
Azithromycin Augments Bacterial Uptake and Anti-Inflammatory Macrophage Polarization in Cystic Fibrosis

 , , ,
, , ,
Abstract
:
1. Introduction
2. Methods
2.1. Study Participants
2.2. Ex Vivo Macrophage Differentiation and Polarization
2.3. AZM Cytotoxicity
2.4. P. aeruginosa Uptake and Killing
2.5. Lysosome Staining
2.6. Western Blot
2.7. ERK1/2 Inhibition
2.8. Flow Cytometric Analysis of Cell Surface Receptors and M1/M2 Markers
2.9. Phagocytosis and Endocytosis
2.10. Cytokine Quantification
2.11. Statistical Analysis
3. Results
3.1. Azithromycin Cytotoxicity
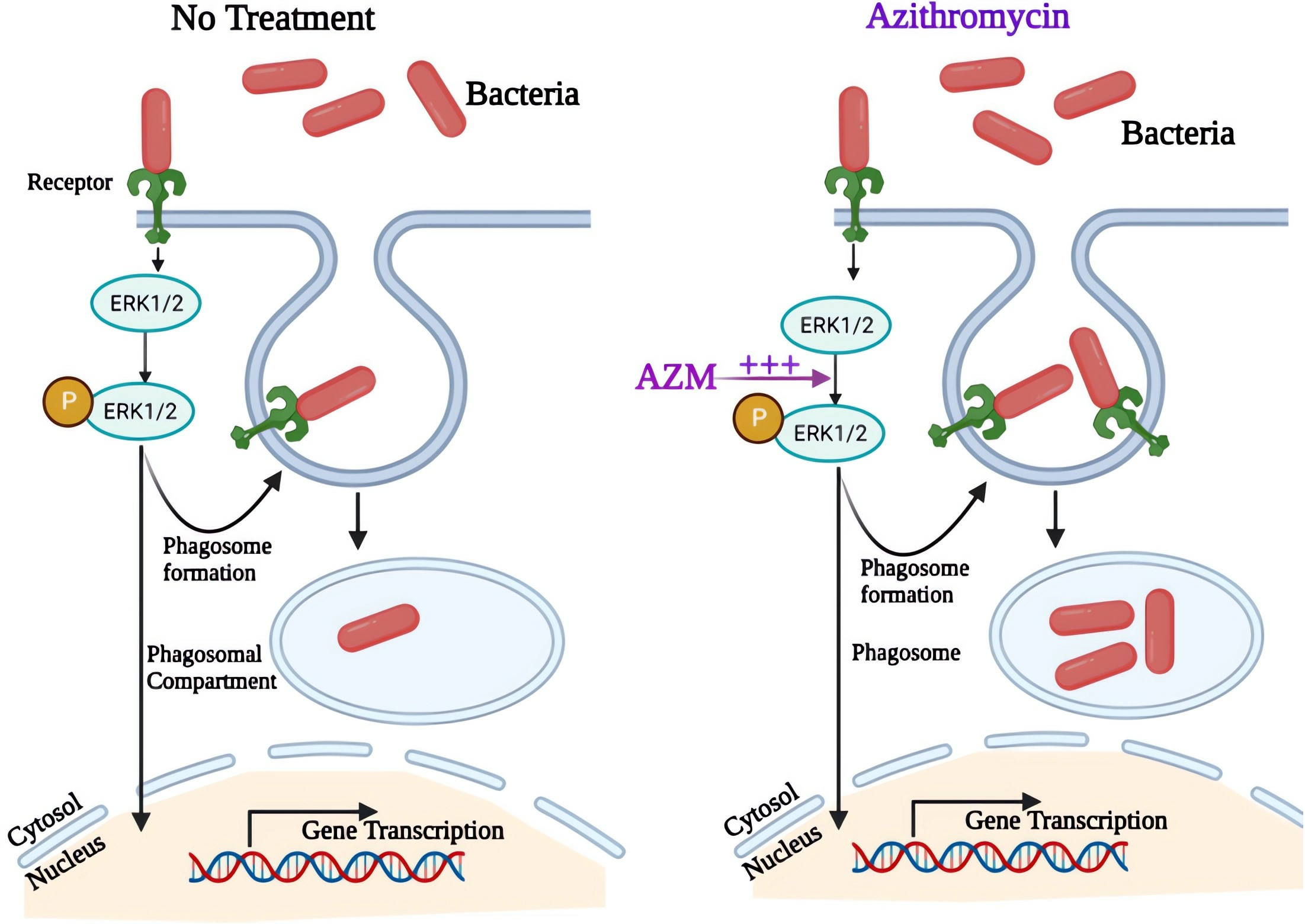
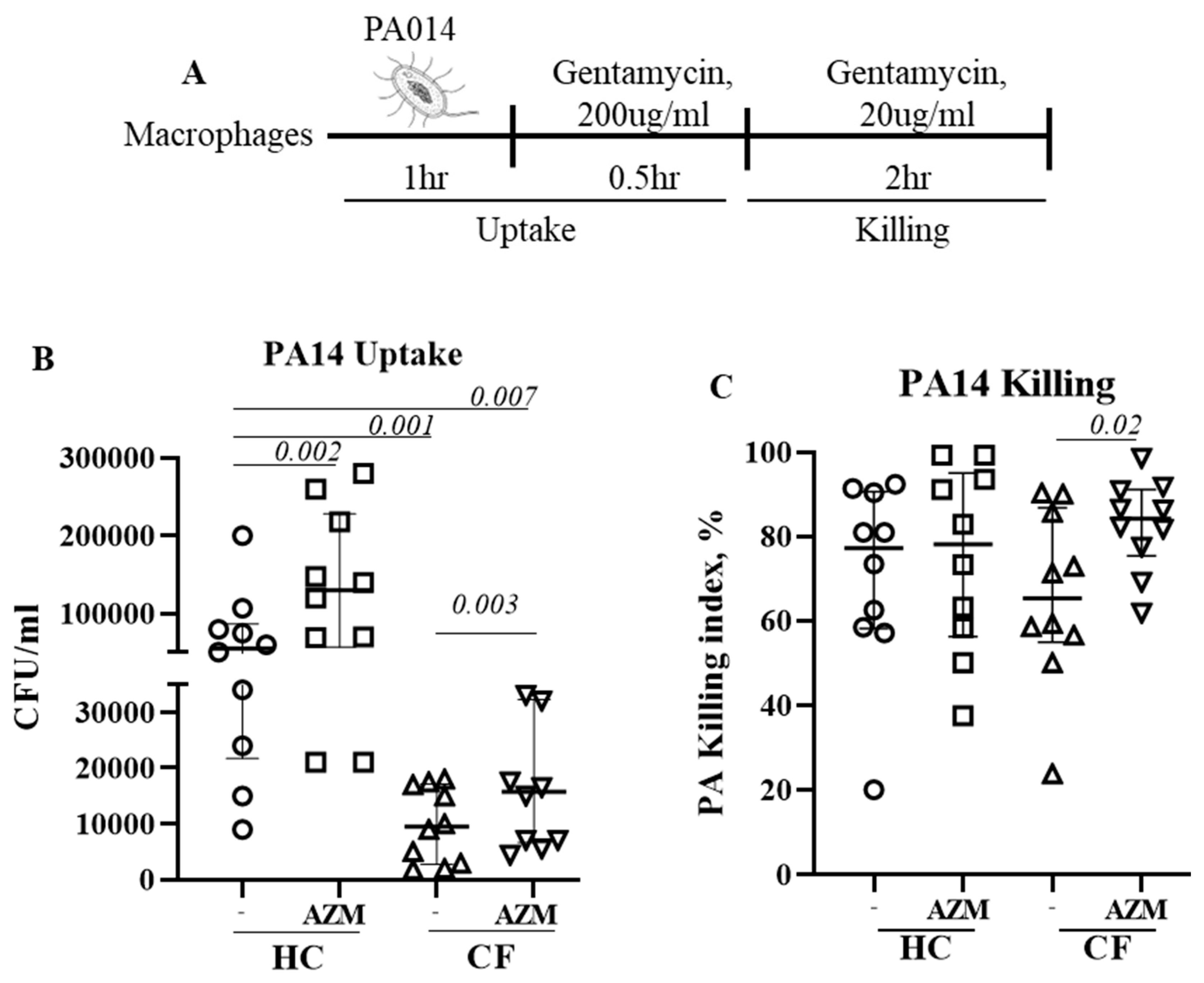
3.2. AZM Increases the Uptake and Killing of P. aeruginosa by CF Macrophages
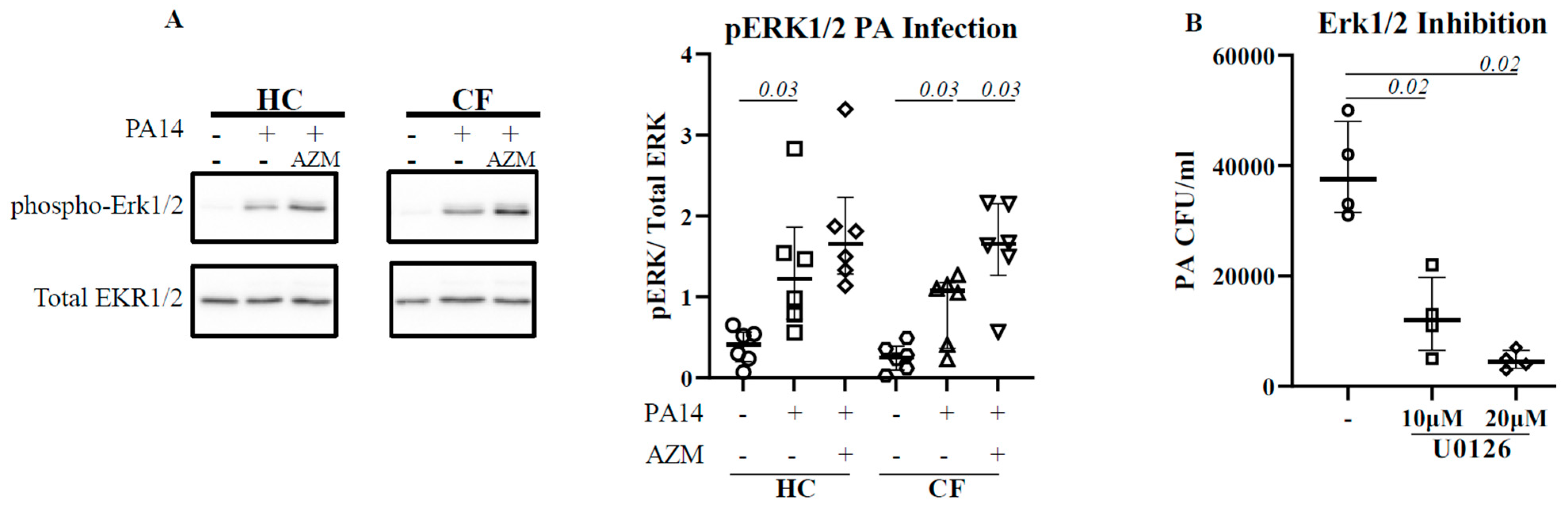
3.3. ERK1/2 Activation Is Pivotal for P. aeruginosa Uptake
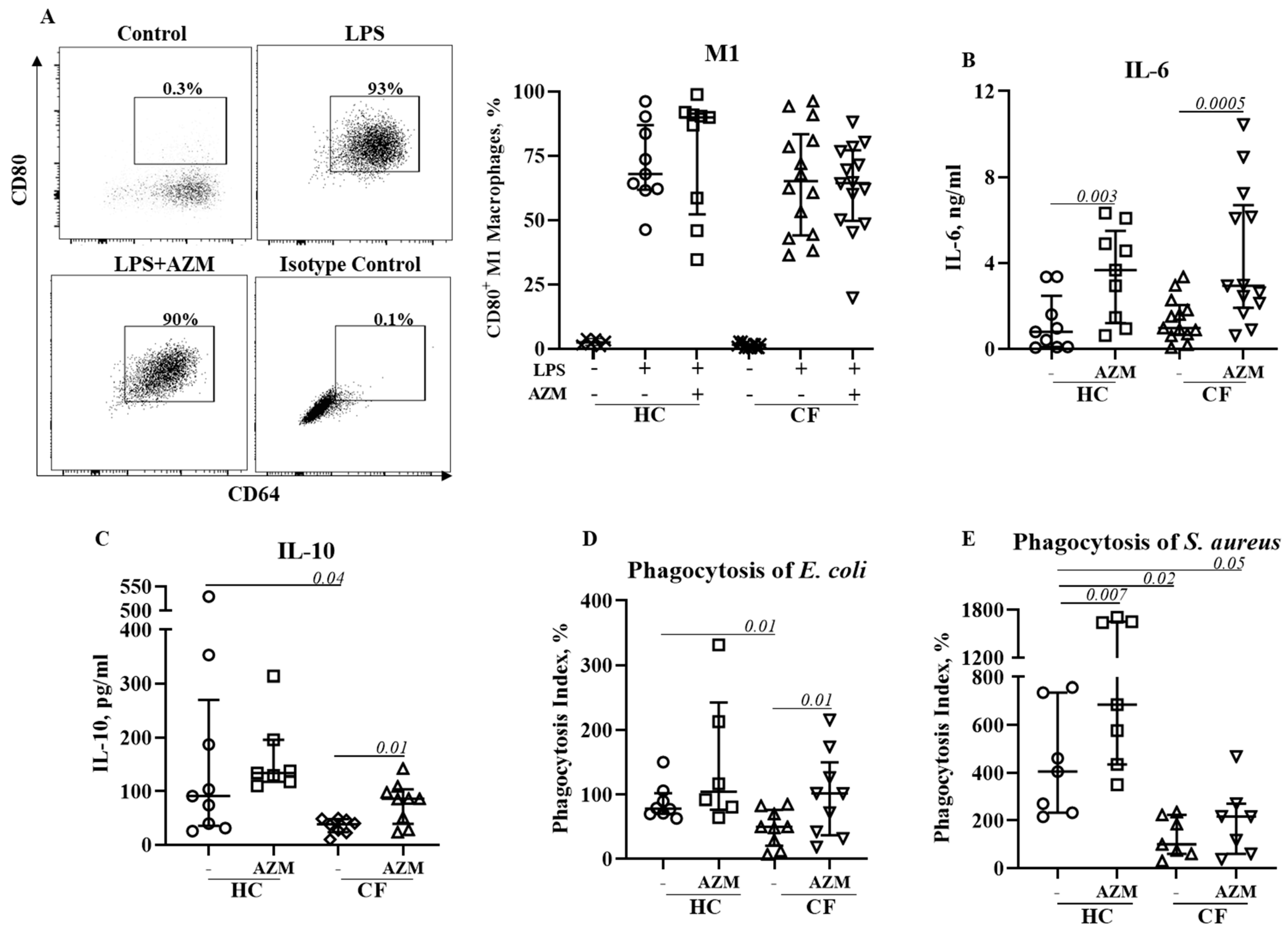
3.4. AZM Neither Reduced Pro-Inflammatory (M1) Macrophage Polarization nor Pro-Inflammatory Cytokine Secretion
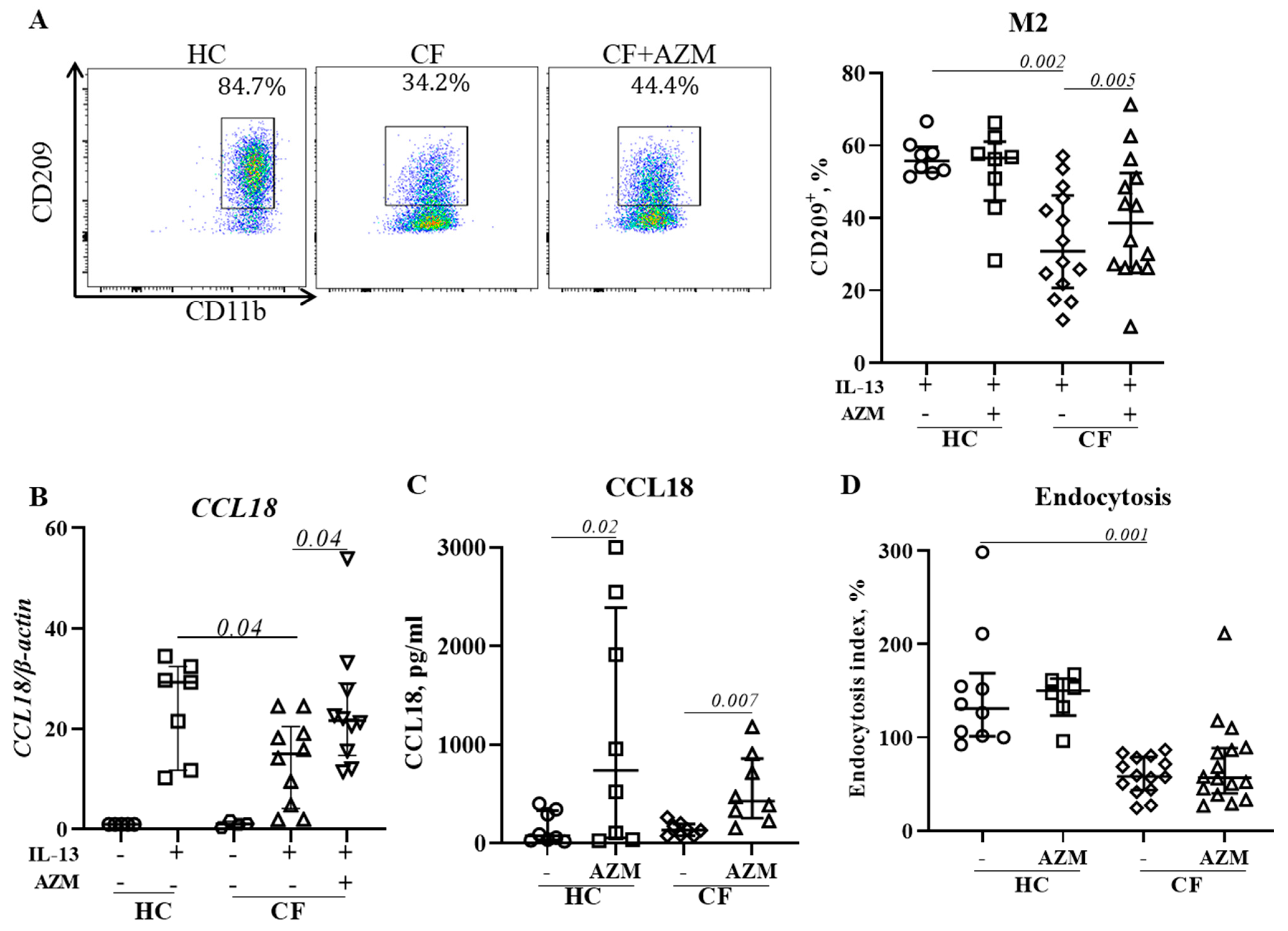
3.5. AZM Promotes Anti-Inflammatory (M2) Macrophage Polarization in CF
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Stick, S.M.; Foti, A.; Ware, R.S.; Tiddens, H.; Clements, B.S.; Armstrong, D.S.; Selvadurai, H.; Tai, A.; Cooper, P.J.; Byrnes, C.A.; et al. The effect of azithromycin on structural lung disease in infants with cystic fibrosis (COMBAT CF): A phase 3, randomised, double-blind, placebo-controlled clinical trial. Lancet Respir. Med. 2022, 10, 776–784. [Google Scholar] [CrossRef]
