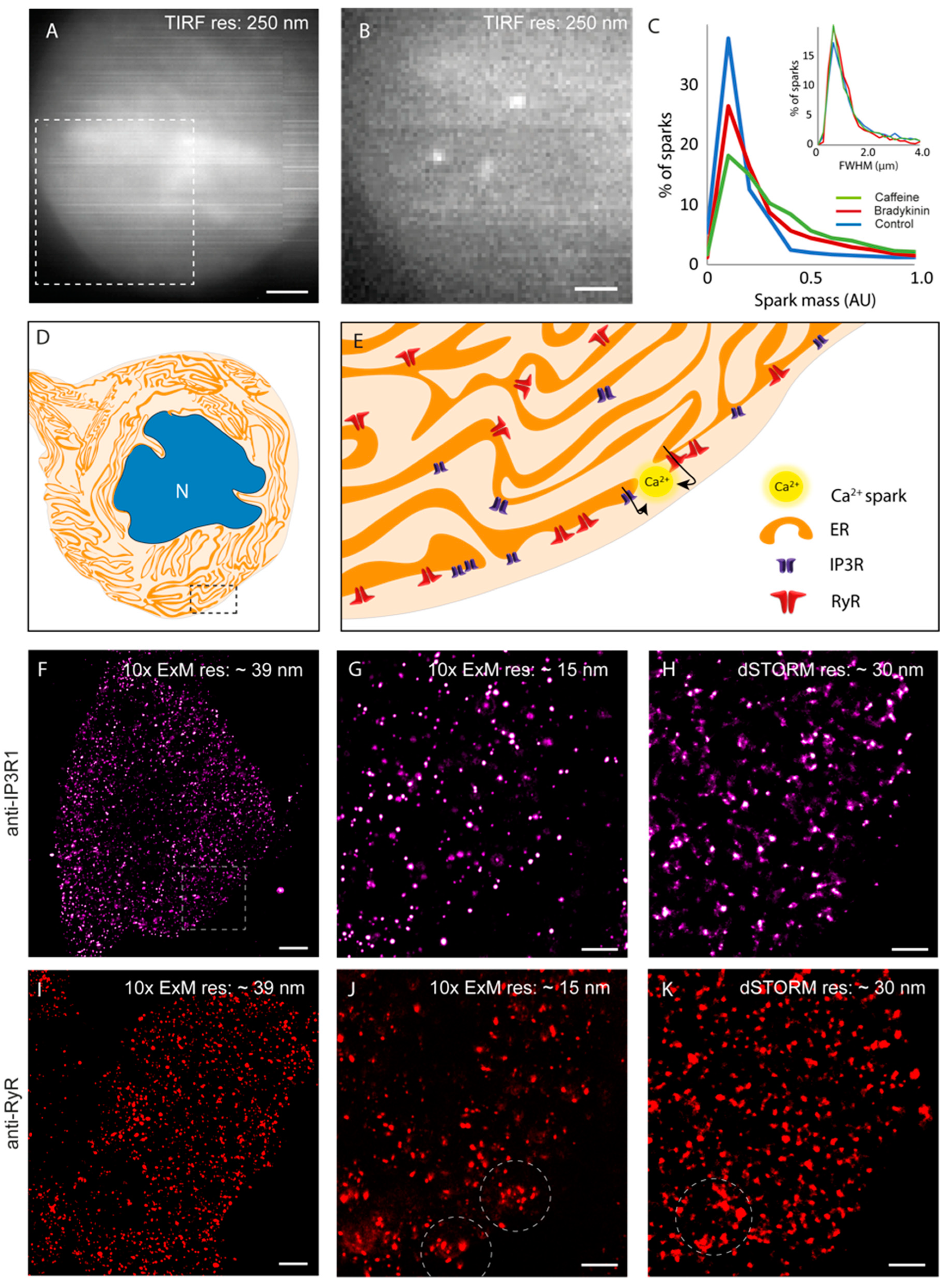
3.1. Optical Visualisation of Fast Ca2+ Sparks and Their Molecular Origins in DRG
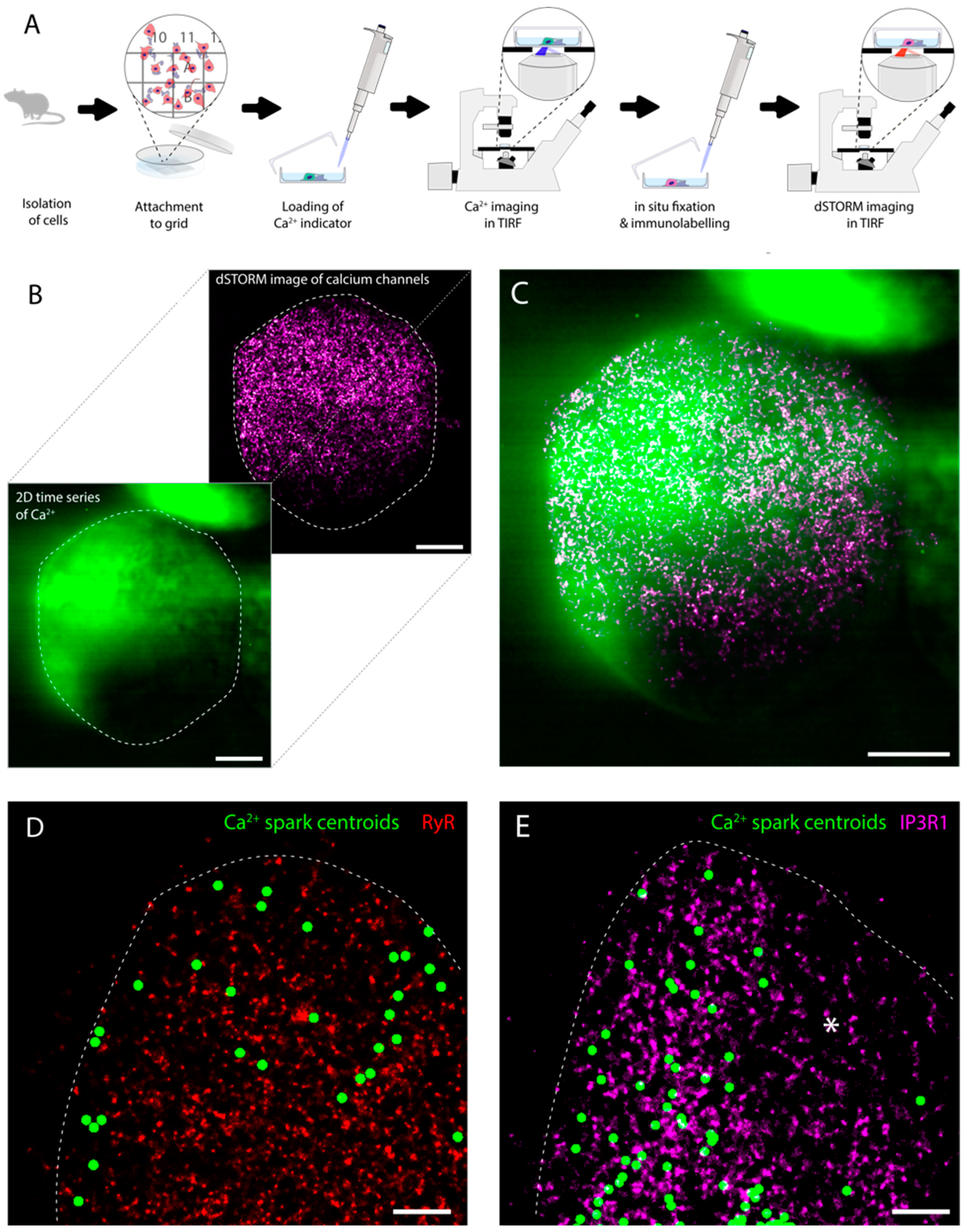
3.2. An Experimental Pipeline to Correlate Ca2+ Sparks with Nanoscale Organisation of RyR3 and IP3R1
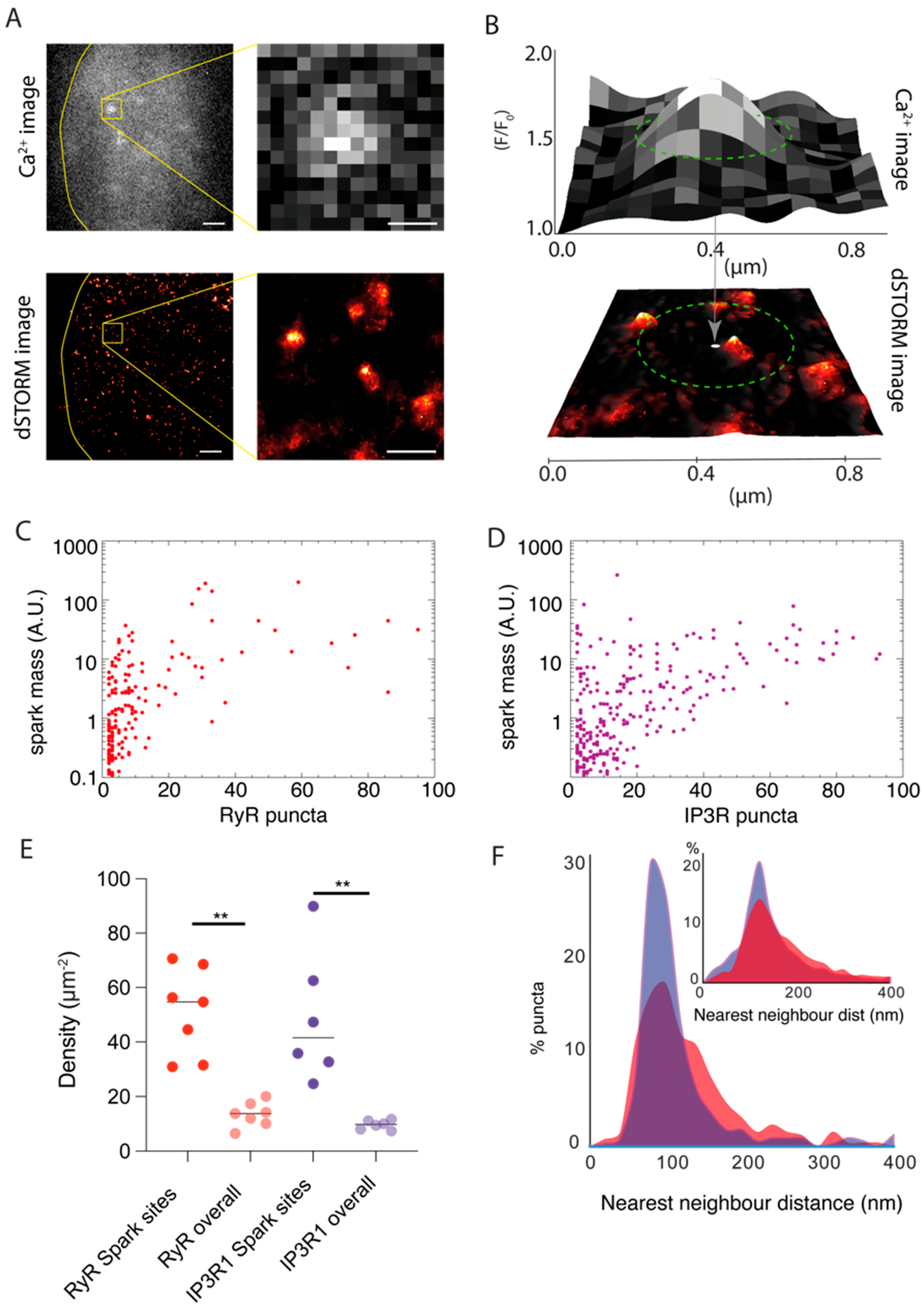
3.3. Local Sampling of RyR3 and IP3R1 Channels Underlying Ca2+ Sparks
4. Discussion
4.1. A Correlative Approach to Probing the Nanoscale Organisation of RyR3 and IP3R1 Underlying Ca2+ Sparks
4.2. Subplasmalemmal Expression Patterns of RyR3 and IP3R1 and Role in the Genesis of Ca2+ Sparks
4.3. Limitations and Uncertainties
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bers, D.M. Cardiac excitation-contraction coupling. Nature 2002, 415, 198–205. [Google Scholar] [CrossRef] [PubMed]
- West, A.E.; Chen, W.G.; Dalva, M.B.; Dolmetsch, R.E.; Kornhauser, J.M.; Shaywitz, A.J.; Takasu, M.A.; Tao, X.; Greenberg, M.E. Calcium regulation of neuronal gene expression. Proc. Natl. Acad. Sci. USA 2001, 98, 11024–11031. [Google Scholar] [CrossRef] [PubMed]
- Miteva, K.T.; Pedicini, L.; Wilson, L.A.; Jayasinghe, I.; Slip, R.G.; Marszalek, K.; Gaunt, H.J.; Bartoli, F.; Deivasigamani, S.; Sobradillo, D.; et al. Rab46 integrates Ca2+ and histamine signaling to regulate selective cargo release from Weibel-Palade bodies. J. Cell Biol. 2019, 218, 2232–2246. [Google Scholar] [CrossRef] [PubMed]
- Ouyang, K.; Zheng, H.; Qin, X.; Zhang, C.; Yang, D.; Wang, X.; Wu, C.; Zhou, Z.; Cheng, H. Ca2+ sparks and secretion in dorsal root ganglion neurons. Proc. Natl. Acad. Sci. USA 2005, 102, 12259–12264. [Google Scholar] [CrossRef]
- Tang, H.; Li, Y.; Wang, S.; Ji, J.; Zhu, X.; Bao, Y.; Huang, C.; Luo, Y.; Huang, L.; Gao, Y.; et al. IP3R-mediated Ca2+ signaling controls B cell proliferation through metabolic reprogramming. iScience 2022, 25, 104209. [Google Scholar] [CrossRef]
