
Dysregulation of Lymphatic Endothelial VEGFR3 Signaling in Disease

 , ,
, ,  and
and
Abstract
:1. Introduction
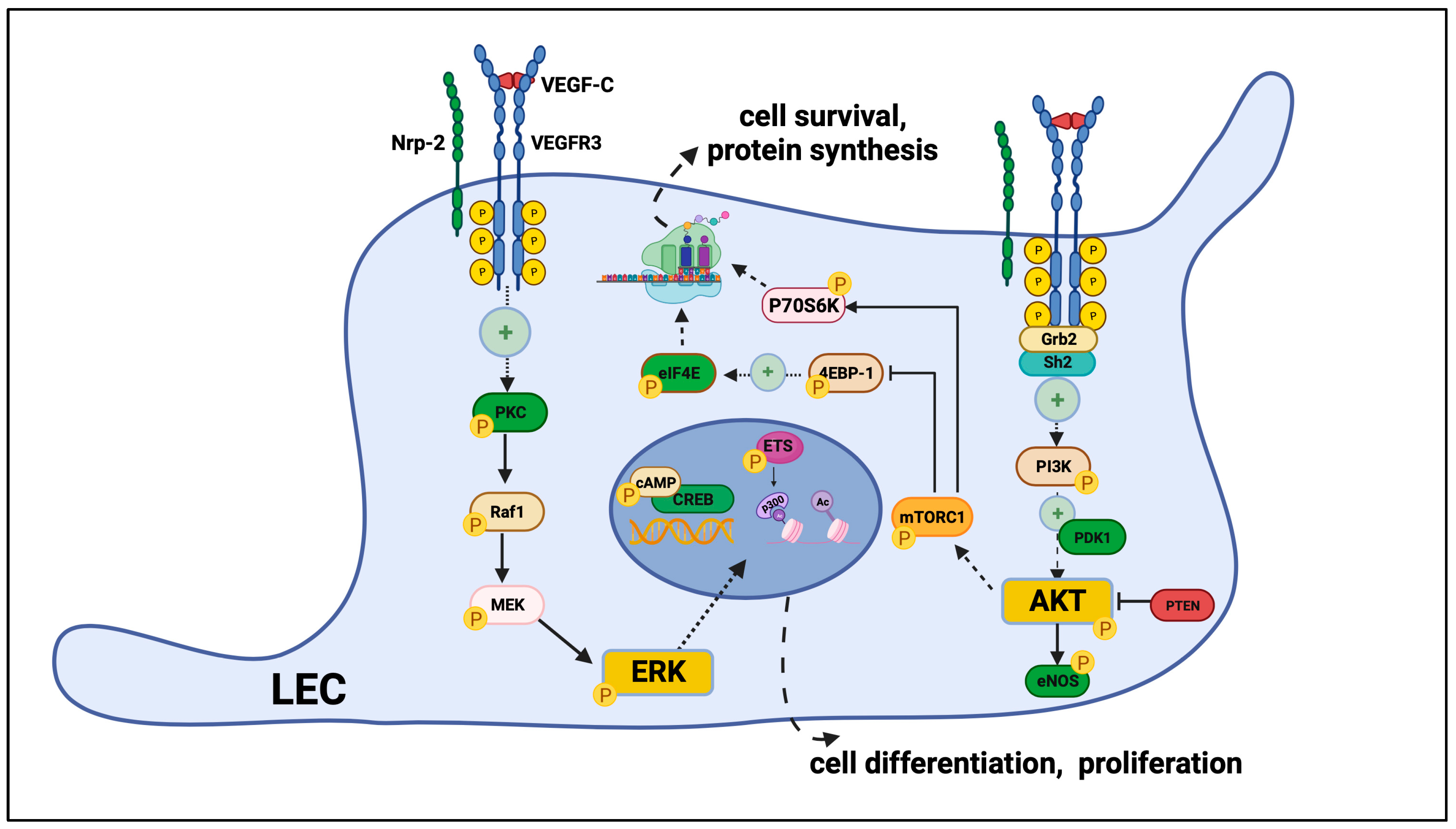
VEGFR3 Signaling in Lymphatic Endothelial Cells
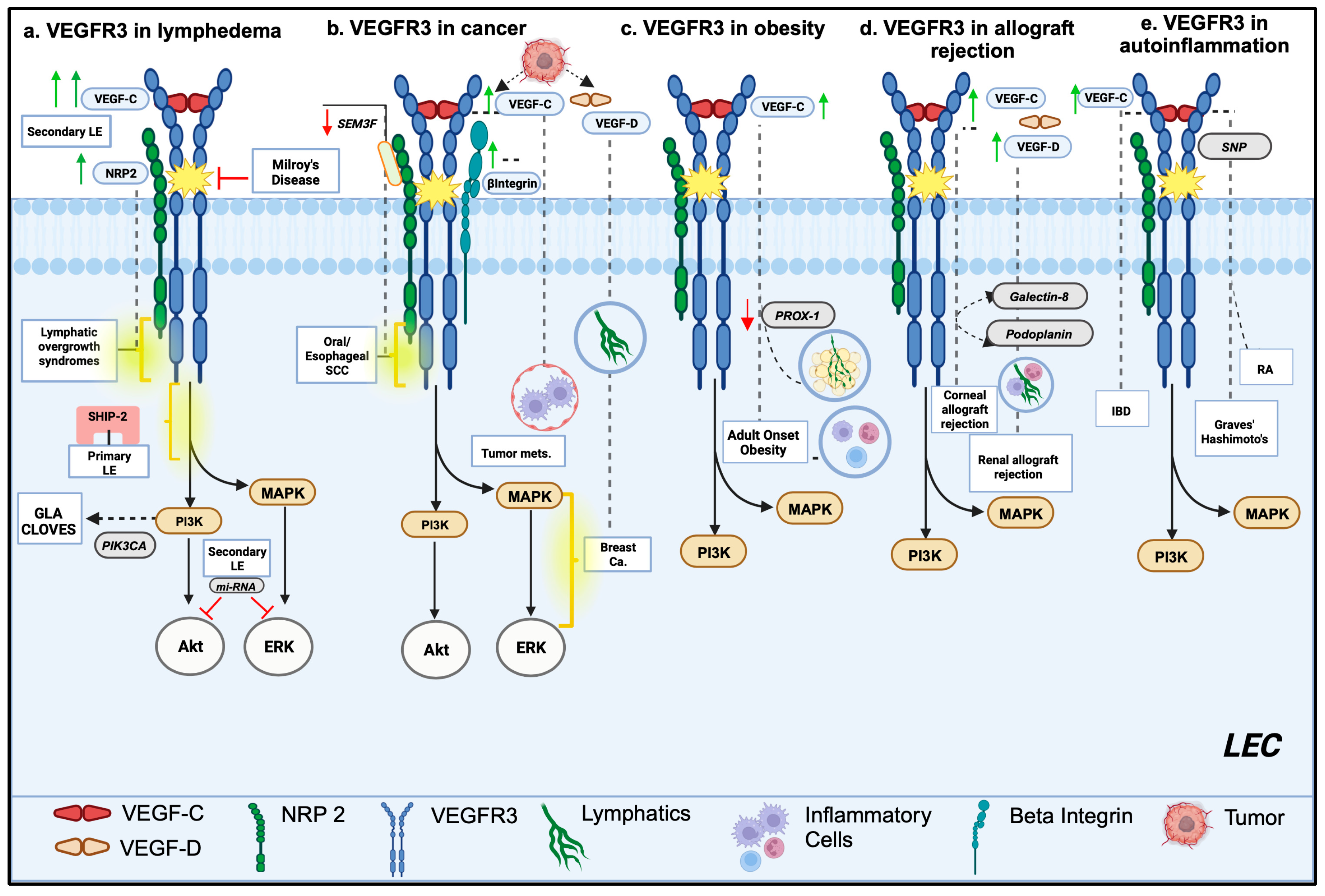
2. Lymphedema and Other Lymphatic Anomalies
2.1. Primary Lymphedema and Other Primary Lymphatic Disorders
2.2. Secondary Lymphedema
3. Tumor Growth and Metastatic Environment
4. Obesity and Metabolic Syndrome
5. Transplant Allograft Rejection
6. Autoimmune and Autoinflammatory Disorders
7. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Petrova, T.V.; Koh, G.Y. Biological functions of lymphatic vessels. Science 2020, 369, eaax4063. [Google Scholar] [CrossRef] [PubMed]
- Wong, B.W.; Zecchin, A.; García-Caballero, M.; Carmeliet, P. Emerging Concepts in Organ-Specific Lymphatic Vessels and Metabolic Regulation of Lymphatic Development. Dev. Cell 2018, 45, 289–301. [Google Scholar] [CrossRef] [PubMed]
- Petrova, T.V.; Koh, G.Y. Organ-specific lymphatic vasculature: From development to pathophysiology. J. Exp. Med. 2018, 215, 35–49. [Google Scholar] [CrossRef] [PubMed]
- Galland, F.; Karamysheva, A.; Pebusque, M.J.; Borg, J.P.; Rottapel, R.; Dubreuil, P.; Rosnet, O.; Birnbaum, D. The FLT4 gene encodes a transmembrane tyrosine kinase related to the vascular endothelial growth factor receptor. Oncogene 1993, 8, 1233–1240. [Google Scholar]
- Kukk, E.; Lymboussaki, A.; Taira, S.; Kaipainen, A.; Jeltsch, M.; Joukov, V.; Alitalo, K. VEGF-C receptor binding and pattern of expression with VEGFR-3 suggests a role in lymphatic vascular development. Development 1996, 122, 3829–3837. [Google Scholar] [CrossRef]
- Zhang, L.; Zhou, F.; Han, W.; Shen, B.; Luo, J.; Shibuya, M.; He, Y. VEGFR-3 ligand-binding and kinase activity are required for lymphangiogenesis but not for angiogenesis. Cell Res. 2010, 20, 1319–1331. [Google Scholar] [CrossRef]
- Salameh, A.; Galvagni, F.; Bardelli, M.; Bussolino, F.; Oliviero, S. Direct recruitment of CRK and GRB2 to VEGFR-3 induces proliferation, migration, and survival of endothelial cells through the activation of ERK, AKT, and JNK pathways. Blood 2005, 106, 3423–3431. [Google Scholar] [CrossRef]
- Wang, J.F.; Zhang, X.; Groopman, J.E. Activation of vascular endothelial growth factor receptor-3 and its downstream signaling promote cell survival under oxidative stress. J. Biol. Chem. 2004, 279, 27088–27097. [Google Scholar] [CrossRef]
- Deng, Y.; Zhang, X.; Simons, M. Molecular controls of lymphatic VEGFR3 signaling. Arterioscler. Thromb. Vasc. Biol. 2015, 35, 421–429. [Google Scholar] [CrossRef]
- Mäkinen, T.; Veikkola, T.; Mustjoki, S.; Karpanen, T.; Catimel, B.; Nice, E.C.; Wise, L.; Mercer, A.; Kowalski, H.; Kerjaschki, D.; et al. Isolated lymphatic endothelial cells transduce growth, survival and migratory signals via the VEGF-C/D receptor VEGFR-3. EMBO J. 2001, 20, 4762–4773. [Google Scholar] [CrossRef]
- Zhou, F.; Chang, Z.; Zhang, L.; Hong, Y.K.; Shen, B.; Wang, B.; Zhang, F.; Lu, G.; Tvorogov, D.; Alitalo, K.; et al. Akt/Protein kinase B is required for lymphatic network formation, remodeling, and valve development. Am. J. Pathol. 2010, 177, 2124–2133. [Google Scholar] [CrossRef] [PubMed]
- Deng, Y.; Atri, D.; Eichmann, A.; Simons, M. Endothelial ERK signaling controls lymphatic fate specification. J. Clin. Investig. 2013, 123, 1202–1215. [Google Scholar] [CrossRef] [PubMed]
- Coso, S.; Zeng, Y.; Opeskin, K.; Williams, E.D. Vascular endothelial growth factor receptor-3 directly interacts with phosphatidylinositol 3-kinase to regulate lymphangiogenesis. PLoS ONE 2012, 7, e39558. [Google Scholar] [CrossRef] [PubMed]
- Harris, T.K. PDK1 and PKB/Akt: Ideal targets for development of new strategies to structure-based drug design. IUBMB Life 2003, 55, 117–126. [Google Scholar] [CrossRef] [PubMed]
- Lahdenranta, J.; Hagendoorn, J.; Padera, T.P.; Hoshida, T.; Nelson, G.; Kashiwagi, S.; Jain, R.K.; Fukumura, D. Endothelial nitric oxide synthase mediates lymphangiogenesis and lymphatic metastasis. Cancer Res. 2009, 69, 2801–2808. [Google Scholar] [CrossRef]
- Huber, S.; Bruns, C.J.; Schmid, G.; Hermann, P.C.; Conrad, C.; Niess, H.; Huss, R.; Graeb, C.; Jauch, K.W.; Heeschen, C.; et al. Inhibition of the mammalian target of rapamycin impedes lymphangiogenesis. Kidney Int. 2007, 71, 771–777. [Google Scholar] [CrossRef]
- Luo, Y.; Liu, L.; Rogers, D.; Su, W.; Odaka, Y.; Zhou, H.; Chen, W.; Shen, T.; Alexander, J.S.; Huang, S. Rapamycin inhibits lymphatic endothelial cell tube formation by downregulating vascular endothelial growth factor receptor 3 protein expression. Neoplasia 2012, 14, 228–237. [Google Scholar] [CrossRef]
- Primo, L.; di Blasio, L.; Roca, C.; Droetto, S.; Piva, R.; Schaffhausen, B.; Bussolino, F. Essential role of PDK1 in regulating endothelial cell migration. J. Cell Biol. 2007, 176, 1035–1047. [Google Scholar] [CrossRef]
- Kataru, R.P.; Baik, J.E.; Park, H.J.; Ly, C.L.; Shin, J.; Schwartz, N.; Lu, T.T.; Ortega, S.; Mehrara, B.J. Lymphatic-specific intracellular modulation of receptor tyrosine kinase signaling improves lymphatic growth and function. Sci. Signal. 2021, 14, eabc0836. [Google Scholar] [CrossRef]
- Bui, K.; Hong, Y.K. Ras Pathways on Prox1 and Lymphangiogenesis: Insights for Therapeutics. Front. Cardiovasc. Med. 2020, 7, 597374. [Google Scholar] [CrossRef]
- Ichise, T.; Yoshida, N.; Ichise, H. Ras/MAPK signaling modulates VEGFR-3 expression through Ets-mediated p300 recruitment and histone acetylation on the Vegfr3 gene in lymphatic endothelial cells. PLoS ONE 2012, 7, e51639. [Google Scholar] [CrossRef]
- Takahashi, T.; Ueno, H.; Shibuya, M. VEGF activates protein kinase C-dependent, but Ras-independent Raf-MEK-MAP kinase pathway for DNA synthesis in primary endothelial cells. Oncogene 1999, 18, 2221–2230. [Google Scholar] [CrossRef] [PubMed]
- Lapinski, P.E.; Kwon, S.; Lubeck, B.A.; Wilkinson, J.E.; Srinivasan, R.S.; Sevick-Muraca, E.; King, P.D. RASA1 maintains the lymphatic vasculature in a quiescent functional state in mice. J. Clin. Investig. 2012, 122, 733–747. [Google Scholar] [CrossRef] [PubMed]
- Datta, S.R.; Brunet, A.; Greenberg, M.E. Cellular survival: A play in three Akts. Genes Dev. 1999, 13, 2905–2927. [Google Scholar] [CrossRef] [PubMed]
- Kandel, E.S.; Hay, N. The regulation and activities of the multifunctional serine/threonine kinase Akt/PKB. Exp. Cell Res. 1999, 253, 210–229. [Google Scholar] [CrossRef] [PubMed]
- Ren, B.; Deng, Y.; Mukhopadhyay, A.; Lanahan, A.A.; Zhuang, Z.W.; Moodie, K.L.; Mulligan-Kehoe, M.J.; Byzova, T.V.; Peterson, R.T.; Simons, M. ERK1/2-Akt1 crosstalk regulates arteriogenesis in mice and zebrafish. J. Clin. Investig. 2010, 120, 1217–1228. [Google Scholar] [CrossRef] [PubMed]
- Uchida, Y.; James, J.M.; Suto, F.; Mukouyama, Y.S. Class 3 semaphorins negatively regulate dermal lymphatic network formation. Biol. Open 2015, 4, 1194–1205. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Yuan, L.; Mak, J.; Pardanaud, L.; Caunt, M.; Kasman, I.; Larrivée, B.; Del Toro, R.; Suchting, S.; Medvinsky, A.; et al. Neuropilin-2 mediates VEGF-C-induced lymphatic sprouting together with VEGFR3. J. Cell Biol. 2010, 188, 115–130. [Google Scholar] [CrossRef]
- Parker, M.W.; Linkugel, A.D.; Goel, H.L.; Wu, T.; Mercurio, A.M.; Vander Kooi, C.W. Structural basis for VEGF-C binding to neuropilin-2 and sequestration by a soluble splice form. Structure 2015, 23, 677–687. [Google Scholar] [CrossRef]
- Wang, Y.; Nakayama, M.; Pitulescu, M.E.; Schmidt, T.S.; Bochenek, M.L.; Sakakibara, A.; Adams, S.; Davy, A.; Deutsch, U.; Lüthi, U.; et al. Ephrin-B2 controls VEGF-induced angiogenesis and lymphangiogenesis. Nature 2010, 465, 483–486. [Google Scholar] [CrossRef]
- Wu, B.; Rockel, J.S.; Lagares, D.; Kapoor, M. Ephrins and Eph Receptor Signaling in Tissue Repair and Fibrosis. Curr. Rheumatol. Rep. 2019, 21, 23. [Google Scholar] [CrossRef] [PubMed]
- Galvagni, F.; Pennacchini, S.; Salameh, A.; Rocchigiani, M.; Neri, F.; Orlandini, M.; Petraglia, F.; Gotta, S.; Sardone, G.L.; Matteucci, G.; et al. Endothelial cell adhesion to the extracellular matrix induces c-Src-dependent VEGFR-3 phosphorylation without the activation of the receptor intrinsic kinase activity. Circ. Res. 2010, 106, 1839–1848. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Groopman, J.E.; Wang, J.F. Extracellular matrix regulates endothelial functions through interaction of VEGFR-3 and integrin alpha5beta1. J. Cell. Physiol. 2005, 202, 205–214. [Google Scholar] [CrossRef] [PubMed]