- Equi, A.; Balfour-Lynn, I.M.; Bush, A.; Rosenthal, M. Long term azithromycin in children with cystic fibrosis: A randomised, placebo-controlled crossover trial. Lancet 2002, 360, 978–984. [Google Scholar] [CrossRef]
- Clement, A.; Tamalet, A.; Leroux, E.; Ravilly, S.; Fauroux, B.; Jais, J.P. Long term effects of azithromycin in patients with cystic fibrosis: A double blind, placebo controlled trial. Thorax 2006, 61, 895–902. [Google Scholar] [CrossRef]
- Zarogoulidis, P.; Papanas, N.; Kioumis, I.; Chatzaki, E.; Maltezos, E.; Zarogoulidis, K. Macrolides: From in vitro anti-inflammatory and immunomodulatory properties to clinical practice in respiratory diseases. Eur. J. Clin. Pharmacol. 2012, 68, 479–503. [Google Scholar] [CrossRef]
- Meyer, M.; Huaux, F.; Gavilanes, X.; van den Brule, S.; Lebecque, P.; Lo Re, S.; Lison, D.; Scholte, B.; Wallemacq, P.; Leal, T. Azithromycin reduces exaggerated cytokine production by M1 alveolar macrophages in cystic fibrosis. Am. J. Respir. Cell Mol. Biol. 2009, 41, 590–602. [Google Scholar] [CrossRef]
- Hodge, S.; Reynolds, P.N. Low-dose azithromycin improves phagocytosis of bacteria by both alveolar and monocyte-derived macrophages in chronic obstructive pulmonary disease subjects. Respirology 2012, 17, 802–807. [Google Scholar] [CrossRef]
- Schupp, J.C.; Khanal, S.; Gomez, J.L.; Sauler, M.; Adams, T.S.; Chupp, G.L.; Poli, S.; Zhao, Y.; Montgomery, R.R.; Rosas, I.O.; et al. Single-Cell Transcriptional Archetypes of Airway Inflammation in Cystic Fibrosis. Am. J. Respir. Crit. Care Med. 2020, 202, 1419–1429. [Google Scholar] [CrossRef]