- Dong, T.X.; Othy, S.; Greenberg, M.L.; Jairaman, A.; Akunwafo, C.; Leverrier, S.; Yu, Y.; Parker, I.; Dynes, J.L.; Cahalan, M.D. Intermittent Ca2+ signals mediated by Orai1 regulate basal T cell motility. eLife 2017, 6, e27827. [Google Scholar] [CrossRef]
- Bootman, M.D.; Chehab, T.; Bultynck, G.; Parys, J.B.; Rietdorf, K. The regulation of autophagy by calcium signals: Do we have a consensus? Cell Calcium 2018, 70, 32–46. [Google Scholar] [CrossRef]
- Sukumaran, P.; Nascimento Da Conceicao, V.; Sun, Y.; Ahamad, N.; Saraiva, L.R.; Selvaraj, S.; Singh, B.B. Calcium Signaling Regulates Autophagy and Apoptosis. Cells 2021, 10, 2125. [Google Scholar] [CrossRef]
- Cheng, H.; Lederer, W.J.; Cannell, M.B. Calcium sparks: Elementary events underlying excitation-contraction coupling in heart muscle. Science 1993, 262, 740–744. [Google Scholar] [CrossRef]
- Tsugorka, A.; Rios, E.; Blatter, L.A. Imaging elementary events of calcium release in skeletal muscle cells. Science 1995, 269, 1723–1726. [Google Scholar] [CrossRef]
- Parker, I.; Ivorra, I. Localized all-or-none calcium liberation by inositol trisphosphate. Science 1990, 250, 977–979. [Google Scholar] [CrossRef] [PubMed]
- Morton-Jones, R.T.; Cannell, M.B.; Jeyakumar, L.H.; Fleischer, S.; Housley, G.D. Differential expression of ryanodine receptors in the rat cochlea. Neuroscience 2006, 137, 275–286. [Google Scholar] [CrossRef] [PubMed]
- Sharp, A.H.; Nucifora, F.C., Jr.; Blondel, O.; Sheppard, C.A.; Zhang, C.; Snyder, S.H.; Russell, J.T.; Ryugo, D.K.; Ross, C.A. Differential cellular expression of isoforms of inositol 1,4,5-triphosphate receptors in neurons and glia in brain. J. Comp. Neurol. 1999, 406, 207–220. [Google Scholar] [CrossRef]