- Kumaravel, S.; Abbey, C.A.; Bayless, K.J.; Chakraborty, S. The β(1)-integrin plays a key role in LEC invasion in an optimized 3-D collagen matrix model. Am. J. Physiol. Cell Physiol. 2020, 319, C1045–C1058. [Google Scholar] [CrossRef] [PubMed]
- Yin, X.; Johns, S.C.; Lawrence, R.; Xu, D.; Reddi, K.; Bishop, J.R.; Varner, J.A.; Fuster, M.M. Lymphatic endothelial heparan sulfate deficiency results in altered growth responses to vascular endothelial growth factor-C (VEGF-C). J. Biol. Chem. 2011, 286, 14952–14962. [Google Scholar] [CrossRef]
- Sun, M.; Puri, S.; Mutoji, K.N.; Coulson-Thomas, Y.M.; Hascall, V.C.; Jackson, D.G.; Gesteira, T.F.; Coulson-Thomas, V.J. Hyaluronan Derived From the Limbus is a Key Regulator of Corneal Lymphangiogenesis. Investig. Ophthalmol. Vis. Sci. 2019, 60, 1050–1062. [Google Scholar] [CrossRef]
- Tan, K.W.; Chong, S.Z.; Angeli, V. Inflammatory lymphangiogenesis: Cellular mediators and functional implications. Angiogenesis 2014, 17, 373–381. [Google Scholar] [CrossRef]
- Kim, H.; Kataru, R.P.; Koh, G.Y. Regulation and implications of inflammatory lymphangiogenesis. Trends Immunol. 2012, 33, 350–356. [Google Scholar] [CrossRef]
- Wu, M.; Matar, D.Y.; Yu, Z.; Chen, Z.; Knoedler, S.; Ng, B.; Darwish, O.; Haug, V.; Friedman, L.; Orgill, D.P.; et al. Modulation of Lymphangiogenesis in Incisional Murine Diabetic Wound Healing using Negative Pressure Wound Therapy. Adv. Wound Care 2022, 12, 483–497. [Google Scholar] [CrossRef]
- Yang, Y.; García-Verdugo, J.M.; Soriano-Navarro, M.; Srinivasan, R.S.; Scallan, J.P.; Singh, M.K.; Epstein, J.A.; Oliver, G. Lymphatic endothelial progenitors bud from the cardinal vein and intersomitic vessels in mammalian embryos. Blood 2012, 120, 2340–2348. [Google Scholar] [CrossRef]
- Oliver, G. Lymphatic endothelial cell fate specification in the mammalian embryo: An historical perspective. Dev. Biol. 2022, 482, 44–54. [Google Scholar] [CrossRef] [PubMed]
- Karkkainen, M.J.; Haiko, P.; Sainio, K.; Partanen, J.; Taipale, J.; Petrova, T.V.; Jeltsch, M.; Jackson, D.G.; Talikka, M.; Rauvala, H.; et al. Vascular endothelial growth factor C is required for sprouting of the first lymphatic vessels from embryonic veins. Nat. Immunol. 2004, 5, 74–80. [Google Scholar] [CrossRef] [PubMed]
- Oliver, G.; Srinivasan, R.S. Endothelial cell plasticity: How to become and remain a lymphatic endothelial cell. Development 2010, 137, 363–372. [Google Scholar] [CrossRef] [PubMed]
- Jerafi-Vider, A.; Bassi, I.; Moshe, N.; Tevet, Y.; Hen, G.; Splittstoesser, D.; Shin, M.; Lawson, N.D.; Yaniv, K. VEGFC/FLT4-induced cell-cycle arrest mediates sprouting and differentiation of venous and lymphatic endothelial cells. Cell Rep. 2021, 35, 109255. [Google Scholar] [CrossRef] [PubMed]
- Srinivasan, R.S.; Escobedo, N.; Yang, Y.; Interiano, A.; Dillard, M.E.; Finkelstein, D.; Mukatira, S.; Gil, H.J.; Nurmi, H.; Alitalo, K.; et al. The Prox1-Vegfr3 feedback loop maintains the identity and the number of lymphatic endothelial cell progenitors. Genes Dev. 2014, 28, 2175–2187. [Google Scholar] [CrossRef] [PubMed]
- Francois, M.; Oszmiana, A.; Harvey, N.L. When form meets function: The cells and signals that shape the lymphatic vasculature during development. Development 2021, 148, dev167098. [Google Scholar] [CrossRef] [PubMed]
- Gordon, K.; Spiden, S.L.; Connell, F.C.; Brice, G.; Cottrell, S.; Short, J.; Taylor, R.; Jeffery, S.; Mortimer, P.S.; Mansour, S.; et al. FLT4/VEGFR3 and Milroy disease: Novel mutations, a review of published variants and database update. Hum. Mutat. 2013, 34, 23–31. [Google Scholar] [CrossRef]
- Irrthum, A.; Karkkainen, M.J.; Devriendt, K.; Alitalo, K.; Vikkula, M. Congenital hereditary lymphedema caused by a mutation that inactivates VEGFR3 tyrosine kinase. Am. J. Hum. Genet. 2000, 67, 295–301. [Google Scholar] [CrossRef]
- Karkkainen, M.J.; Ferrell, R.E.; Lawrence, E.C.; Kimak, M.A.; Levinson, K.L.; McTigue, M.A.; Alitalo, K.; Finegold, D.N. Missense mutations interfere with VEGFR-3 signalling in primary lymphoedema. Nat. Genet. 2000, 25, 153–159. [Google Scholar] [CrossRef]
- Ferrell, R.E.; Levinson, K.L.; Esman, J.H.; Kimak, M.A.; Lawrence, E.C.; Barmada, M.M.; Finegold, D.N. Hereditary lymphedema: Evidence for linkage and genetic heterogeneity. Hum. Mol. Genet. 1998, 7, 2073–2078. [Google Scholar] [CrossRef]
- Gordon, K.; Schulte, D.; Brice, G.; Simpson, M.A.; Roukens, M.G.; van Impel, A.; Connell, F.; Kalidas, K.; Jeffery, S.; Mortimer, P.S.; et al. Mutation in vascular endothelial growth factor-C, a ligand for vascular endothelial growth factor receptor-3, is associated with autosomal dominant milroy-like primary lymphedema. Circ. Res. 2013, 112, 956–960. [Google Scholar] [CrossRef] [PubMed]
- Balboa-Beltran, E.; Fernández-Seara, M.J.; Pérez-Muñuzuri, A.; Lago, R.; García-Magán, C.; Couce, M.L.; Sobrino, B.; Amigo, J.; Carracedo, A.; Barros, F. A novel stop mutation in the vascular endothelial growth factor-C gene (VEGFC) results in Milroy-like disease. J. Med. Genet. 2014, 51, 475–478. [Google Scholar] [CrossRef] [PubMed]
- Le Guen, L.; Karpanen, T.; Schulte, D.; Harris, N.C.; Koltowska, K.; Roukens, G.; Bower, N.I.; van Impel, A.; Stacker, S.A.; Achen, M.G.; et al. Ccbe1 regulates Vegfc-mediated induction of Vegfr3 signaling during embryonic lymphangiogenesis. Development 2014, 141, 1239–1249. [Google Scholar] [CrossRef] [PubMed]
- Frosk, P.; Chodirker, B.; Simard, L.; El-Matary, W.; Hanlon-Dearman, A.; Schwartzentruber, J.; Majewski, J.; Rockman-Greenberg, C. A novel CCBE1 mutation leading to a mild form of hennekam syndrome: Case report and review of the literature. BMC Med. Genet. 2015, 16, 28. [Google Scholar] [CrossRef]
- Jeltsch, M.; Jha, S.K.; Tvorogov, D.; Anisimov, A.; Leppänen, V.M.; Holopainen, T.; Kivelä, R.; Ortega, S.; Kärpanen, T.; Alitalo, K. CCBE1 enhances lymphangiogenesis via A disintegrin and metalloprotease with thrombospondin motifs-3-mediated vascular endothelial growth factor-C activation. Circulation 2014, 129, 1962–1971. [Google Scholar] [CrossRef]
- Brouillard, P.; Dupont, L.; Helaers, R.; Coulie, R.; Tiller, G.E.; Peeden, J.; Colige, A.; Vikkula, M. Loss of ADAMTS3 activity causes Hennekam lymphangiectasia-lymphedema syndrome 3. Hum. Mol. Genet. 2017, 26, 4095–4104. [Google Scholar] [CrossRef]
- Alders, M.; Al-Gazali, L.; Cordeiro, I.; Dallapiccola, B.; Garavelli, L.; Tuysuz, B.; Salehi, F.; Haagmans, M.A.; Mook, O.R.; Majoie, C.B.; et al. Hennekam syndrome can be caused by FAT4 mutations and be allelic to Van Maldergem syndrome. Hum. Genet. 2014, 133, 1161–1167. [Google Scholar] [CrossRef]
- Ghalamkarpour, A.; Holnthoner, W.; Saharinen, P.; Boon, L.M.; Mulliken, J.B.; Alitalo, K.; Vikkula, M. Recessive primary congenital lymphoedema caused by a VEGFR3 mutation. J. Med. Genet. 2009, 46, 399–404. [Google Scholar] [CrossRef]
- Leppänen, V.M.; Brouillard, P.; Korhonen, E.A.; Sipilä, T.; Jha, S.K.; Revencu, N.; Labarque, V.; Fastré, E.; Schlögel, M.; Ravoet, M.; et al. Characterization of ANGPT2 mutations associated with primary lymphedema. Sci. Transl. Med. 2020, 12, eaax8013. [Google Scholar] [CrossRef]
- Maltese, P.E.; Michelini, S.; Ricci, M.; Maitz, S.; Fiorentino, A.; Serrani, R.; Lazzerotti, A.; Bruson, A.; Paolacci, S.; Benedetti, S.; et al. Increasing evidence of hereditary lymphedema caused by CELSR1 loss-of-function variants. Am. J. Med. Genet. A 2019, 179, 1718–1724. [Google Scholar] [CrossRef]
- Finegold, D.N.; Schacht, V.; Kimak, M.A.; Lawrence, E.C.; Foeldi, E.; Karlsson, J.M.; Baty, C.J.; Ferrell, R.E. HGF and MET mutations in primary and secondary lymphedema. Lymphat. Res. Biol. 2008, 6, 65–68. [Google Scholar] [CrossRef] [PubMed]
- Gordon, K.; Varney, R.; Keeley, V.; Riches, K.; Jeffery, S.; Van Zanten, M.; Mortimer, P.; Ostergaard, P.; Mansour, S. Update and audit of the St George’s classification algorithm of primary lymphatic anomalies: A clinical and molecular approach to diagnosis. J. Med. Genet. 2020, 57, 653–659. [Google Scholar] [CrossRef] [PubMed]