- Xue, J.; Schmidt, S.V.; Sander, J.; Draffehn, A.; Krebs, W.; Quester, I.; De Nardo, D.; Gohel, T.D.; Emde, M.; Schmidleithner, L.; et al. Transcriptome-Based Network Analysis Reveals a Spectrum Model of Human Macrophage Activation. Immunity 2014, 40, 274–288. [Google Scholar] [CrossRef]
- Tarique, A.A.; Logan, J.; Thomas, E.; Holt, P.G.; Sly, P.D.; Fantino, E. Phenotypic, functional, and plasticity features of classical and alternatively activated human macrophages. Am. J. Respir. Cell Mol. Biol. 2015, 53, 676–688. [Google Scholar] [CrossRef]
- Tarique, A.A.; Sly, P.D.; Holt, P.G.; Bosco, A.; Ware, R.S.; Logan, J.; Bell, S.C.; Wainwright, C.E.; Fantino, E. CFTR-dependent defect in alternatively-activated macrophages in cystic fibrosis. J. Cyst. Fibros. 2017, 16, 475–482. [Google Scholar] [CrossRef]
- Hodge, S.; Tran, H.B.; Hamon, R.; Roscioli, E.; Hodge, G.; Jersmann, H.; Ween, M.; Reynolds, P.N.; Yeung, A.; Treiberg, J.; et al. Nonantibiotic macrolides restore airway macrophage phagocytic function with potential anti-inflammatory effects in chronic lung diseases. Am. J. Physiol. Lung Cell. Mol. Physiol. 2017, 312, L678–L687. [Google Scholar] [CrossRef]
- Baldwin, D.R.; Wise, R.; Andrews, J.; Ashby, J.; Honeybourne, D. Azithromycin concentrations at the sites of pulmonary infection. Eur. Respir. J. 1990, 3, 886–890. [Google Scholar] [CrossRef]