- Cheng, L.Z.; Lu, N.; Zhang, Y.Q.; Zhao, Z.Q. Ryanodine receptors contribute to the induction of nociceptive input-evoked long-term potentiation in the rat spinal cord slice. Mol. Pain 2010, 6, 1744–8069. [Google Scholar] [CrossRef] [PubMed]
- Ruiz, A.; Matute, C.; Alberdi, E. Endoplasmic reticulum Ca2+ release through ryanodine and IP3 receptors contributes to neuronal excitotoxicity. Cell Calcium 2009, 46, 273–281. [Google Scholar] [CrossRef]
- Pelisch, N.; Gomes, C.; Nally, J.M.; Petruska, J.C.; Stirling, D.P. Differential expression of ryanodine receptor isoforms after spinal cord injury. Neurosci. Lett. 2017, 660, 51–56. [Google Scholar] [CrossRef]
- Jin, X.; Shah, S.; Liu, Y.; Zhang, H.; Lees, M.; Fu, Z.; Lippiat, J.D.; Beech, D.J.; Sivaprasadarao, A.; Baldwin, S.A.; et al. Activation of the Cl− channel ANO1 by localized calcium signals in nociceptive sensory neurons requires coupling with the IP3 receptor. Sci. Signal 2013, 6, ra73. [Google Scholar] [CrossRef]
- Shah, S.; Carver, C.M.; Mullen, P.; Milne, S.; Lukacs, V.; Shapiro, M.S.; Gamper, N. Local Ca2+ signals couple activation of TRPV1 and ANO1 sensory ion channels. Sci. Signal. 2020, 13, eaaw7963. [Google Scholar] [CrossRef]
- Kushnireva, L.; Korkotian, E.; Segal, M. Calcium Sensors STIM1 and STIM2 Regulate Different Calcium Functions in Cultured Hippocampal Neurons. Front. Synaptic Neurosci. 2020, 12, 573714. [Google Scholar] [CrossRef]
- Hogea, A.; Shah, S.; Jones, F.; Carver, C.M.; Hao, H.; Liang, C.; Huang, D.; Du, X.; Gamper, N. Junctophilin-4 facilitates inflammatory signalling at plasma membrane-endoplasmic reticulum junctions in sensory neurons. J. Physiol. 2021, 599, 2103–2123. [Google Scholar] [CrossRef]