- Ostergaard, P.; Simpson, M.A.; Mendola, A.; Vasudevan, P.; Connell, F.C.; van Impel, A.; Moore, A.T.; Loeys, B.L.; Ghalamkarpour, A.; Onoufriadis, A.; et al. Mutations in KIF11 cause autosomal-dominant microcephaly variably associated with congenital lymphedema and chorioretinopathy. Am. J. Hum. Genet. 2012, 90, 356–362. [Google Scholar] [CrossRef] [PubMed]
- Partanen, T.A.; Vuola, P.; Jauhiainen, S.; Lohi, J.; Salminen, P.; Pitkäranta, A.; Häkkinen, S.K.; Honkonen, K.; Alitalo, K.; Ylä-Herttuala, S. Neuropilin-2 and vascular endothelial growth factor receptor-3 are up-regulated in human vascular malformations. Angiogenesis 2013, 16, 137–146. [Google Scholar] [CrossRef] [PubMed]
- Qin, L.; Zhang, H.; Li, B.; Jiang, Q.; Lopez, F.; Min, W.; Zhou, J.H. CCM3 Loss-Induced Lymphatic Defect Is Mediated by the Augmented VEGFR3-ERK1/2 Signaling. Arterioscler. Thromb. Vasc. Biol. 2021, 41, 2943–2960. [Google Scholar] [CrossRef] [PubMed]
- Monaghan, R.M.; Page, D.J.; Ostergaard, P.; Keavney, B.D. The physiological and pathological functions of VEGFR3 in cardiac and lymphatic development and related diseases. Cardiovasc. Res. 2021, 117, 1877–1890. [Google Scholar] [CrossRef] [PubMed]
- Jin, S.C.; Homsy, J.; Zaidi, S.; Lu, Q.; Morton, S.; DePalma, S.R.; Zeng, X.; Qi, H.; Chang, W.; Sierant, M.C.; et al. Contribution of rare inherited and de novo variants in 2,871 congenital heart disease probands. Nat. Genet. 2017, 49, 1593–1601. [Google Scholar] [CrossRef] [PubMed]
- Reuter, M.S.; Jobling, R.; Chaturvedi, R.R.; Manshaei, R.; Costain, G.; Heung, T.; Curtis, M.; Hosseini, S.M.; Liston, E.; Lowther, C.; et al. Haploinsufficiency of vascular endothelial growth factor related signaling genes is associated with tetralogy of Fallot. Genet. Med. 2019, 21, 1001–1007. [Google Scholar] [CrossRef]
- Page, D.J.; Miossec, M.J.; Williams, S.G.; Monaghan, R.M.; Fotiou, E.; Cordell, H.J.; Sutcliffe, L.; Topf, A.; Bourgey, M.; Bourque, G.; et al. Whole Exome Sequencing Reveals the Major Genetic Contributors to Nonsyndromic Tetralogy of Fallot. Circ. Res. 2019, 124, 553–563. [Google Scholar] [CrossRef]
- Ramirez-Suarez, K.I.; Tierradentro-García, L.O.; Biko, D.M.; Otero, H.J.; White, A.M.; Dori, Y.; Smith, C.L.; Vatsky, S.; Rapp, J.B. Lymphatic anomalies in congenital heart disease. Pediatr. Radiol. 2022, 52, 1862–1876. [Google Scholar] [CrossRef]
- Itkin, M.; Chidekel, A.; Ryan, K.A.; Rabinowitz, D. Abnormal pulmonary lymphatic flow in patients with paediatric pulmonary lymphatic disorders: Diagnosis and treatment. Paediatr. Respir. Rev. 2020, 36, 15–24. [Google Scholar] [CrossRef] [PubMed]
- Schneider, S.; Köllges, R.; Stegmann, J.D.; Thieme, F.; Hilger, A.C.; Waffenschmidt, L.; Fazaal, J.; Kalanithy, J.C.; Geipel, A.; Strizek, B.; et al. Resequencing of VEGFR3 pathway genes implicate GJC2 and FLT4 in the formation of primary congenital chylothorax. Am. J. Med. Genet. A 2022, 188, 1607–1611. [Google Scholar] [CrossRef] [PubMed]
- Agollah, G.D.; Gonzalez-Garay, M.L.; Rasmussen, J.C.; Tan, I.C.; Aldrich, M.B.; Darne, C.; Fife, C.E.; Guilliod, R.; Maus, E.A.; King, P.D.; et al. Evidence for SH2 domain-containing 5’-inositol phosphatase-2 (SHIP2) contributing to a lymphatic dysfunction. PLoS ONE 2014, 9, e112548. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Laguna, L.; Agra, N.; Ibañez, K.; Oliva-Molina, G.; Gordo, G.; Khurana, N.; Hominick, D.; Beato, M.; Colmenero, I.; Herranz, G.; et al. Somatic activating mutations in PIK3CA cause generalized lymphatic anomaly. J. Exp. Med. 2018, 216, 407–418. [Google Scholar] [CrossRef] [PubMed]
- Brouillard, P.; Boon, L.; Vikkula, M. Genetics of lymphatic anomalies. J. Clin. Investig. 2014, 124, 898–904. [Google Scholar] [CrossRef] [PubMed]
- Osborn, A.J.; Dickie, P.; Neilson, D.E.; Glaser, K.; Lynch, K.A.; Gupta, A.; Dickie, B.H. Activating PIK3CA alleles and lymphangiogenic phenotype of lymphatic endothelial cells isolated from lymphatic malformations. Hum. Mol. Genet. 2015, 24, 926–938. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Corral, I.; Zhang, Y.; Petkova, M.; Ortsäter, H.; Sjöberg, S.; Castillo, S.D.; Brouillard, P.; Libbrecht, L.; Saur, D.; Graupera, M.; et al. Blockade of VEGF-C signaling inhibits lymphatic malformations driven by oncogenic PIK3CA mutation. Nat. Commun. 2020, 11, 2869. [Google Scholar] [CrossRef]
- Zhou, X.P.; Marsh, D.J.; Hampel, H.; Mulliken, J.B.; Gimm, O.; Eng, C. Germline and germline mosaic PTEN mutations associated with a Proteus-like syndrome of hemihypertrophy, lower limb asymmetry, arteriovenous malformations and lipomatosis. Hum. Mol. Genet. 2000, 9, 765–768. [Google Scholar] [CrossRef]
- Lindhurst, M.J.; Sapp, J.C.; Teer, J.K.; Johnston, J.J.; Finn, E.M.; Peters, K.; Turner, J.; Cannons, J.L.; Bick, D.; Blakemore, L.; et al. A mosaic activating mutation in AKT1 associated with the Proteus syndrome. N. Engl. J. Med. 2011, 365, 611–619. [Google Scholar] [CrossRef]
- Sevick-Muraca, E.M.; King, P.D. Lymphatic vessel abnormalities arising from disorders of Ras signal transduction. Trends Cardiovasc. Med. 2014, 24, 121–127. [Google Scholar] [CrossRef]
- Nozawa, A.; Ozeki, M.; Niihori, T.; Suzui, N.; Miyazaki, T.; Aoki, Y. A somatic activating KRAS variant identified in an affected lesion of a patient with Gorham-Stout disease. J. Hum. Genet. 2020, 65, 995–1001. [Google Scholar] [CrossRef] [PubMed]
- Homayun-Sepehr, N.; McCarter, A.L.; Helaers, R.; Galant, C.; Boon, L.M.; Brouillard, P.; Vikkula, M.; Dellinger, M.T. KRAS-driven model of Gorham-Stout disease effectively treated with trametinib. JCI Insight 2021, 6, e149831. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; March, M.E.; Gutierrez-Uzquiza, A.; Kao, C.; Seiler, C.; Pinto, E.; Matsuoka, L.S.; Battig, M.R.; Bhoj, E.J.; Wenger, T.L.; et al. ARAF recurrent mutation causes central conducting lymphatic anomaly treatable with a MEK inhibitor. Nat. Med. 2019, 25, 1116–1122. [Google Scholar] [CrossRef] [PubMed]
- Ekvall, S.; Wilbe, M.; Dahlgren, J.; Legius, E.; van Haeringen, A.; Westphal, O.; Annerén, G.; Bondeson, M.L. Mutation in NRAS in familial Noonan syndrome--case report and review of the literature. BMC Med. Genet. 2015, 16, 95. [Google Scholar] [CrossRef] [PubMed]
- Sarkozy, A.; Carta, C.; Moretti, S.; Zampino, G.; Digilio, M.C.; Pantaleoni, F.; Scioletti, A.P.; Esposito, G.; Cordeddu, V.; Lepri, F.; et al. Germline BRAF mutations in Noonan, LEOPARD, and cardiofaciocutaneous syndromes: Molecular diversity and associated phenotypic spectrum. Hum. Mutat. 2009, 30, 695–702. [Google Scholar] [CrossRef]
- Tartaglia, M.; Zampino, G.; Gelb, B.D. Noonan syndrome: Clinical aspects and molecular pathogenesis. Mol. Syndromol. 2010, 1, 2–26. [Google Scholar] [CrossRef]
- Kim, T.; Tafoya, E.; Chelliah, M.P.; Lekwuttikarn, R.; Li, J.; Sarin, K.Y.; Teng, J. Alterations of the MEK/ERK, BMP, and Wnt/β-catenin pathways detected in the blood of individuals with lymphatic malformations. PLoS ONE 2019, 14, e0213872. [Google Scholar] [CrossRef]
- Li, D.; Wenger, T.L.; Seiler, C.; March, M.E.; Gutierrez-Uzquiza, A.; Kao, C.; Bhoj, E.; Tian, L.; Rosenbach, M.; Liu, Y.; et al. Pathogenic variant in EPHB4 results in central conducting lymphatic anomaly. Hum. Mol. Genet. 2018, 27, 3233–3245. [Google Scholar] [CrossRef]
- Kawasaki, J.; Aegerter, S.; Fevurly, R.D.; Mammoto, A.; Mammoto, T.; Sahin, M.; Mably, J.D.; Fishman, S.J.; Chan, J. RASA1 functions in EPHB4 signaling pathway to suppress endothelial mTORC1 activity. J. Clin. Investig. 2014, 124, 2774–2784. [Google Scholar] [CrossRef]
- Miaskowski, C.; Dodd, M.; Paul, S.M.; West, C.; Hamolsky, D.; Abrams, G.; Cooper, B.A.; Elboim, C.; Neuhaus, J.; Schmidt, B.L.; et al. Lymphatic and angiogenic candidate genes predict the development of secondary lymphedema following breast cancer surgery. PLoS ONE 2013, 8, e60164. [Google Scholar] [CrossRef]
- Leung, G.; Baggott, C.; West, C.; Elboim, C.; Paul, S.M.; Cooper, B.A.; Abrams, G.; Dhruva, A.; Schmidt, B.L.; Kober, K.; et al. Cytokine candidate genes predict the development of secondary lymphedema following breast cancer surgery. Lymphat. Res. Biol. 2014, 12, 10–22. [Google Scholar] [CrossRef] [PubMed]