- Di, A.; Brown, M.E.; Deriy, L.V.; Li, C.; Szeto, F.L.; Chen, Y.; Huang, P.; Tong, J.; Naren, A.P.; Bindokas, V.; et al. CFTR regulates phagosome acidification in macrophages and alters bactericidal activity. Nat. Cell Biol. 2006, 8, 933–944. [Google Scholar] [CrossRef]
- Botelho, R.J.; Harrison, R.E.; Stone, J.C.; Hancock, J.F.; Philips, M.R.; Jongstra-Bilen, J.; Mason, D.; Plumb, J.; Gold, M.R.; Grinstein, S. Localized diacylglycerol-dependent stimulation of Ras and Rap1 during phagocytosis. J. Biol. Chem. 2009, 284, 28522–28532. [Google Scholar] [CrossRef]
- Xin, C.; Kim, J.; Quan, H.; Yin, M.; Jeong, S.; Choi, J.I.; Jang, E.-A.; Lee, C.-H.; Kim, D.-H.; Bae, H.-B. Ginsenoside Rg3 promotes Fc gamma receptor-mediated phagocytosis of bacteria by macrophages via an extracellular signal-regulated kinase 1/2 and p38 mitogen-activated protein kinase-dependent mechanism. Int. Immunopharmacol. 2019, 77, 105945. [Google Scholar] [CrossRef]
- Farias, R.; Rousseau, S. The TAK1-->IKKbeta-->TPL2-->MKK1/MKK2 Signaling Cascade Regulates IL-33 Expression in Cystic Fibrosis Airway Epithelial Cells Following Infection by Pseudomonas aeruginosa. Front. Cell Dev. Biol. 2015, 3, 87. [Google Scholar]
- Sly, P.D.; Gangell, C.L.; Chen, L.; Ware, R.; Ranganathan, S.; Mott, L.S.; Murray, C.P.; Stick, S. Risk factors for bronchiectasis in children with cystic fibrosis. N. Engl. J. Med. 2013, 368, 1963–1970. [Google Scholar] [CrossRef]
- Lagasse, H.A.; Anidi, I.U.; Craig, J.M.; Limjunyawong, N.; Poupore, A.K.; Mitzner, W.; Scott, A.L. Recruited monocytes modulate malaria-induced lung injury through CD36-mediated clearance of sequestered infected erythrocytes. J. Leukoc. Biol. 2016, 99, 659–671. [Google Scholar] [CrossRef]