- Buijs, T.J.; Vilar, B.; Tan, C.H.; McNaughton, P.A. STIM1 and ORAI1 form a novel cold transduction mechanism in sensory and sympathetic neurons. EMBO J. 2023, 42, e111348. [Google Scholar] [CrossRef] [PubMed]
- Lokuta, A.J.; Komai, H.; McDowell, T.S.; Valdivia, H.H. Functional properties of ryanodine receptors from rat dorsal root ganglia. FEBS Lett. 2002, 511, 90–96. [Google Scholar] [CrossRef]
- Jayasinghe, I.; Clowsley, A.H.; de Langen, O.; Sali, S.S.; Crossman, D.J.; Soeller, C. Shining New Light on the Structural Determinants of Cardiac Couplon Function: Insights From Ten Years of Nanoscale Microscopy. Front. Physiol. 2018, 9, 1472. [Google Scholar] [CrossRef] [PubMed]
- Hurley, M.E.; Sheard, T.M.D.; Norman, R.; Kirton, H.M.; Shah, S.S.; Pervolaraki, E.; Yang, Z.; Gamper, N.; White, E.; Steele, D.; et al. A correlative super-resolution protocol to visualise structural underpinnings of fast second-messenger signalling in primary cell types. Methods 2021, 193, 27–37. [Google Scholar] [CrossRef] [PubMed]
- Marin, Z.; Graff, M.; Barentine, A.E.S.; Soeller, C.; Chung, K.K.H.; Fuentes, L.A.; Baddeley, D. PYMEVisualize: An open-source tool for exploring 3D super-resolution data. Nat. Methods 2021, 18, 582–584. [Google Scholar] [CrossRef] [PubMed]
- Kirton, H.M.; Pettinger, L.; Gamper, N. Transient overexpression of genes in neurons using nucleofection. Methods Mol. Biol. 2013, 998, 55–64. [Google Scholar] [CrossRef]
- Grillo, M.A.; Grillo, S.L.; Gerdes, B.C.; Kraus, J.G.; Koulen, P. Control of Neuronal Ryanodine Receptor-Mediated Calcium Signaling by Calsenilin. Mol. Neurobiol. 2019, 56, 525–534. [Google Scholar] [CrossRef]
- Steele, E.M.; Steele, D.S. Automated detection and analysis of Ca2+ sparks in x-y image stacks using a thresholding algorithm implemented within the open-source image analysis platform ImageJ. Biophys. J. 2014, 106, 566–576. [Google Scholar] [CrossRef]