- Hespe, G.E.; Ly, C.L.; Kataru, R.P.; Mehrara, B.J. Baseline Lymphatic Dysfunction Amplifies the Negative Effects of Lymphatic Injury. Plast. Reconstr. Surg. 2019, 143, 77e–87e. [Google Scholar] [CrossRef] [PubMed]
- Jensen, M.R.; Simonsen, L.; Karlsmark, T.; Lanng, C.; Bülow, J. Higher vascular endothelial growth factor-C concentration in plasma is associated with increased forearm capillary filtration capacity in breast cancer-related lymphedema. Physiol. Rep. 2015, 3, e12403. [Google Scholar] [CrossRef] [PubMed]
- Rutkowski, J.M.; Moya, M.; Johannes, J.; Goldman, J.; Swartz, M.A. Secondary lymphedema in the mouse tail: Lymphatic hyperplasia, VEGF-C upregulation, and the protective role of MMP-9. Microvasc. Res. 2006, 72, 161–171. [Google Scholar] [CrossRef] [PubMed]
- Zampell, J.C.; Avraham, T.; Yoder, N.; Fort, N.; Yan, A.; Weitman, E.S.; Mehrara, B.J. Lymphatic function is regulated by a coordinated expression of lymphangiogenic and anti-lymphangiogenic cytokines. Am. J. Physiol. Cell Physiol. 2012, 302, C392–C404. [Google Scholar] [CrossRef] [PubMed]
- Gousopoulos, E.; Proulx, S.T.; Bachmann, S.B.; Dieterich, L.C.; Scholl, J.; Karaman, S.; Bianchi, R.; Detmar, M. An Important Role of VEGF-C in Promoting Lymphedema Development. J. Investig. Dermatol. 2017, 137, 1995–2004. [Google Scholar] [CrossRef] [PubMed]
- Plc, H.P. Herantis Announces Inconclusive Results from Phase II Study with Lymfactin in Breast Cancer Related Lymphedema®; Herantis Pharma Plc.: Espoo, Finland, 2021. [Google Scholar]
- Yusof, K.M.; Groen, K.; Rosli, R.; Avery-Kiejda, K.A. Crosstalk Between microRNAs and the Pathological Features of Secondary Lymphedema. Front. Cell Dev. Biol. 2021, 9, 732415. [Google Scholar] [CrossRef] [PubMed]
- Jones, D.; Li, Y.; He, Y.; Xu, Z.; Chen, H.; Min, W. Mirtron microRNA-1236 inhibits VEGFR-3 signaling during inflammatory lymphangiogenesis. Arterioscler. Thromb. Vasc. Biol. 2012, 32, 633–642. [Google Scholar] [CrossRef]
- Zhou, J.; He, Z.; Guo, L.; Zeng, J.; Liang, P.; Ren, L.; Zhang, M.; Zhang, P.; Huang, X. MiR-128-3p directly targets VEGFC/VEGFR3 to modulate the proliferation of lymphatic endothelial cells through Ca2+ signaling. Int. J. Biochem. Cell Biol. 2018, 102, 51–58. [Google Scholar] [CrossRef]
- Yusof, K.M.; Groen, K.; Rosli, R.; Abdullah, M.; Mahmud, R.; Avery-Kiejda, K.A. Evaluation of Circulating MicroRNAs and Adipokines in Breast Cancer Survivors with Arm Lymphedema. Int. J. Mol. Sci. 2022, 23, 11359. [Google Scholar] [CrossRef]
- Ji, R.C. Lymphatic endothelial cells, tumor lymphangiogenesis and metastasis: New insights into intratumoral and peritumoral lymphatics. Cancer Metastasis Rev. 2006, 25, 677–694. [Google Scholar] [CrossRef] [PubMed]
- Franchi, A.; Gallo, O.; Massi, D.; Baroni, G.; Santucci, M. Tumor lymphangiogenesis in head and neck squamous cell carcinoma: A morphometric study with clinical correlations. Cancer 2004, 101, 973–978. [Google Scholar] [CrossRef] [PubMed]
- Kitadai, Y.; Kodama, M.; Cho, S.; Kuroda, T.; Ochiumi, T.; Kimura, S.; Tanaka, S.; Matsumura, S.; Yasui, W.; Chayama, K. Quantitative analysis of lymphangiogenic markers for predicting metastasis of human gastric carcinoma to lymph nodes. Int. J. Cancer 2005, 115, 388–392. [Google Scholar] [CrossRef] [PubMed]
- Valtola, R.; Salven, P.; Heikkilä, P.; Taipale, J.; Joensuu, H.; Rehn, M.; Pihlajaniemi, T.; Weich, H.; deWaal, R.; Alitalo, K. VEGFR-3 and its ligand VEGF-C are associated with angiogenesis in breast cancer. Am. J. Pathol. 1999, 154, 1381–1390. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.C.; Ni, X.J.; Li, Y.; Dai, M.; Yuan, Z.X.; Zhu, Y.Y.; Luo, C.Y. Peritumoral lymphangiogenesis induced by vascular endothelial growth factor C and D promotes lymph node metastasis in breast cancer patients. World J. Surg. Oncol. 2012, 10, 165. [Google Scholar] [CrossRef] [PubMed]
- Kigure, W.; Fujii, T.; Sutoh, T.; Morita, H.; Katoh, T.; Yajima, R.N.; Yamaguchi, S.; Tsutsumi, S.; Asao, T.; Kuwano, H. The association of VEGF-C expression with tumor lymphatic vessel density and lymph node metastasis in patients with gastric cancer and gastrointestinal stromal tumor. Hepatogastroenterology 2013, 60, 277–280. [Google Scholar] [PubMed]
- Miyahara, M.; Tanuma, J.; Sugihara, K.; Semba, I. Tumor lymphangiogenesis correlates with lymph node metastasis and clinicopathologic parameters in oral squamous cell carcinoma. Cancer 2007, 110, 1287–1294. [Google Scholar] [CrossRef] [PubMed]
- He, M.; He, Q.; Cai, X.; Chen, Z.; Lao, S.; Deng, H.; Liu, X.; Zheng, Y.; Liu, X.; Liu, J.; et al. Role of lymphatic endothelial cells in the tumor microenvironment-a narrative review of recent advances. Transl. Lung Cancer Res. 2021, 10, 2252–2277. [Google Scholar] [CrossRef]
- Cao, R.; Ji, H.; Feng, N.; Zhang, Y.; Yang, X.; Andersson, P.; Sun, Y.; Tritsaris, K.; Hansen, A.J.; Dissing, S.; et al. Collaborative interplay between FGF-2 and VEGF-C promotes lymphangiogenesis and metastasis. Proc. Natl. Acad. Sci. USA 2012, 109, 15894–15899. [Google Scholar] [CrossRef]
- Holopainen, T.; Saharinen, P.; D’Amico, G.; Lampinen, A.; Eklund, L.; Sormunen, R.; Anisimov, A.; Zarkada, G.; Lohela, M.; Heloterä, H.; et al. Effects of angiopoietin-2-blocking antibody on endothelial cell-cell junctions and lung metastasis. J. Natl. Cancer Inst. 2012, 104, 461–475. [Google Scholar] [CrossRef]
- Jeon, B.H.; Jang, C.; Han, J.; Kataru, R.P.; Piao, L.; Jung, K.; Cha, H.J.; Schwendener, R.A.; Jang, K.Y.; Kim, K.S.; et al. Profound but dysfunctional lymphangiogenesis via vascular endothelial growth factor ligands from CD11b+ macrophages in advanced ovarian cancer. Cancer Res. 2008, 68, 1100–1109. [Google Scholar] [CrossRef] [PubMed]
- Volk-Draper, L.; Patel, R.; Bhattarai, N.; Yang, J.; Wilber, A.; DeNardo, D.; Ran, S. Myeloid-Derived Lymphatic Endothelial Cell Progenitors Significantly Contribute to Lymphatic Metastasis in Clinical Breast Cancer. Am. J. Pathol. 2019, 189, 2269–2292. [Google Scholar] [CrossRef] [PubMed]
- Elder, A.M.; Tamburini, B.A.J.; Crump, L.S.; Black, S.A.; Wessells, V.M.; Schedin, P.J.; Borges, V.F.; Lyons, T.R. Semaphorin 7A Promotes Macrophage-Mediated Lymphatic Remodeling during Postpartum Mammary Gland Involution and in Breast Cancer. Cancer Res. 2018, 78, 6473–6485. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Gao, Z.; Sun, M.; Li, H.; Fan, H.; Chen, D.; Zheng, J. Prognostic significance of VEGF-C, semaphorin 3F, and neuropilin-2 expression in oral squamous cell carcinomas and their relationship with lymphangiogenesis. J. Surg. Oncol. 2015, 111, 382–388. [Google Scholar] [CrossRef] [PubMed]
- Xie, Z.; Li, T.; Huang, B.; Liu, S.; Zhang, L.; Zhang, Q. Semaphorin 3F Serves as a Tumor Suppressor in Esophageal Squamous Cell Carcinoma and is Associated With Lymph Node Metastasis in Disease Progression. Technol. Cancer Res. Treat. 2020, 19, 1533033820928117. [Google Scholar] [CrossRef] [PubMed]
- Ernst, B.P.; Mikstas, C.; Stöver, T.; Stauber, R.; Strieth, S. Association of eIF4E and SPARC Expression with Lymphangiogenesis and Lymph Node Metastasis in Hypopharyngeal Cancer. Anticancer Res. 2018, 38, 699–706. [Google Scholar] [CrossRef]
- Lin, Y.C.; Ohbayashi, N.; Hongu, T.; Katagiri, N.; Funakoshi, Y.; Lee, H.; Kanaho, Y. Arf6 in lymphatic endothelial cells regulates lymphangiogenesis by controlling directional cell migration. Sci. Rep. 2017, 7, 11431. [Google Scholar] [CrossRef]
- Karnezis, T.; Farnsworth, R.H.; Harris, N.C.; Williams, S.P.; Caesar, C.; Byrne, D.J.; Herle, P.; Macheda, M.L.; Shayan, R.; Zhang, Y.F.; et al. CCL27/CCL28-CCR10 Chemokine Signaling Mediates Migration of Lymphatic Endothelial Cells. Cancer Res. 2019, 79, 1558–1572. [Google Scholar] [CrossRef]
- Zhuo, W.; Jia, L.; Song, N.; Lu, X.A.; Ding, Y.; Wang, X.; Song, X.; Fu, Y.; Luo, Y. The CXCL12-CXCR4 chemokine pathway: A novel axis regulates lymphangiogenesis. Clin. Cancer Res. 2012, 18, 5387–5398. [Google Scholar] [CrossRef]
- Chen, J.Y.; Lai, Y.S.; Chu, P.Y.; Chan, S.H.; Wang, L.H.; Hung, W.C. Cancer-Derived VEGF-C Increases Chemokine Production in Lymphatic Endothelial Cells to Promote CXCR2-Dependent Cancer Invasion and MDSC Recruitment. Cancers 2019, 11, 1120. [Google Scholar] [CrossRef]