- Florentin, J.; Coppin, E.; Vasamsetti, S.B.; Zhao, J.; Tai, Y.Y.; Tang, Y.; Zhang, Y.; Watson, A.; Sembrat, J.; Rojas, M.; et al. Inflammatory Macrophage Expansion in Pulmonary Hypertension Depends upon Mobilization of Blood-Borne Monocytes. J. Immunol. 2018, 200, 3612–3625. [Google Scholar] [CrossRef]
- Shin, O.S.; Miller, L.S.; Modlin, R.L.; Akira, S.; Uematsu, S.; Hu, L.T. Downstream signals for MyD88-mediated phagocytosis of Borrelia burgdorferi can be initiated by TRIF and are dependent on PI3K. J. Immunol. 2009, 183, 491–498. [Google Scholar] [CrossRef]
- Parnham, M.J.; Haber, V.E.; Giamarellos-Bourboulis, E.J.; Perletti, G.; Verleden, G.M.; Vos, R. Azithromycin: Mechanisms of action and their relevance for clinical applications. Pharmacol. Ther. 2014, 143, 225–245. [Google Scholar] [CrossRef]
- Vrančić, M.; Banjanac, M.; Nujić, K.; Bosnar, M.; Murati, T.; Munić, V.; Polančec, D.S.; Belamarić, D.; Parnham, M.; Haber, V.E. Azithromycin distinctively modulates classical activation of human monocytes in vitro. Br. J. Pharmacol. 2012, 165, 1348–1360. [Google Scholar] [CrossRef]
- Haydar, D.; Cory, T.J.; Birket, S.E.; Murphy, B.S.; Pennypacker, K.R.; Sinai, A.P.; Feola, D.J. Azithromycin Polarizes Macrophages to an M2 Phenotype via Inhibition of the STAT1 and NF-kappaB Signaling Pathways. J. Immunol. 2019, 203, 1021–1030. [Google Scholar] [CrossRef]