- Hurley, M.E.; White, E.; Sheard, T.M.D.; Steele, D.; Jayasinghe, I. Correlative super-resolution analysis of cardiac calcium sparks and their molecular origins in health and disease. R. Soc. Open Biol. 2023, 13, 230045. [Google Scholar] [CrossRef]
- Hollingworth, S.; Peet, J.; Chandler, W.K.; Baylor, S.M. Calcium sparks in intact skeletal muscle fibers of the frog. J. Gen. Physiol. 2001, 118, 653–678. [Google Scholar] [CrossRef]
- Sheard, T.M.D.; Hurley, M.E.; Colyer, J.; White, E.; Norman, R.; Pervolaraki, E.; Narayanasamy, K.K.; Hou, Y.; Kirton, H.M.; Yang, Z.; et al. Three-Dimensional and Chemical Mapping of Intracellular Signaling Nanodomains in Health and Disease with Enhanced Expansion Microscopy. ACS Nano 2019, 13, 2143–2157. [Google Scholar] [CrossRef] [PubMed]
- Jin, X.; Amoni, M.; Gilbert, G.; Dries, E.; Donate Puertas, R.; Tomar, A.; Nagaraju, C.K.; Pradhan, A.; Yule, D.I.; Martens, T.; et al. InsP3R-RyR Ca2+ channel crosstalk facilitates arrhythmias in the failing human ventricle. Basic Res. Cardiol. 2022, 117, 60. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Zhang, J.; Sharma, M.R.; Li, P.; Chen, S.R.; Wagenknecht, T. Three-dimensional reconstruction of the recombinant type 3 ryanodine receptor and localization of its amino terminus. Proc. Natl. Acad. Sci. USA 2001, 98, 6104–6109. [Google Scholar] [CrossRef] [PubMed]
- Perni, S.; Beam, K. Neuronal junctophilins recruit specific CaV and RyR isoforms to ER-PM junctions and functionally alter CaV2.1 and CaV2.2. eLife 2021, 10, e64249. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Whiteus, C.; Xu, C.S.; Hayworth, K.J.; Weinberg, R.J.; Hess, H.F.; De Camilli, P. Contacts between the endoplasmic reticulum and other membranes in neurons. Proc. Natl. Acad. Sci. USA 2017, 114, E4859–E4867. [Google Scholar] [CrossRef] [PubMed]