- Tutunea-Fatan, E.; Majumder, M.; Xin, X.; Lala, P.K. The role of CCL21/CCR7 chemokine axis in breast cancer-induced lymphangiogenesis. Mol. Cancer 2015, 14, 35. [Google Scholar] [CrossRef] [PubMed]
- Nykänen, A.I.; Sandelin, H.; Krebs, R.; Keränen, M.A.I.; Tuuminen, R.; Kärpänen, T.; Wu, Y.; Pytowski, B.; Koskinen, P.K.; Ylä-Herttuala, S.; et al. Targeting Lymphatic Vessel Activation and CCL21 Production by Vascular Endothelial Growth Factor Receptor-3 Inhibition Has Novel Immunomodulatory and Antiarteriosclerotic Effects in Cardiac Allografts. Circulation 2010, 121, 1413–1422. [Google Scholar] [CrossRef] [PubMed]
- Van de Velde, M.; Ebroin, M.; Durré, T.; Joiret, M.; Gillot, L.; Blacher, S.; Geris, L.; Kridelka, F.; Noel, A. Tumor exposed-lymphatic endothelial cells promote primary tumor growth via IL6. Cancer Lett. 2021, 497, 154–164. [Google Scholar] [CrossRef] [PubMed]
- Nandi, P.; Girish, G.V.; Majumder, M.; Xin, X.; Tutunea-Fatan, E.; Lala, P.K. PGE2 promotes breast cancer-associated lymphangiogenesis by activation of EP4 receptor on lymphatic endothelial cells. BMC Cancer 2017, 17, 11. [Google Scholar] [CrossRef] [PubMed]
- Lala, P.K.; Nandi, P.; Majumder, M. Roles of prostaglandins in tumor-associated lymphangiogenesis with special reference to breast cancer. Cancer Metastasis Rev. 2018, 37, 369–384. [Google Scholar] [CrossRef] [PubMed]
- Kimura, T.; Sugaya, M.; Oka, T.; Blauvelt, A.; Okochi, H.; Sato, S. Lymphatic dysfunction attenuates tumor immunity through impaired antigen presentation. Oncotarget 2015, 6, 18081–18093. [Google Scholar] [CrossRef] [PubMed]
- Lund, A.W.; Wagner, M.; Fankhauser, M.; Steinskog, E.S.; Broggi, M.A.; Spranger, S.; Gajewski, T.F.; Alitalo, K.; Eikesdal, H.P.; Wiig, H.; et al. Lymphatic vessels regulate immune microenvironments in human and murine melanoma. J. Clin. Investig. 2016, 126, 3389–3402. [Google Scholar] [CrossRef] [PubMed]
- Kataru, R.P.; Ly, C.L.; Shin, J.; Park, H.J.; Baik, J.E.; Rehal, S.; Ortega, S.; Lyden, D.; Mehrara, B.J. Tumor Lymphatic Function Regulates Tumor Inflammatory and Immunosuppressive Microenvironments. Cancer Immunol. Res. 2019, 7, 1345–1358. [Google Scholar] [CrossRef]
- Song, E.; Mao, T.; Dong, H.; Boisserand, L.S.B.; Antila, S.; Bosenberg, M.; Alitalo, K.; Thomas, J.L.; Iwasaki, A. VEGF-C-driven lymphatic drainage enables immunosurveillance of brain tumours. Nature 2020, 577, 689–694. [Google Scholar] [CrossRef]
- Hu, X.; Deng, Q.; Ma, L.; Li, Q.; Chen, Y.; Liao, Y.; Zhou, F.; Zhang, C.; Shao, L.; Feng, J.; et al. Meningeal lymphatic vessels regulate brain tumor drainage and immunity. Cell Res. 2020, 30, 229–243. [Google Scholar] [CrossRef]
- Zhou, C.; Ma, L.; Xu, H.; Huo, Y.; Luo, J. Meningeal lymphatics regulate radiotherapy efficacy through modulating anti-tumor immunity. Cell Res. 2022, 32, 543–554. [Google Scholar] [CrossRef] [PubMed]
- Sasso, M.S.; Mitrousis, N.; Wang, Y.; Briquez, P.S.; Hauert, S.; Ishihara, J.; Hubbell, J.A.; Swartz, M.A. Lymphangiogenesis-inducing vaccines elicit potent and long-lasting T cell immunity against melanomas. Sci. Adv. 2021, 7, eabe4362. [Google Scholar] [CrossRef] [PubMed]
- Bordry, N.; Broggi, M.A.S.; de Jonge, K.; Schaeuble, K.; Gannon, P.O.; Foukas, P.G.; Danenberg, E.; Romano, E.; Baumgaertner, P.; Fankhauser, M.; et al. Lymphatic vessel density is associated with CD8(+) T cell infiltration and immunosuppressive factors in human melanoma. Oncoimmunology 2018, 7, e1462878. [Google Scholar] [CrossRef] [PubMed]
- Lund, A.W.; Duraes, F.V.; Hirosue, S.; Raghavan, V.R.; Nembrini, C.; Thomas, S.N.; Issa, A.; Hugues, S.; Swartz, M.A. VEGF-C promotes immune tolerance in B16 melanomas and cross-presentation of tumor antigen by lymph node lymphatics. Cell Rep. 2012, 1, 191–199. [Google Scholar] [CrossRef]
- Kataru, R.P.; Park, H.J.; Baik, J.E.; Li, C.; Shin, J.; Mehrara, B.J. Regulation of Lymphatic Function in Obesity. Front. Physiol. 2020, 11, 459. [Google Scholar] [CrossRef]
- Escobedo, N.; Oliver, G. The Lymphatic Vasculature: Its Role in Adipose Metabolism and Obesity. Cell Metab. 2017, 26, 598–609. [Google Scholar] [CrossRef]
- Norden, P.R.; Kume, T. The Role of Lymphatic Vascular Function in Metabolic Disorders. Front. Physiol. 2020, 11, 404. [Google Scholar] [CrossRef]
- Nitti, M.D.; Hespe, G.E.; Kataru, R.P.; García Nores, G.D.; Savetsky, I.L.; Torrisi, J.S.; Gardenier, J.C.; Dannenberg, A.J.; Mehrara, B.J. Obesity-induced lymphatic dysfunction is reversible with weight loss. J. Physiol. 2016, 594, 7073–7087. [Google Scholar] [CrossRef]
- Mehrara, B.J.; Greene, A.K. Lymphedema and obesity: Is there a link? Plast. Reconstr. Surg. 2014, 134, 154e–160e. [Google Scholar] [CrossRef]
- Zafar, M.I.; Mills, K.; Ye, X.; Blakely, B.; Min, J.; Kong, W.; Zhang, N.; Gou, L.; Regmi, A.; Hu, S.Q.; et al. Association between the expression of vascular endothelial growth factors and metabolic syndrome or its components: A systematic review and meta-analysis. Diabetol. Metab. Syndr. 2018, 10, 62. [Google Scholar] [CrossRef]
- Gómez-Ambrosi, J.; Catalán, V.; Rodríguez, A.; Ramírez, B.; Silva, C.; Gil, M.J.; Salvador, J.; Frühbeck, G. Involvement of serum vascular endothelial growth factor family members in the development of obesity in mice and humans. J. Nutr. Biochem. 2010, 21, 774–780. [Google Scholar] [CrossRef]
- Wada, H.; Ura, S.; Kitaoka, S.; Satoh-Asahara, N.; Horie, T.; Ono, K.; Takaya, T.; Takanabe-Mori, R.; Akao, M.; Abe, M.; et al. Distinct characteristics of circulating vascular endothelial growth factor-a and C levels in human subjects. PLoS ONE 2011, 6, e29351. [Google Scholar] [CrossRef]
- Rockson, S.G.; Zhou, X.; Zhao, L.; Hosseini, D.K.; Jiang, X.; Sweatt, A.J.; Kim, D.; Tian, W.; Snyder, M.P.; Nicolls, M.R. Exploring disease interrelationships in patients with lymphatic disorders: A single center retrospective experience. Clin. Transl. Med. 2022, 12, e760. [Google Scholar] [CrossRef]
- Harvey, N.L.; Srinivasan, R.S.; Dillard, M.E.; Johnson, N.C.; Witte, M.H.; Boyd, K.; Sleeman, M.W.; Oliver, G. Lymphatic vascular defects promoted by Prox1 haploinsufficiency cause adult-onset obesity. Nat. Genet. 2005, 37, 1072–1081. [Google Scholar] [CrossRef]
- Rutkowski, J.M.; Markhus, C.E.; Gyenge, C.C.; Alitalo, K.; Wiig, H.; Swartz, M.A. Dermal Collagen and Lipid Deposition Correlate with Tissue Swelling and Hydraulic Conductivity in Murine Primary Lymphedema. Am. J. Pathol. 2010, 176, 1122–1129. [Google Scholar] [CrossRef]
- Karkkainen, M.J.; Saaristo, A.; Jussila, L.; Karila, K.A.; Lawrence, E.C.; Pajusola, K.; Bueler, H.; Eichmann, A.; Kauppinen, R.; Kettunen, M.I.; et al. A model for gene therapy of human hereditary lymphedema. Proc. Natl. Acad Sci. USA 2001, 98, 12677–12682. [Google Scholar] [CrossRef]
- Escobedo, N.; Proulx, S.T.; Karaman, S.; Dillard, M.E.; Johnson, N.; Detmar, M.; Oliver, G. Restoration of lymphatic function rescues obesity in Prox1-haploinsufficient mice. JCI Insight 2016, 1, e85096. [Google Scholar] [CrossRef]
- Aschen, S.; Zampell, J.C.; Elhadad, S.; Weitman, E.; De Brot Andrade, M.; Mehrara, B.J. Regulation of adipogenesis by lymphatic fluid stasis: Part II. Expression of adipose differentiation genes. Plast. Reconstr. Surg. 2012, 129, 838–847. [Google Scholar] [CrossRef] [PubMed]
- Chakraborty, A.; Barajas, S.; Lammoglia, G.M.; Reyna, A.J.; Morley, T.S.; Johnson, J.A.; Scherer, P.E.; Rutkowski, J.M. Vascular Endothelial Growth Factor-D (VEGF-D) Overexpression and Lymphatic Expansion in Murine Adipose Tissue Improves Metabolism in Obesity. Am. J. Pathol. 2019, 189, 924–939. [Google Scholar] [CrossRef] [PubMed]
- Nurmi, H.; Saharinen, P.; Zarkada, G.; Zheng, W.; Robciuc, M.R.; Alitalo, K. VEGF-C is required for intestinal lymphatic vessel maintenance and lipid absorption. EMBO Mol. Med. 2015, 7, 1418–1425. [Google Scholar] [CrossRef] [PubMed]
- Martel, C.; Li, W.; Fulp, B.; Platt, A.M.; Gautier, E.L.; Westerterp, M.; Bittman, R.; Tall, A.R.; Chen, S.H.; Thomas, M.J.; et al. Lymphatic vasculature mediates macrophage reverse cholesterol transport in mice. J. Clin. Investig. 2013, 123, 1571–1579. [Google Scholar] [CrossRef] [PubMed]
- Milasan, A.; Smaani, A.; Martel, C. Early rescue of lymphatic function limits atherosclerosis progression in Ldlr(-/-) mice. Atherosclerosis 2019, 283, 106–119. [Google Scholar] [CrossRef] [PubMed]