- Bouhamdan, M.; Bauerfeld, C.; Talreja, J.; Beuret, L.; Charron, J.; Samavati, L. MEK1 dependent and independent ERK activation regulates IL-10 and IL-12 production in bone marrow derived macrophages. Cell. Signal. 2015, 27, 2068–2076. [Google Scholar] [CrossRef]
- Hoetzenecker, W.; Echtenacher, B.; Guenova, E.; Hoetzenecker, K.; Woelbing, F.; Brück, J.; Teske, A.; Valtcheva, N.; Fuchs, K.; Kneilling, M.; et al. ROS-induced ATF3 causes susceptibility to secondary infections during sepsis-associated immunosuppression. Nat. Med. 2011, 18, 128–134. [Google Scholar] [CrossRef]
- Akoumianaki, T.; Vaporidi, K.; Diamantaki, E.; Pène, F.; Beau, R.; Gresnigt, M.S.; Gkountzinopulou, M.; Venichaki, M.; Drakos, E.; El-Benna, J.; et al. Uncoupling of IL-6 signaling and LC3-associated phagocytosis drives immunoparalysis during sepsis. Cell Host Microbe 2021, 29, 1277–1293.e6. [Google Scholar] [CrossRef]
- Pechkovsky, D.V.; Prasse, A.; Kollert, F.; Engel, K.M.; Dentler, J.; Luttmann, W.; Friedrich, K.; Müller-Quernheim, J.; Zissel, G. Alternatively activated alveolar macrophages in pulmonary fibrosis-mediator production and intracellular signal transduction. Clin. Immunol. 2010, 137, 89–101. [Google Scholar] [CrossRef]
- Tyteca, D.; Van Der Smissen, P.; Mettlen, M.; Van Bambeke, F.; Tulkens, P.M.; Mingeot-Leclercq, M.P.; Courtoy, P.J. Azithromycin, a lysosomotropic antibiotic, has distinct effects on fluid-phase and receptor-mediated endocytosis, but does not impair phagocytosis in J774 macrophages. Exp. Cell Res. 2002, 281, 86–100. [Google Scholar] [CrossRef]