- Chung, J.; Tilunaite, A.; Ladd, D.; Hunt, H.; Soeller, C.; Crampin, E.J.; Johnston, S.T.; Roderick, H.L.; Rajagopal, V. IP3R activity increases propensity of RyR-mediated sparks by elevating dyadic [Ca2+]. Math. Biosci. 2022, 355, 108923. [Google Scholar] [CrossRef] [PubMed]
- Ooashi, N.; Futatsugi, A.; Yoshihara, F.; Mikoshiba, K.; Kamiguchi, H. Cell adhesion molecules regulate Ca2+-mediated steering of growth cones via cyclic AMP and ryanodine receptor type 3. J. Cell Biol. 2005, 170, 1159–1167. [Google Scholar] [CrossRef]
- Jung, M.; Dourado, M.; Maksymetz, J.; Jacobson, A.; Laufer, B.I.; Baca, M.; Foreman, O.; Hackos, D.H.; Riol-Blanco, L.; Kaminker, J.S. Cross-species transcriptomic atlas of dorsal root ganglia reveals species-specific programs for sensory function. Nat. Commun. 2023, 14, 366. [Google Scholar] [CrossRef]
- Kupari, J.; Usoskin, D.; Parisien, M.; Lou, D.; Hu, Y.; Fatt, M.; Lonnerberg, P.; Spangberg, M.; Eriksson, B.; Barkas, N.; et al. Single cell transcriptomics of primate sensory neurons identifies cell types associated with chronic pain. Nat. Commun. 2021, 12, 1510. [Google Scholar] [CrossRef]
- Nguyen, M.Q.; von Buchholtz, L.J.; Reker, A.N.; Ryba, N.J.; Davidson, S. Single-nucleus transcriptomic analysis of human dorsal root ganglion neurons. eLife 2021, 10, e71752. [Google Scholar] [CrossRef]
- Katona, M.; Bartok, A.; Nichtova, Z.; Csordas, G.; Berezhnaya, E.; Weaver, D.; Ghosh, A.; Varnai, P.; Yule, D.I.; Hajnoczky, G. Capture at the ER-mitochondrial contacts licenses IP3 receptors to stimulate local Ca2+ transfer and oxidative metabolism. Nat. Commun. 2022, 13, 6779. [Google Scholar] [CrossRef] [PubMed]