- Cifarelli, V.; Appak-Baskoy, S.; Peche, V.S.; Kluzak, A.; Shew, T.; Narendran, R.; Pietka, K.M.; Cella, M.; Walls, C.W.; Czepielewski, R.; et al. Visceral obesity and insulin resistance associate with CD36 deletion in lymphatic endothelial cells. Nat. Commun. 2021, 12, 3350. [Google Scholar] [CrossRef] [PubMed]
- Savetsky, I.L.; Torrisi, J.S.; Cuzzone, D.A.; Ghanta, S.; Albano, N.J.; Gardenier, J.C.; Joseph, W.J.; Mehrara, B.J. Obesity increases inflammation and impairs lymphatic function in a mouse model of lymphedema. Am. J. Physiol. Heart Circ. Physiol. 2014, 307, H165–H172. [Google Scholar] [CrossRef]
- Rehal, S.; Kataru, R.P.; Hespe, G.E.; Baik, J.E.; Park, H.J.; Ly, C.; Shin, J.; Mehrara, B.J. Regulation of lymphatic function and injury by nitrosative stress in obese mice. Mol. Metab. 2020, 42, 101081. [Google Scholar] [CrossRef]
- Cao, E.; Watt, M.J.; Nowell, C.J.; Quach, T.; Simpson, J.S.; De Melo Ferreira, V.; Agarwal, S.; Chu, H.; Srivastava, A.; Anderson, D.; et al. Mesenteric lymphatic dysfunction promotes insulin resistance and represents a potential treatment target in obesity. Nat. Metab. 2021, 3, 1175–1188. [Google Scholar] [CrossRef]
- Karaman, S.; Hollmén, M.; Yoon, S.Y.; Alkan, H.F.; Alitalo, K.; Wolfrum, C.; Detmar, M. Transgenic overexpression of VEGF-C induces weight gain and insulin resistance in mice. Sci. Rep. 2016, 6, 31566. [Google Scholar] [CrossRef]
- García Nores, G.D.; Cuzzone, D.A.; Albano, N.J.; Hespe, G.E.; Kataru, R.P.; Torrisi, J.S.; Gardenier, J.C.; Savetsky, I.L.; Aschen, S.Z.; Nitti, M.D.; et al. Obesity but not high-fat diet impairs lymphatic function. Int. J. Obes. 2016, 40, 1582–1590. [Google Scholar] [CrossRef]
- Montgomery, M.K.; Hallahan, N.L.; Brown, S.H.; Liu, M.; Mitchell, T.W.; Cooney, G.J.; Turner, N. Mouse strain-dependent variation in obesity and glucose homeostasis in response to high-fat feeding. Diabetologia 2013, 56, 1129–1139. [Google Scholar] [CrossRef]
- McPherron, A.C.; Lee, S.J. Suppression of body fat accumulation in myostatin-deficient mice. J. Clin. Investig. 2002, 109, 595–601. [Google Scholar] [CrossRef]
- Hou, Y.; Bock, F.; Hos, D.; Cursiefen, C. Lymphatic Trafficking in the Eye: Modulation of Lymphatic Trafficking to Promote Corneal Transplant Survival. Cells 2021, 10, 1661. [Google Scholar] [CrossRef]
- Inomata, T.; Mashaghi, A.; Di Zazzo, A.; Lee, S.M.; Chiang, H.; Dana, R. Kinetics of Angiogenic Responses in Corneal Transplantation. Cornea 2017, 36, 491–496. [Google Scholar] [CrossRef]
- Zhang, H.; Grimaldo, S.; Yuen, D.; Chen, L. Combined blockade of VEGFR-3 and VLA-1 markedly promotes high-risk corneal transplant survival. Investig. Ophthalmol. Vis. Sci. 2011, 52, 6529–6535. [Google Scholar] [CrossRef]
- Hou, Y.; Le, V.N.H.; Tóth, G.; Siebelmann, S.; Horstmann, J.; Gabriel, T.; Bock, F.; Cursiefen, C. UV light crosslinking regresses mature corneal blood and lymphatic vessels and promotes subsequent high-risk corneal transplant survival. Am. J. Transpl. 2018, 18, 2873–2884. [Google Scholar] [CrossRef]
- Hos, D.; Bock, F.; Dietrich, T.; Onderka, J.; Kruse, F.E.; Thierauch, K.H.; Cursiefen, C. Inflammatory corneal (lymph)angiogenesis is blocked by VEGFR-tyrosine kinase inhibitor ZK 261991, resulting in improved graft survival after corneal transplantation. Investig. Ophthalmol. Vis. Sci. 2008, 49, 1836–1842. [Google Scholar] [CrossRef]
- Dietrich, T.; Bock, F.; Yuen, D.; Hos, D.; Bachmann, B.O.; Zahn, G.; Wiegand, S.; Chen, L.; Cursiefen, C. Cutting Edge: Lymphatic Vessels, Not Blood Vessels, Primarily Mediate Immune Rejections After Transplantation. J. Immunol. 2010, 184, 535. [Google Scholar] [CrossRef]
- Chen, W.S.; Cao, Z.; Sugaya, S.; Lopez, M.J.; Sendra, V.G.; Laver, N.; Leffler, H.; Nilsson, U.J.; Fu, J.; Song, J.; et al. Pathological lymphangiogenesis is modulated by galectin-8-dependent crosstalk between podoplanin and integrin-associated VEGFR-3. Nat. Commun. 2016, 7, 11302. [Google Scholar] [CrossRef]
- Lin, J.; Chen, Y.; Zhu, H.; Cheng, K.; Wang, H.; Yu, X.; Tang, M.; Chen, J. Lymphatic Reconstruction in Kidney Allograft Aggravates Chronic Rejection by Promoting Alloantigen Presentation. Front. Immunol. 2021, 12, 796260. [Google Scholar] [CrossRef] [PubMed]
- Palin, N.K.; Savikko, J.; Koskinen, P.K. Sirolimus inhibits lymphangiogenesis in rat renal allografts, a novel mechanism to prevent chronic kidney allograft injury. Transpl. Int. 2013, 26, 195–205. [Google Scholar] [CrossRef] [PubMed]
- Dashkevich, A.; Raissadati, A.; Syrjälä, S.O.; Zarkada, G.; Keränen, M.A.; Tuuminen, R.; Krebs, R.; Anisimov, A.; Jeltsch, M.; Leppänen, V.M.; et al. Ischemia-Reperfusion Injury Enhances Lymphatic Endothelial VEGFR3 and Rejection in Cardiac Allografts. Am. J. Transpl. 2016, 16, 1160–1172. [Google Scholar] [CrossRef] [PubMed]
- Edwards, L.A.; Nowocin, A.K.; Jafari, N.V.; Meader, L.L.; Brown, K.; Sarde, A.; Lam, C.; Murray, A.; Wong, W. Chronic Rejection of Cardiac Allografts Is Associated With Increased Lymphatic Flow and Cellular Trafficking. Circulation 2018, 137, 488–503. [Google Scholar] [CrossRef] [PubMed]
- Iwami, D.; Brinkman, C.C.; Bromberg, J.S. Vascular endothelial growth factor c/vascular endothelial growth factor receptor 3 signaling regulates chemokine gradients and lymphocyte migration from tissues to lymphatics. Transplantation 2015, 99, 668–677. [Google Scholar] [CrossRef] [PubMed]
- Cui, Y.; Liu, K.; Monzon-Medina, M.E.; Padera, R.F.; Wang, H.; George, G.; Toprak, D.; Abdelnour, E.; D’Agostino, E.; Goldberg, H.J.; et al. Therapeutic lymphangiogenesis ameliorates established acute lung allograft rejection. J. Clin. Investig. 2015, 125, 4255–4268. [Google Scholar] [CrossRef] [PubMed]
- Gao, C.; Zhu, J.; Qin, Q.; Yang, X.; Jiang, Y.; Zhang, J. The Relationship between VEGFC Gene Polymorphisms and Autoimmune Thyroiditis. Biomed. Res. Int. 2022, 2022, 2603519. [Google Scholar] [CrossRef] [PubMed]
- Alunno, A.; Ibba-Manneschi, L.; Bistoni, O.; Rosa, I.; Caterbi, S.; Gerli, R.; Manetti, M. Mobilization of lymphatic endothelial precursor cells and lymphatic neovascularization in primary Sjögren’s syndrome. J. Cell. Mol. Med. 2016, 20, 613–622. [Google Scholar] [CrossRef] [PubMed]
- Sertoglu, E.; Yücel, Ç.; Omma, A.; Hayran, Y.; Colak, S.; Sandıkçı, S.C.; Durukan, A.H.; Ozgurtas, T. Determination of serum vascular endothelial growth factor (VEGF) and VEGF receptor levels with VEGF gene polymorphisms in patients with Behçet’s uveitis. Adv. Clin. Exp. Med. 2022, 31, 231–240. [Google Scholar] [CrossRef] [PubMed]
- William, G.A.; Mir, H.; Madhavi Latha, S.C.; Ethan, S.S.; JiHyun, S.; Jinyeon, S.; Noa, S.; William, D.S., III; Dragos, D.; Camila, B.C.; et al. Lymphatic dysfunction in lupus contributes to cutaneous photosensitivity and lymph node B cell responses. bioRxiv, 2022; bioRxiv:2022.06.13.495930. [Google Scholar] [CrossRef]
- Honda, N.; Jinnin, M.; Kajihara, I.; Makino, T.; Fukushima, S.; Ihn, H. Impaired lymphangiogenesis due to excess vascular endothelial growth factor-D/Flt-4 signalling in the skin of patients with systemic sclerosis. Br. J. Dermatol. 2010, 163, 776–780. [Google Scholar] [CrossRef]
- Akhmetshina, A.; Beer, J.; Zwerina, K.; Englbrecht, M.; Palumbo, K.; Dees, C.; Reich, N.; Zwerina, J.; Szucs, G.; Gusinde, J.; et al. Decreased lymphatic vessel counts in patients with systemic sclerosis: Association with fingertip ulcers. Arthritis Rheum. 2010, 62, 1513–1522. [Google Scholar] [CrossRef]
- Manetti, M.; Romano, E.; Rosa, I.; Fioretto, B.S.; Guiducci, S.; Bellando-Randone, S.; Pigatto, E.; Cozzi, F.; Ibba-Manneschi, L.; Matucci-Cerinic, M. Systemic Sclerosis Serum Significantly Impairs the Multi-Step Lymphangiogenic Process: In Vitro Evidence. Int. J. Mol. Sci. 2019, 20, 6189. [Google Scholar] [CrossRef]
- Kunstfeld, R.; Hirakawa, S.; Hong, Y.K.; Schacht, V.; Lange-Asschenfeldt, B.; Velasco, P.; Lin, C.; Fiebiger, E.; Wei, X.; Wu, Y.; et al. Induction of cutaneous delayed-type hypersensitivity reactions in VEGF-A transgenic mice results in chronic skin inflammation associated with persistent lymphatic hyperplasia. Blood 2004, 104, 1048–1057. [Google Scholar] [CrossRef]