- Saint-Criq, V.; Rebeyrol, C.; Ruffin, M.; Roque, T.; Guillot, L.; Jacquot, J.; Clement, A.; Tabary, O. Restoration of chloride efflux by azithromycin in airway epithelial cells of cystic fibrosis patients. Antimicrob. Agents Chemother. 2011, 55, 1792–1793. [Google Scholar] [CrossRef]
- Misharin, A.V.; Morales-Nebreda, L.; Reyfman, P.A.; Cuda, C.M.; Walter, J.M.; McQuattie-Pimentel, A.C.; Chen, C.-I.; Anekalla, K.R.; Joshi, N.; Williams, K.J.N.; et al. Monocyte-derived alveolar macrophages drive lung fibrosis and persist in the lung over the life span. J. Exp. Med. 2017, 214, 2387–2404. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Adults | Children | |
|---|---|---|
| n | 13 | 10 |
| Age, years (range) | 23–55 | 8–12 |
| Gender, female | 4 (30.8%) | 5 (50%) |
| Genotype, n (%) | ||
| Phe508del homozygous | 7 (54%) | 5 (50%) |
| Phe508del heterozygous | 6 (46%) | 5 (50%) |
| Lung function *, (mean ± SD) | ||
| FEV1, L | 1.8 ± 0.85 | 1.8 ± 0.36 * |
| FVC, L | 3.5 ± 1.36 | 2.0 ± 0.4 * |
| Pseudomonas aeruginosa infection status, n (%) | ||
| Chronic | 12 (92.3%) | 6 (50%) |
| Intermittent | 1 (7.7%) | 2 (25%) |
| Never | 0 | 2 (25%) |
| Antibody | Cat# | Supplier | Used in |
|---|---|---|---|
| TLR4 AF488 | #539917 | eBioscience | Flow cytometry |
| IL-13Rα1 APC | #360406 | BioLegend | Flow cytometry |
| IL-4Rα PE Cy 7 | #355008 | BioLegend | Flow cytometry |
| CD80 PE | #305208 | BioLegend | Flow cytometry |
| CD64 PE Cy7 | #305022 | BioLegend | Flow cytometry |
| CD209 BV421 | #330117 | BD | Flow cytometry |
| CD11b APC | #550019 | BD | Flow cytometry |