- Hiess, F.; Detampel, P.; Nolla-Colomer, C.; Vallmitjana, A.; Ganguly, A.; Amrein, M.; Ter Keurs, H.; Benitez, R.; Hove-Madsen, L.; Chen, S.R.W. Dynamic and Irregular Distribution of RyR2 Clusters in the Periphery of Live Ventricular Myocytes. Biophys. J. 2018, 114, 343–354. [Google Scholar] [CrossRef] [PubMed]
- Sage, D.; Unser, M. Teaching image-processing programming in Java. IEEE Signal Process. Mag. 2003, 20, 43–52. [Google Scholar] [CrossRef]
- Nieuwenhuizen, R.P.; Lidke, K.A.; Bates, M.; Puig, D.L.; Grunwald, D.; Stallinga, S.; Rieger, B. Measuring image resolution in optical nanoscopy. Nat. Methods 2013, 10, 557–562. [Google Scholar] [CrossRef]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hurley, M.E.; Shah, S.S.; Sheard, T.M.D.; Kirton, H.M.; Steele, D.S.; Gamper, N.; Jayasinghe, I. Super-Resolution Analysis of the Origins of the Elementary Events of ER Calcium Release in Dorsal Root Ganglion Neurons. Cells 2024, 13, 38. https://doi.org/10.3390/cells13010038
Hurley ME, Shah SS, Sheard TMD, Kirton HM, Steele DS, Gamper N, Jayasinghe I. Super-Resolution Analysis of the Origins of the Elementary Events of ER Calcium Release in Dorsal Root Ganglion Neurons. Cells. 2024; 13(1):38. https://doi.org/10.3390/cells13010038
Chicago/Turabian StyleHurley, Miriam E., Shihab S. Shah, Thomas M. D. Sheard, Hannah M. Kirton, Derek S. Steele, Nikita Gamper, and Izzy Jayasinghe. 2024. "Super-Resolution Analysis of the Origins of the Elementary Events of ER Calcium Release in Dorsal Root Ganglion Neurons" Cells 13, no. 1: 38. https://doi.org/10.3390/cells13010038
APA StyleHurley, M. E., Shah, S. S., Sheard, T. M. D., Kirton, H. M., Steele, D. S., Gamper, N., & Jayasinghe, I. (2024). Super-Resolution Analysis of the Origins of the Elementary Events of ER Calcium Release in Dorsal Root Ganglion Neurons. Cells, 13(1), 38. https://doi.org/10.3390/cells13010038