- Sundberg, J.P.; Pratt, C.H.; Silva, K.A.; Kennedy, V.E.; Stearns, T.M.; Sundberg, B.A.; King, L.E.; HogenEsch, H. Dermal lymphatic dilation in a mouse model of alopecia areata. Exp. Mol. Pathol. 2016, 100, 332–336. [Google Scholar] [CrossRef] [PubMed]
- Yoon, S.Y.; Dieterich, L.C.; Karaman, S.; Proulx, S.T.; Bachmann, S.B.; Sciaroni, C.; Detmar, M. An important role of cutaneous lymphatic vessels in coordinating and promoting anagen hair follicle growth. PLoS ONE 2019, 14, e0220341. [Google Scholar] [CrossRef] [PubMed]
- Yoon, S.Y.; Detmar, M. Sostdc1 Secreted from Cutaneous Lymphatic Vessels Acts as a Paracrine Factor for Hair Follicle Growth. Curr. Issues Mol. Biol. 2022, 44, 2167–2174. [Google Scholar] [CrossRef] [PubMed]
- Gur-Cohen, S.; Yang, H.; Baksh, S.C.; Miao, Y.; Levorse, J.; Kataru, R.P.; Liu, X.; de la Cruz-Racelis, J.; Mehrara, B.J.; Fuchs, E. Stem cell-driven lymphatic remodeling coordinates tissue regeneration. Science 2019, 366, 1218–1225. [Google Scholar] [CrossRef] [PubMed]
- Dai, C.; Kuo, S.J.; Hu, S.L.; Tsai, C.H.; Huang, Y.L.; Huang, C.C.; Wang, L.; Xu, G.; Su, C.M.; Tang, C.H. VEGF-C Gene Polymorphisms Increase Susceptibility to Rheumatoid Arthritis. Int. J. Med. Sci. 2019, 16, 1397–1403. [Google Scholar] [CrossRef] [PubMed]
- Cha, H.-S.; Bae, E.-K.; Koh, J.-H.; Chai, J.-Y.; Jeon, C.H.; Ahn, K.-S.; Kim, J.; Koh, E.-M. Tumor necrosis factor-alpha induces vascular endothelial growth factor-C expression in rheumatoid synoviocytes. J. Rheumatol. 2007, 34, 16–19. [Google Scholar] [PubMed]
- Bell, R.D.; Rahimi, H.; Kenney, H.M.; Lieberman, A.A.; Wood, R.W.; Schwarz, E.M.; Ritchlin, C.T. Altered Lymphatic Vessel Anatomy and Markedly Diminished Lymph Clearance in Affected Hands of Patients With Active Rheumatoid Arthritis. Arthritis Rheumatol. 2020, 72, 1447–1455. [Google Scholar] [CrossRef]
- Shi, J.X.; Liang, Q.Q.; Wang, Y.J.; Mooney, R.A.; Boyce, B.F.; Xing, L. Use of a whole-slide imaging system to assess the presence and alteration of lymphatic vessels in joint sections of arthritic mice. Biotech. Histochem. 2013, 88, 428–439. [Google Scholar] [CrossRef]
- Guo, R.; Zhou, Q.; Proulx, S.T.; Wood, R.; Ji, R.C.; Ritchlin, C.T.; Pytowski, B.; Zhu, Z.; Wang, Y.J.; Schwarz, E.M.; et al. Inhibition of lymphangiogenesis and lymphatic drainage via vascular endothelial growth factor receptor 3 blockade increases the severity of inflammation in a mouse model of chronic inflammatory arthritis. Arthritis Rheum. 2009, 60, 2666–2676. [Google Scholar] [CrossRef]
- Zhou, Q.; Guo, R.; Wood, R.; Boyce, B.F.; Liang, Q.; Wang, Y.J.; Schwarz, E.M.; Xing, L. Vascular endothelial growth factor C attenuates joint damage in chronic inflammatory arthritis by accelerating local lymphatic drainage in mice. Arthritis Rheum. 2011, 63, 2318–2328. [Google Scholar] [CrossRef]
- Chen, X.; Hu, Q.Y.; Wang, M.; Jia, J.; Teng, J.; Sun, Y.; Cheng, X.; Ye, J.; Su, Y.; Shi, H.; et al. Serum VEGF-C as an evaluation marker of disease activity in adult-onset Still’s disease. Rheumatol. Int. 2022, 42, 149–157. [Google Scholar] [CrossRef] [PubMed]
- Sato, H.; Higashiyama, M.; Hozumi, H.; Sato, S.; Furuhashi, H.; Takajo, T.; Maruta, K.; Yasutake, Y.; Narimatsu, K.; Yoshikawa, K.; et al. Platelet interaction with lymphatics aggravates intestinal inflammation by suppressing lymphangiogenesis. Am. J. Physiol. Gastrointest. Liver Physiol. 2016, 311, G276–G285. [Google Scholar] [CrossRef] [PubMed]
- Rahier, J.F.; De Beauce, S.; Dubuquoy, L.; Erdual, E.; Colombel, J.F.; Jouret-Mourin, A.; Geboes, K.; Desreumaux, P. Increased lymphatic vessel density and lymphangiogenesis in inflammatory bowel disease. Aliment Pharmacol. Ther. 2011, 34, 533–543. [Google Scholar] [CrossRef] [PubMed]
- D’Alessio, S.; Correale, C.; Tacconi, C.; Gandelli, A.; Pietrogrande, G.; Vetrano, S.; Genua, M.; Arena, V.; Spinelli, A.; Peyrin-Biroulet, L.; et al. VEGF-C-dependent stimulation of lymphatic function ameliorates experimental inflammatory bowel disease. J. Clin. Investig. 2014, 124, 3863–3878. [Google Scholar] [CrossRef] [PubMed]
- Ocansey, D.K.W.; Pei, B.; Xu, X.; Zhang, L.; Olovo, C.V.; Mao, F. Cellular and molecular mediators of lymphangiogenesis in inflammatory bowel disease. J. Transl. Med. 2021, 19, 254. [Google Scholar] [CrossRef] [PubMed]
- Jurisic, G.; Sundberg, J.P.; Detmar, M. Blockade of VEGF receptor-3 aggravates inflammatory bowel disease and lymphatic vessel enlargement. Inflamm. Bowel Dis. 2013, 19, 1983–1989. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.L.; Zhao, J.; Qin, L.; Qiao, M. Promoting inflammatory lymphangiogenesis by vascular endothelial growth factor-C (VEGF-C) aggravated intestinal inflammation in mice with experimental acute colitis. Braz. J. Med. Biol. Res. 2016, 49, e4738. [Google Scholar] [CrossRef]
- Becker, F.; Yi, P.; Al-Kofahi, M.; Ganta, V.C.; Morris, J.; Alexander, J.S. Lymphatic dysregulation in intestinal inflammation: New insights into inflammatory bowel disease pathomechanisms. Lymphology 2014, 47, 3–27. [Google Scholar]
- Boucher, J.; Kleinridders, A.; Kahn, C.R. Insulin receptor signaling in normal and insulin-resistant states. Cold Spring Harb. Perspect. Biol. 2014, 6, a009191. [Google Scholar] [CrossRef]
- Ghalamkarpour, A.; Morlot, S.; Raas-Rothschild, A.; Utkus, A.; Mulliken, J.B.; Boon, L.M.; Vikkula, M. Hereditary lymphedema type I associated with VEGFR3 mutation: The first de novo case and atypical presentations. Clin. Genet. 2006, 70, 330–335. [Google Scholar] [CrossRef]
- Keppler-Noreuil, K.M.; Rios, J.J.; Parker, V.E.; Semple, R.K.; Lindhurst, M.J.; Sapp, J.C.; Alomari, A.; Ezaki, M.; Dobyns, W.; Biesecker, L.G. PIK3CA-related overgrowth spectrum (PROS): Diagnostic and testing eligibility criteria, differential diagnosis, and evaluation. Am. J. Med. Genet. A 2015, 167, 287–295. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.; Smith, C.L.; Biko, D.M.; Li, D.; Pinto, E.; O’Connor, N.; Skraban, C.; Zackai, E.H.; Hakonarson, H.; Dori, Y.; et al. Genetics etiologies and genotype phenotype correlations in a cohort of individuals with central conducting lymphatic anomaly. Eur. J. Hum. Genet. 2022, 30, 1022–1028. [Google Scholar] [CrossRef] [PubMed]
- Roy, S.; Kumaravel, S.; Banerjee, P.; White, T.K.; O’Brien, A.; Seelig, C.; Chauhan, R.; Ekser, B.; Bayless, K.J.; Alpini, G.; et al. Tumor Lymphatic Interactions Induce CXCR2-CXCL5 Axis and Alter Cellular Metabolism and Lymphangiogenic Pathways to Promote Cholangiocarcinoma. Cells 2021, 10, 3093. [Google Scholar] [CrossRef] [PubMed]
- Di Tommaso, L.; Battista, S.; Destro, A.; Sciarra, A.; Morenghi, E.; Roncalli, M. Cracking spaces in Hashimoto Thyroiditis are lymphatic and prelymphatic vessels: A gift of immunohistochemistry for the centenary of Hashimoto’s description. Am. J. Surg. Pathol. 2010, 34, 1857–1861. [Google Scholar] [CrossRef]
- Choi, I.; Lee, S.; Kyoung Chung, H.; Suk Lee, Y.; Eui Kim, K.; Choi, D.; Park, E.K.; Yang, D.; Ecoiffier, T.; Monahan, J.; et al. 9-cis retinoic acid promotes lymphangiogenesis and enhances lymphatic vessel regeneration: Therapeutic implications of 9-cis retinoic acid for secondary lymphedema. Circulation 2012, 125, 872–882. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Disease | Clinical Manifestations | Histopathologic Findings | Proposed VEGFR3-Related Molecular Pathology | Selected References |
|---|---|---|---|---|
| Milroy/Milroy-like primary lymphedema | Lower extremity swelling | Hypoplastic lymphatics | VEGF-C, VEGFR3, INPPL1 gene mutations | [48,49,51,52,58,73,202] |
| Hennekam’s syndrome | Generalized lymphedema and lymphangiectasia, variable intellectual disability, characteristic facial dysmorphic features | Lymphatic vessel dysplasia | CCBE-1, ADAMTS3 gene mutations | [54,55,56] |
| Isolated lymphovenous malformations | Congenital lymphatic malformations, cerebral cavernous malformations (CCM) | Hyperplastic lymphatics | Increased cellular VEGFR3, neuropilin-2, ERK 1/2 activity | [64,65] |
| Generalized lymphatic anomaly (GLA) | Diffuse or multi-focal lymphatic malformations; cutaneous/soft tissue edema; chylous thoracic effusions, ascites, lymphorrhea | Hyperproliferative, dilated lymphatics | PIK3CA mutation (hyperactivating) | [74] |