| 7-AAD | #559925 | BD | Flow cytometry |
| GAPDH | #2118 | CST | Western blot |
| Phospho-p65 | #3031 | CST | Western blot |
| Phospho-ERK1/2 | #9272 | CST | Western blot |
| Total ERK1/2 | #9102 | CST | Western blot |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tarique, A.A.; Tuladhar, N.; Kelk, D.; Begum, N.; Lucas, R.M.; Luo, L.; Stow, J.L.; Wainwright, C.E.; Bell, S.C.; Sly, P.D.; et al. Azithromycin Augments Bacterial Uptake and Anti-Inflammatory Macrophage Polarization in Cystic Fibrosis. Cells 2024, 13, 166. https://doi.org/10.3390/cells13020166
Tarique AA, Tuladhar N, Kelk D, Begum N, Lucas RM, Luo L, Stow JL, Wainwright CE, Bell SC, Sly PD, et al. Azithromycin Augments Bacterial Uptake and Anti-Inflammatory Macrophage Polarization in Cystic Fibrosis. Cells. 2024; 13(2):166. https://doi.org/10.3390/cells13020166
Chicago/Turabian StyleTarique, Abdullah A., Neeraj Tuladhar, Dean Kelk, Nelufa Begum, Richard M. Lucas, Lin Luo, Jennifer L. Stow, Claire E. Wainwright, Scott C. Bell, Peter D. Sly, and et al. 2024. "Azithromycin Augments Bacterial Uptake and Anti-Inflammatory Macrophage Polarization in Cystic Fibrosis" Cells 13, no. 2: 166. https://doi.org/10.3390/cells13020166
APA StyleTarique, A. A., Tuladhar, N., Kelk, D., Begum, N., Lucas, R. M., Luo, L., Stow, J. L., Wainwright, C. E., Bell, S. C., Sly, P. D., & Fantino, E. (2024). Azithromycin Augments Bacterial Uptake and Anti-Inflammatory Macrophage Polarization in Cystic Fibrosis. Cells, 13(2), 166. https://doi.org/10.3390/cells13020166