| CLOVES syndrome | Capillary, venous, lymphatic vascular malformations; thoracic lipomatous hyperplasia; asymmetric limb growth | Macrocystic and microcystic malformations, or combined lymphatic lesions w/disorganized channels | PIK3CA mutation (hyperactivating) | [76,203] |
| Proteus syndrome | Cutaneous, Musculoskeletal, and vascular tissue overgrowth lesions | Hyperplastic lymphatics, abnormal lymphovascular channels | AKT1 mutation (hyperactivating) PTEN (inactivating) | [78,79] |
| Gorham–Stout disease | Spontaneous, progressive osteolysis; soft tissue lymphangioma; chylothorax | Proliferative ectopic lymphatics within bony structures | KRAS mutation (activating) | [81,82] |
| Noonan syndrome | Lymphedema of bilateral lower limb and genitals; posterior cervical hygroma | Dilated hyperplastic lymphatics | Ras-Raf, PTPN11, SHP2 mutations | [84,85,86] |
| Central conducting lymphatic anomaly (CCLA) | Abnormal lymphatic drainage by large vessels within the trunk | Dilated channels, central lymphatic obstruction | ARAF, EPHB4 mutations | [83,88,204] |
| Tumor Metastasis | Clinical Manifestations | Histopathologic Findings | Proposed VEGFR3-Related Molecular Pathology | Selected References |
| Breast cancer | Local tissue infiltration and metastasis to distant organs | Peri-tumoral lymphatic growth and tumor invasion of neighboring lymph nodes | PGE2-EP4 axis stimulation, CLEC2A activation, VEGF-C overexpression | [114,125,126] |
| Melanoma | Local tissue infiltration and metastasis to distant organs | Aberrant distribution of melanocytes, Pagetoid spread, dyscohesive nests of melanocytes | ARF6 upregulation, integrin B1 upregulation, k-Cyclin deletion | [118,127,128,133,134,135] |
| Oral SCC | Local tissue infiltration and metastasis to distant organs | Tumor budding, perineural and vascular invasion, sarcolemma spread, tumor-infiltrating T-lymphocytes, CD68+-tumor-associated macrophages, tumor-associated tissue eosinophilia, cellular cannibalism | Neuropilin-2 upregulation, SEMA3F downregulation, VEGF-C overexpression | [108,115] |
| Hypopharyngeal cancer | Local tissue infiltration and metastasis to distant organs | Squamous cell (most common), submucosal tumor extension | EIF4E activation | [117] |
| Esophageal SCC | Local tissue infiltration and metastasis to distant organs | Keratin pearls, individual cell keratinization, intercellular bridges | SEMA3F downregulation | [116] |
| Cholangiocarcinoma | Local tissue infiltration and metastasis to distant organs | Abundant fibrous desmoplastic stroma | CXCR2-CXCL5 axis activation | [205] |
| Autoimmune Disorders | Clinical Manifestations | Histopathologic Findings | Proposed VEGFR3-Related Molecular Pathology | Selected References |
| Autoimmune thyroid disease | Grave’s disease; Hashimoto’s thyroiditis; dysregulated thyroid hormone secretion | Increased lymphatic vessel density | VEGF-C polymorphisms | [175,206] |
| Sjogren’s syndrome | Diminished lacrimal and salivary gland function, xerostomia, keratoconjunctivitis sicca, parotid gland enlargement | Expansion of lymphatic capillaries, periductal inflammatory cell infiltration | VEGFR3, VEGF-C polymorphisms | [176] |
| Behcet’s disease | Mucocutaneous lesions, recurrent genital ulcerations, uveitis, skin lesions | Expansion of lymphatic capillaries, periductal inflammatory cell infiltration | Increased circulating sVEGFR3, dysregulated VEGF-C/ VEGFR3 ratio | [177] |
| Alopecia areata | Discrete, round patches of hair loss | Dilated dermal lymphatics | Elevated vascular endothelial growth factor expression | [183] |
| Systemic lupus erythematosis (SLE) | Progressive multi-organ tissue fibrosis, vascular damage and inflammation | SLE: Dilated cutaneous lymphatic vessels without changes in lymphatic density | Increased VEGF-C, VEGF-D, VEGFR2, VEGFR3 activity | [178] |
| Psoriasis | Well circumscribed, circular, erythematous papules and plaques with dry scaling | Perivascular and dermal inflammatory cell infiltration, vascular dilatation, edema of dermal papillae, parakeratosis | Increased VEGF-A expression, VEGFR2 activation | [182] |
| Rheumatoid arthritis | Systemic polyarthritis, articular destruction | Chronic perilymphatic inflammation, lymphatic leakiness | VEGF-C polymorphisms, impaired VEGF-C/VEGFR3 signaling | [187,188,189,190] |
| Still’s disease | Autoinflammatory fever, arthralgias, lymphadenopathy, joint pain, persistent papules and plaques | Upper keratinocyte dyskeratosis, scattered superficial dermal neutrophils, vacuolar interface changes, apoptotic keratinocytes | Increased VEGF-C expression | [193] |
| Inflammatory bowel disease | Crohn’s disease, ulcerative colitis | Increased lymphatic vessel density | Impaired VEGFC/VEGFR3 axis signaling | [194,195,196,197,198,199,200] |
| Allograft Transplant Rejection | Clinical Manifestations | Histopathologic Findings | Proposed VEGFR3 Related Molecular Pathology | Selected References |
| Corneal transplant rejection | Progressive end-organ dysfunction, corneal edema, anterior chamber inflammation, increased intraocular pressure | Increased pathologic lymphangiogenesis | Increased VEGFR3 activation; galectin-8-mediated integrin- podoplanin-VEGFR3 crosstalk | [162,163,166,167,168] |
| Renal allograft rejection | Malaise, fever, oliguria, graft pain or tenderness, progressive end-organ dysfunction | Endothelial cell swelling and inflammation, dilated peri-tubular capillaries, thrombotic microangiopathy, subendothelial widening (acute), tubular hypertrophy, interstitial fibrosis, mononuclear inflammatory cell infiltrate (chronic) | Increased VEGFR3, VEGF-D, mTORC1 activation | [169,170] |
| Cardiac allograft rejection | Arterial luminal occlusion, diminished cardiac output, hypotension, mechanical abnormalities | CD4+ T cell, CD8+ T cell, ED1+ macrophage, myeloperoxidase +, neutrophil infiltration, perivascular infiltration, interstitial inflammatory cells Decreased pulmonary lymphatic vessel density, increased hyluronan, fragment accumulation, lymphocytic infiltrate | Increased VEGF-C/VEGFR3 activation | [123,171,172] |
| Pulmonary allograft rejection | Dyspnea, cough, sputum production, respiratory distress (acute) | Insufficient VEGF-C/VEGFR3 activation | [174] | |
| Obesity/metabolic syndrome | Hypertension, hyperglycemia, visceral adiposity, dyslipidemia | Leaky lymphatics, impaired lymph drainage, lymphatic fluid stasis | Prox-1 mutations, increased adipose tissue and serum VEGF-C levels, reduced VEGFR3-AKT-eNOS activation, impaired LEC CD36 activity | [141,142,143,144,150,154,157,158] |
| Secondary/Acquired Lymphedema | Clinical Manifestations | Histopathologic Findings | Proposed VEGFR3-Related Molecular Pathology | Selected References |
| Breast-cancer-related lymphedema (BCRL) | Subcutaneous lymphatic fluid stasis and accumulation, fibroadipose skin deposition, dermal thickening | CD4+ T cell infiltration, fibrosis, immature hyperplastic, leaky lymphatics | Prox-1, VEGF-C, Neuropilin-2 mutations, elevated tissue and systemic VEGF-C expression, SHIP2 mutation, epigenetic microRNA alterations | [90,91,92,93,94,96,97,98,207] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kuonqui, K.; Campbell, A.-C.; Sarker, A.; Roberts, A.; Pollack, B.L.; Park, H.J.; Shin, J.; Brown, S.; Mehrara, B.J.; Kataru, R.P. Dysregulation of Lymphatic Endothelial VEGFR3 Signaling in Disease. Cells 2024, 13, 68. https://doi.org/10.3390/cells13010068
Kuonqui K, Campbell A-C, Sarker A, Roberts A, Pollack BL, Park HJ, Shin J, Brown S, Mehrara BJ, Kataru RP. Dysregulation of Lymphatic Endothelial VEGFR3 Signaling in Disease. Cells. 2024; 13(1):68. https://doi.org/10.3390/cells13010068
Chicago/Turabian StyleKuonqui, Kevin, Adana-Christine Campbell, Ananta Sarker, Arielle Roberts, Bracha L. Pollack, Hyeung Ju Park, Jinyeon Shin, Stav Brown, Babak J. Mehrara, and Raghu P. Kataru. 2024. "Dysregulation of Lymphatic Endothelial VEGFR3 Signaling in Disease" Cells 13, no. 1: 68. https://doi.org/10.3390/cells13010068
APA StyleKuonqui, K., Campbell, A. -C., Sarker, A., Roberts, A., Pollack, B. L., Park, H. J., Shin, J., Brown, S., Mehrara, B. J., & Kataru, R. P. (2024). Dysregulation of Lymphatic Endothelial VEGFR3 Signaling in Disease. Cells, 13(1), 68. https://doi.org/10.3390/cells13010068